
ABSTRACT
Cages prepared using irreversible reactions in the cage-forming step typically give relatively low yields, but are more robust than cages formed through reversible bond formation reactions. The irreversible reactions are also well-suited to preparing functionalised and/or low symmetry species that are difficult to prepare using self – assembly. In this work, we prepare two hexanitrile-functionalised cages and a hexabromo-functionalised cage using Eglinton coupling reactions, with both hexanitrile-functionalised cages structurally characterised by X-ray crystallography. Yields for the functionalised cages (3–28%) were significantly lower than those previously reported for analogous unfunctionalised cages. We also prepared three cages using Sonogashira coupling reactions, including a tri-nitrile functionalised lower symmetry cage. Unfortunately, these reactions also suffered from low yields (3–11%), with the reaction to give the functionalised cage particularly low-yielding (3%). While the low yields meant we were unable to obtain sufficient amounts of compounds to measure their gas sorption properties, we were able to demonstrate that a hexanitrile cage could be post-synthetically modified to give a hexa-amidinium species.
GRAPHICAL ABSTRACT
Introduction
Cage molecules are three-dimensional molecules containing internal cavities, and are currently the subject of intense research interest [Citation1]. The internal cavities can be used for solution phase host – guest chemistry or in homogeneous catalysis where host – guest binding forces substrates into unusual conformations or proximity to reactive species [Citation2,Citation3]. Another possible application of cages is as porous solid state materials [Citation1,Citation4], with the large internal cavities offering the potential for very low density materials containing large voids [Citation5–7]. In practice, preparing permanently porous cages is not trivial, as the cages need to pack in such away that their cavities align to give channels. [Citation8–11]
The vast majority of cages are prepared through either metal – ligand coordination bonds [Citation2,Citation3,Citation12], or reversible covalent bond-forming reactions such as imine or boronate ester condensation [Citation6,Citation7,Citation13–19]. Both of these approaches have the advantage that the cage-forming reaction is reversible allowing for an error-correction mechanism and typically leading to high yields. There are some disadvantages to these approaches however: firstly, the cages are often not particularly stable [Citation20]. This is a disadvantage in itself, and also limits the subsequent chemistry that can be conducted on the cage, although notable examples of post-synthetic modification of cages assembled using reversible bonds have been reported recently [Citation21–24]. Secondly, the reversibility of the reactions and use of self – assembly under thermodynamic control mean it is very difficult to produce low symmetry species through this approach. While sterling efforts to produce low symmetry metal – organic cages have been reported [Citation25–29], this still remains a considerable challenge.
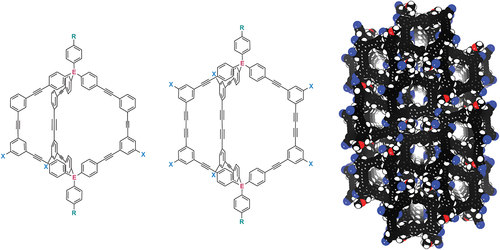
Cages prepared using irreversible covalent bond-forming reactions are fundamentally more difficult to prepare: the reactions to form the cages do not have the capacity to error correct and so typically give low yields. Furthermore, the precursors used in the cage-forming reaction often require multi-step syntheses. In 2007, Zhang and Chen reported that organic cages could be prepared from the Eglinton coupling reaction of two triptycene-based tris-alkyne ‘half-cage’ compounds [Citation30]. Subsequently, the Sumby and Doonan groups have reported a number of robust, large organic cages formed via Eglinton coupling of two tris-alkyne half-cage precursors based on tetraphenylmethane scaffolds () [Citation31–33]. Some of these, and other alkyne-based cages [Citation34–36] are permanently porous. However, others are not, which arises from the different packing arrangements of the cages in the solid state.
Scheme 1. Family of butadiyne cages reported by Sumby, Doonan and co-workers [Citation31–33], and by us [Citation37].
![Scheme 1. Family of butadiyne cages reported by Sumby, Doonan and co-workers [Citation31–33], and by us [Citation37].](/cms/asset/5bd16df8-1e76-4b11-bdab-78617e29df94/gsch_a_2324822_sch0001_oc.jpg)
Notably, Sumby and Doonan showed that incorporating bromine substituents on the tetraphenylmethane groups did not affect the yield of the cage formation reaction (20%) [Citation33], however introducing endohedral nitrogen atoms resulted in a very low yield (4%) of cage when the tris-alkyne half-cages were coupled directly (higher yields were obtained by a multi-step coupling route involving coupling of a partially-protected tris-alkyne) [Citation33]. Inspired by their work, we recently reported the related porous cage SiH≡⋅≡SiH (), and showed that it could be reacted with Co2(CO)8 to give the Co24(CO)72 adduct of the cage in one-pot, although this organometallic complex had poor stability [Citation37]. We were able to obtain yields of up to 64% for the cage-formation reaction, although found that the reactions scaled poorly, with yields decreasing rapidly on larger scales.
In the current work, we investigate the use of irreversible Eglinton [Citation38] and Sonogashira reactions to prepare functionalised and low symmetry organic cages. Incorporating additional functionality to the cages may allow greater control of crystallisation and potentially better control of porosity, while low symmetry cages may have enhanced guest binding properties. While both of these approaches were hampered by low yields, a small family of cages was produced successfully.
Results
Synthesis of cages
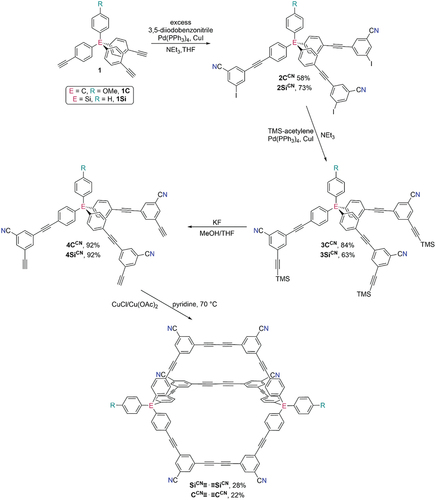
Compound numbering: To keep compound numbering as simple as possible the following convention has been used. The building blocks used in the synthesis of the half-cages have been numbered 1–4, where compounds 2 and 4 represent the tripodal tris-iodide and tris-alkyne half-cages, respectively (). A C or Si indicates whether the atom at the centre of the tetraphenyl group is carbon or silicon. In all cases, the C-containing building blocks have methoxy groups on the phenyl group while the Si-containing building blocks do not (for reasons of synthetic convenience). A superscript annotation describes the functional groups attached at the ‘shoulders’ of the half-cages. So for example, 4SiCN refers to a silicon-centred tris-alkyne with nitrile substituents. Cages are referred to without numbers but indicating whether they contain di-alkyne or mono-alkyne linkages between the two half-cages (i.e. cages formed using Eglinton or Sonogashira reactions, respectively). For example, SiCN≡·≡SiCN refers to the cage formed from the Eglinton coupling of two molecules of 4SiCN.
Scheme 2. Synthesis of tris-iodide half-cages 2, tris-alkyne half-cages 4, and hexanitrile-functionalised cages CCN≡·≡CCN and SiCN≡·≡SiCN.
Synthesis of hexanitrile cages using Eglinton reactions
We targeted nitrile-containing cages due to the synthetic versatility of this functionality. Nitrile groups are competent ligands and have been used to prepare numerous coordination polymers [Citation39,Citation40], and can be readily transformed into carboxylic acid/carboxylate or amidinium groups. Carboxylate groups are also good ligands [Citation41], and both the carboxylic acid/carboxylate and amidinium functionalities are useful for preparing hydrogen bonded frameworks [Citation42–45]. We prepared the two new half-cages 4CCN and 4SiCN by reaction of the appropriate tetraphenyl tris-alkyne with excess 3,5-diiodobenzonitrile, followed by reaction with TMS-acetylene and deprotection. In both cases, these reaction sequences proceeded smoothly. It was important to use KF in methanol to deprotect the TMS groups in 3CCN/3SiCN as more conventional K2CO3 in methanol appeared to cause partial degradation of the nitrile groups.
Having prepared the nitrile-functionalised half-cages 4CCN and 4SiCN, we next studied cage formation using Eglinton coupling reactions. We have previously shown that cages can be prepared from the unfunctionalised half-cage 4SiH in yields of up to 64%. Initial investigations of this coupling reaction indicated that yields were significantly lower when the nitrile-functionalised precursors were used. No unreacted starting material was observed, suggesting the lower yield is not due difficulties in promoting the reaction. Indeed, it is unclear why addition of nitrile substituents caused a significant decrease in the yields of cage formation, although we note that a similar result was observed by Sumby and Doonan when they synthesised related cages containing endohedral pyridyl substituents [Citation33].
We investigated various conditions to optimise this reaction focussing on the reaction of 4SiCN to give SiCN≡·≡SiCN (see Supporting Information for details of optimisation). We found that the best yield was obtained by adding a solution of 4SiCN to a large excess of CuCl/Cu(OAc)2 at 70°C, which resulted in a 28% yield of the hexanitrile-functionalised cage after purification by preparative TLC when the reaction was conducted on a small scale (tens of mgs). We then used these conditions to prepare the carbon-centred cage CCN≡·≡CCN, which was isolated in 22% yield. Both cages were characterised by 1H and 13C{1H} NMR spectroscopy and high resolution ESI mass spectrometry (HRESI-MS). We note that purification conditions other than preparative TLC would be necessary if the cages were prepared on larger scales.
Synthesis of hexabromo cage SiBr≡·≡SiBr using Eglinton reactions: We also prepared an analogous half-cage containing bromo substituents in place of the nitrile substituents in 4SiCN, i.e. 4SiBr, via similar strategies. We attempted to synthesise the hexabromo-functionalised cage SiBr≡·≡SiBr from 4SiBr using an analogous coupling reaction with the hope that post-functionalisation reactions of these bromo groups could be used to generate a family of porous cages. Unfortunately, only a very low yield of SiBr≡·≡SiBr (3%) could be isolated from these reactions. While it was possible to characterise this product by1H NMR spectroscopy and HRESI-MS, the trace amounts of material were not sufficient to investigate any post-functionalisation reactions (the synthesis and characterisation of 4SiBr and SiBr≡·≡SiBr are detailed in the Supporting Information).
Synthesis of cages using Sonogashira reactions: We next investigated the use of Sonogashira reactions to prepare cages as these offer the potential to prepare low symmetry cages. Before attempting to prepare low symmetry cages, we investigated the formation of the symmetric cage SiH≡SiH from 2SiH [Citation37] and 4SiH [Citation37] () as these half-cages are simple to prepare. Initial investigations revealed that the reaction was low-yielding and so a range of reactions conditions were again trialled to improve the yield. After considerable optimisation (see Supporting Information), it was found that using Pd(PPh3)4 as catalyst without an added copper co-catalyst and conducting the reaction at elevated temperatures minimised the formation of insoluble by-products and allowed the isolation of pure SiH≡SiH, albeit in only 9% yield after purification by preparative TLC.
Scheme 3. Synthesis of cages using Sonogashira reactions.
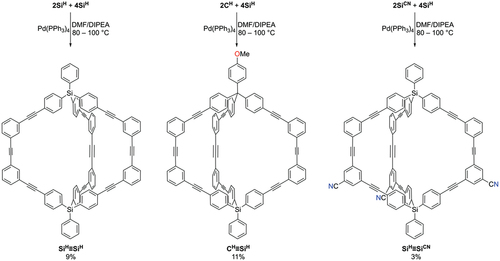
Having demonstrated that cages could be prepared using this Sonogashira reaction, we next turned attention to the synthesis of the lower symmetry cage CH≡SiH. Using the same conditions as those optimised when preparing SiH≡SiH, we were able to prepare CH≡SiH from 2CH [Citation31] and 4SiH [Citation37] in 11% yield, again after purification using preparative TLC. Both of these cages were characterised by1H and13C{1H} NMR spectroscopy as well as HRESI-MS.
We next investigated whether low symmetry cages containing nitrile functional groups could be prepared. To this end we reacted nitrile-functionalised tris-iodide half-cage 2SiCN with unfunctionalised tris-alkyne half-cage 4SiH. While we were able to isolate the desired cage SiH≡SiCN, and characterise it by1H NMR spectroscopy and HRESI-MS, unfortunately the yields for this reaction were very low (3%).
It is unclear why these yields are quite so low, and may be due to steric difficulties conducting Sonogashira reactions on the second and third ‘arms’ of the cage, i.e. that it is difficult for the partially-formed cage to distort to allow the catalyst to complete subsequent bond-forming reactions. We note that the geometries required for Sonogashira reactions to take place are likely to impose more strain on the cage than those for Eglinton cages, and that the resulting cages are smaller due to the presence of one alkyne group rather than two. This steric strain suggestion is consistent with the need to use higher temperatures than are typically required for Sonogashira reactions on aryl iodides. Oligomerisation/polymerisation would not face these steric difficulties and so would presumably occur rapidly.
An additional/alternative explanation is that while relatively large amounts of catalyst were used (60 mol%, i.e. 20 mol% per reaction site), this is considerably less than the copper catalyst used in the Eglinton reactions (30–40-fold excess) and so the reaction likely occurred more slowly resulting in an increased concentration of the half-cages in solution (i.e. not as highly dilute). It was not practical to use higher catalyst loadings for the Sonogashira reactions due to the cost of palladium reagents and the difficulty of separating the product from organic ligand groups. Adding nitrile functional groups seems to lower the yields of the Sonogashira reactions still further, as was observed with cages formed via Eglinton reactions.
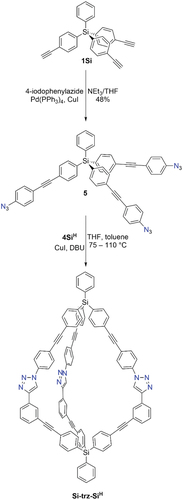
Synthesis of cages using CuAAC reactions: An alternative reaction that could give access to low symmetry cages is the copper(I)-catalysed azide alkyne coupling (CuAAC) reaction. Low level computational calculations (see Supporting Information) suggested a cage (Si-trz-SiH) that would result from the reaction of tris-alkyne 4SiH with the tris-azide 5 would be geometrically sensible. Tris-azide 5 was prepared from 1Si and 4-iodophenylazide, and CuAAC reactions of this compound with 4SiH were undertaken (). Several different catalyst, ligand and solvent combinations were investigated (see Supporting Information) but generally these resulted in either unreacted starting material or insoluble precipitates. When we used CuI in toluene/THF at elevated temperatures, Si-trz-SiH appeared to form. While the1H NMR spectrum of this compound was broad, evidence for product formation was provided by HRESI-MS. However, due to the cage’s poor solubility we were unable to fully purify it (TLC indicated the presence of more than one compound, but we were unable to achieve separation due to streaking and co-elution).
Scheme 4. Synthesis of tris-azide 5 and tris-triazole cage Si-trz-SiH. The cage could not be fully purified, so an exact yield could not be calculated. However the yield of soluble unpurified material was 17%; based on1H NMR spectroscopic analysis, we estimate the yield of cage formation is approximately 10%.
X-ray crystal structures of cages
We were able to obtain single crystals of both of the hexanitrile cages SiCN≡·≡SiCN and CCN≡·≡CCN. Crystals of SiCN≡·≡SiCN were obtained by vapour diffusion of diethyl ether into a chloroform solution of the cage. Crystals of CCN≡·≡CCN were grown from two different solvent mixtures, either by vapour diffusion of pentane into a chloroform solution or by vapour diffusion of methanol into a solution of the cage in a mixture of THF/toluene. In all cases crystals were small and required the use of synchrotron radiation to collect data. Despite these challenges, the overall connectivity and packing of the cages can be determined unambiguously, and the structures reveal that all three cages show significant distortion from idealised high symmetry geometries ().
Figure 1. X-ray crystal structures of SiCN≡·≡SiCN, CCN≡·≡CCN crystallised from CHCl3/pentane, and CCN≡·≡CCN crystallised from THF/toluene/methanol. In each case packing diagrams with a space-filling representation of the structures are shown. PLATON-SQUEEZE was used in the refinement of all structures [Citation46].
![Figure 1. X-ray crystal structures of SiCN≡·≡SiCN, CCN≡·≡CCN crystallised from CHCl3/pentane, and CCN≡·≡CCN crystallised from THF/toluene/methanol. In each case packing diagrams with a space-filling representation of the structures are shown. PLATON-SQUEEZE was used in the refinement of all structures [Citation46].](/cms/asset/c3e0164c-34e7-4e4f-9565-647272b1c5f9/gsch_a_2324822_f0001_oc.jpg)
All three structures contain large amounts of diffuse electron density, which we attribute to poorly ordered solvent molecules located within the large cavities of the cages, as well as between the cages, which cannot pack efficiently. This electron density was included in the crystallographic refinement using PLATON-SQUEEZE [Citation46], which removed electron density from volumes corresponding to 55–59% of the unit cell volumes. Similar void volumes were calculated using Mercury [Citation47] to calculate contact surface areas (51–56%), however when solvent-accessible surface areas were calculated, there are distinct differences between the structures. These surface areas were calculated in Mercury using a probe radius of 1.82 Å (corresponding to the kinetic radius of nitrogen gas) and reveal that SiCN≡·≡SiCN and CCN≡·≡CCN crystallised from THF/toluene/methanol have solvent-accessible channels comprising 18 and 17% of their volume, respectively. In contrast, CCN≡·≡CCN crystallised from chloroform/pentane has solvent-accessible channels comprising 53% of the unit cell volume.
This illustrates the importance of crystal packing in obtaining high porosity cage structures, with the crystal structure of CCN≡·≡CCN crystallised from chloroform/pentane showing much better alignment of the cage pores than the other two structures. It is important to note that these are ‘virtual’ porosities, determined based solely on X-ray crystal structures. Unfortunately, due to the low yield of cage formation, we were unable to conduct gas sorption experiments to determine if the materials are permanently porous.
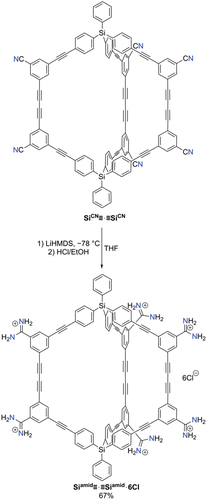
Post-synthetic modification of hexanitrile cage SiCN≡·≡SiCN: Various post-synthetic modifications of porous organic cages have been reported, in particular by Mastalerz and colleagues who have shown that a wide range of transformations can be carried out on imine-based cages [Citation21–23]. Our group has prepared numerous supramolecular frameworks using the hydrogen-bonded assembly of amidinium and carboxylate building blocks [Citation45,Citation48], and we were interested to see whether the nitrile groups of a hexa-nitrile cages could be converted to amidinium groups, for subsequent synthesis of hydrogen bonded cage networks. Adding a solution of LiHMDS in THF to a cooled solution of SiCN≡·≡SiCN in THF followed by work-up with ethanolic HCl gave the hexacationic cage Siamid≡·≡Siamid·6Cl in 67% yield after washing with water to remove salt by-products ().
Scheme 5. Post-synthetic modification of SiCN≡·≡SiCN to give hexa-cationic cage Siamid≡·≡Siamid·6Cl.
We attempted to crystallise this hexacationic cage with a range of dicarboxylates, tricarboxylates and tetracarboxylates in a wide variety of solvent systems (Siamid≡·≡Siamid 6+ was also converted to its tetraphenylborate salt to give solubility in a wider range of solvents). Unfortunately, we were unable to obtain singly crystalline products under any conditions, with addition of polycarboxylate anions to Siamid≡·≡Siamid 6+ typically resulting in rapid precipitation of amorphous solids, presumably arising from formation of hydrogen bonded networks.
Conclusions
Eglinton reactions were used to prepare two hexanitrile cages and a hexabromo cage. Yields for the nitrile-containing cages were slightly under half of those for the parent unsubstituted cages [Citation31,Citation37], while yields for the bromo-containing cages were lower by another order of magnitude. Sonogashira reactions were used to prepare three cages, one of which was functionalised with three nitrile groups. While yields for these cages were modest (3–11%), it was possible to prepare cages with lowered symmetry. We were able to obtain X-ray crystal structures of the hexanitrile cages, which showed they contain large solvent-filled voids, and in one case high virtual porosities, although due to the low amounts of material obtained, we could not study their gas sorption properties. One of the hexanitrile cages was post-synthetically modified to give a hexa-amidinium cage. This work shows that irreversible reactions can be used to give functionalised and low symmetry cages with very open structures, however work is needed to discover synthetic pathways to access useful quantities of cage.
Experimental
General remarks
Compounds 4-iodophenylazide [Citation49], 1C [Citation31], 1Si [Citation37], 2CH [Citation31], 2SiH [Citation37] and 4SiH [Citation37] were prepared as previously described. Compound 3,5-diiodobenzonitrile [Citation50] was prepared from 3,5-diiodobenzoic acid [Citation51], which was itself prepared from 3,5-diaminobenzoic acid. ‘Dry’ pyridine was dried over 3 Å molecular sieves before use. ‘Dry’ THF was distilled from sodium and then stored over 3 Å molecular sieves before use. Other compounds were bought from commercial suppliers and used as received. The preparation of 4SiBr and SiBr≡·≡SiBr are described in the Supporting Information. Details of instrumentation, NMR spectra, and details of optimisation of reactions to form cages are provided in the Supporting Information. Due to the (very) low yields obtained in several of the cage formation reactions, it was not possible to obtain useful13C{1H} NMR data for SiH≡SiCN and SiBr≡⋅≡SiBr. For some other cages, the small amount of material obtained and the large number of carbon atoms in very similar chemical environments mean that13C{1H} data are of limited quality.
Data for all three crystals were collected on the MX2beamline [Citation52] at the Australian Synchrotron. Raw frame data (including data reduction, interframe scaling and unit cell refinement) were processed using XDS [Citation53]. The structures were solved using Superflip [Citation54] and refined using full-matrix least-squares on F2 within the Crystals suite [Citation55]. More details are provided in the Supporting Information. Deposition Numbers 2304996–2304998 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre.
Synthesis of organic precursors
Tris-iodide 2CCN:3,5-Diiodobenzonitrile (0.82 g, 2.3 mmol), tris-alkyne 1C (0.10 g, 0.23 mmol), Pd(PPh3)4 (23 mg, 0.020 mmol) and CuI (3.8 mg, 0.020 mmol) were stirred overnight at room temperature in NEt3 (10 mL). The solvent was removed under vacuum and the solid was dissolved in dichloromethane (200 mL). The organic layer was washed with aqueous HCl(aq) (1.0 M, 50 mL) and then brine (50 mL). The organic fraction was dried over MgSO4 and the residue obtained after removal of solvent was purified on a silica column (1:1 DCM:pet. spirits) to give 2CCN as a yellow powder. Yield: 0.14 g (0.13 mmol, 58%).
1H NMR (CDCl3): δ 8.07 (t, J = 1.6 Hz, 3 H), 7.90 (t, J = 1.6 Hz, 3 H), 7.74 (t, J = 1.6 Hz, 3 H), 7.42 (d, J = 8.5 Hz, 6 H), 7.23 (d, J = 8.5 Hz, 6 H), 7.09 (d, J = 8.8 Hz, 2 H), 6.83 (d, J = 8.8 Hz, 2 H), 3.81 (s, 3 H). 13C{1H} NMR (CDCl3): δ 158.1, 147.5, 144.2, 139.7, 137.3, 134.0, 132.0, 131.4, 131.1, 126.5, 120.0, 116.6, 114.4, 113.5, 93.7, 92.9, 85.9, 64.6, 55.4. HRESI-MS (pos.): found 1125.9266, calculated for [C53H28N3OI3·Na]+ i.e. [M+Na]+ = 1125.9264 Da.
Tris-TMS-alkyne 3CCN: Compound 2CCN (30 mg, 20 μmol), trimethylsilylacetylene (20 μL, 0.10 mmol), Pd(PPh3)4 (3.0 mg, 0.0030 mmol) and CuI (1.0 mg, 0.0030 mmol) were stirred overnight at room temperature in NEt3 (3 mL). The solvent was removed under vacuum and the solid was dissolved in dichloromethane (75 mL). The organic layer was washed with HCl(aq) (1.0 M, 25 mL) and then brine (25 mL). The organic layer was dried over MgSO4 and the residue obtained after removal of solvent was purified on a silica column (3:2 DCM:pet. spirits) to give 3CCN as yellow powder. Yield: 18 mg (17 μmol, 84%).
1H NMR (CDCl3): δ 7.79 (t, J = 1.6 Hz, 3 H), 7.69 (t, J = 1.6 Hz, 3 H), 7.66 (t, J = 1.6 Hz, 3 H), 7.43 (d, J = 8.6 Hz, 6 H), 7.22 (d, J = 8.6 Hz, 6 H), 7.09 (d, J = 8.9 Hz, 2 H), 6.83 (d, J = 8.9 Hz, 2 H), 3.81 (s, 3 H), 0.26 (s, 27 H). 13C{1H} NMR (CDCl3): δ 158.2, 147.5, 138.8, 134.5, 134.4, 132.1, 131.5, 131.2, 125.4, 125.4, 120.3, 117.6, 113.5, 113.5, 101.7, 98.6, 92.2, 86.8, 64.6, 55.5, 0.0 (one expected peak not observed). HRESI-MS (pos.) found 1036.3553, calculated for [C68H55N3OSi3·Na]+, i.e. [M+Na]+ = 1036.3551 Da.
Tris-alkyne 4CCN:To a solution of 3CCN (0.14 g, 0.14 mmol) in 1:1 THF:MeOH (10 mL) KF (4.9 mg, 0.84 mmol) was added and stirred at room temperature for 3 hours under a nitrogen atmosphere. Solvent was removed in vacuo and the solid was dissolved in DCM (40 mL) and washed with water (2 × 30 mL) and brine (30 mL). The organic layer was dried over MgSO4 and concentrated to give 4CCN as a fine yellow powder. Yield: 96 mg, (0.12 mmol, 92%).
1H NMR (CDCl3): δ 7.81 (t, J = 1.6 Hz, 3 H), 7.74 (t, J = 1.6 Hz, 3 H), 7.68 (t, J = 1.6 Hz, 3 H), 7.44 (d, J = 8.5 Hz, 6 H), 7.23 (d, J = 8.5 Hz, 6 H), 7.09 (d, J = 8.9 Hz, 2 H), 6.83 (d, J = 8.9 Hz, 2 H), 3.81 (s, 3 H), 3.21 (s, 3 H). 13C{1H} NMR (CDCl3): δ 158.5, 147.8, 140.0, 139.2, 137.7, 135.0, 134.9, 132.3, 131.7, 131.4, 125.8, 124.6, 120.4, 117.7, 113.9, 113.8, 92.7, 86.9, 80.9, 64.9, 55.7. HRESI-MS (pos.): found 820.2360, calculated for [C59H31N3ONa]+, i.e. [M+Na]+ = 820.2365 Da.
Tris-iodide 2SiCN: 3,5-Diiodobenzonitrile (2.6 g, 7.3 mmol), Pd(PPh3)4 (23 mg, 0.020 mmol), CuI (3.8 mg, 0.020 mol) and NEt3 (10 mL) were cooled to 0°C. The tris-alkyne 1Si (0.30 g, 0.73 mmol) dissolved in THF (5 mL) was added dropwise to the reaction mixture, which was allowed to warm to room temperature and stirred overnight under a nitrogen atmosphere. The solvent was removed under reduced pressure, the solid was dissolved in dichloromethane (100 mL) and washed with water (40 mL) and then brine (40 mL). The organic layer was dried over MgSO4 and the residue obtained after the removal of solvent was purified on a silica column (1:1 DCM:pet. spirits) to give 2SiCN as a white powder. Yield: 0.59 g (0.53 mmol, 73%).
1H NMR (CDCl3): δ 8.10 (t, J = 1.6 Hz, 3 H), 7.93 (t, J = 1.6 Hz, 3 H), 7.77 (t, J = 1.6 Hz, 3 H), 7.41–7.59 (m, 17 H). 13C{1H} NMR (CDCl3): δ 144.4, 139.9, 136.4, 136.4, 135.2, 134.1, 132.4, 131.3, 130.5, 128.4, 126.4, 123.6, 116.6, 114.5, 93.7, 93.0, 86.8. HRESI-MS (pos.): found 1111.8922, calculated for [C51H26N3SiI3· Na]+, i.e. [M+Na]+ = 1111.8928 Da.
Tris-TMS-alkyne 3SiCN: Tris-iodo compound 2SiCN (0.21 g, 0.19 mmol), Pd(PPh3)4 (11 mg, 0.010 mmol), and CuI (1.9 mg, 0.010 mmol) were stirred in NEt3 (10 mL). Trimethylsilylacetylene (0.12 mL, 0.80 mmol) was added to the reaction mixture, which was stirred overnight at room temperature under a nitrogen atmosphere. The solvent was removed under reduced pressure, the solid was dissolved in dichloromethane (60 mL) and washed with water (20 mL) and then brine (20 mL). The organic extract was dried over MgSO4 and the residue obtained after the removal of solvent was purified on a silica column (1:15 EtOAc:pet. spirits) to give 3SiCN as a fine white powder. Yield: 0.12 g (0.12 mmol, 63%).
1H NMR (CDCl3): δ 7.82 (t, J = 1.6 Hz, 3 H), 7.72 (t, J = 1.6 Hz, 3 H), 7.67 (t, J = 1.6 Hz, 3 H), 7.41–7.58 (m, 17 H), 0.26 (s, 27 H). 13C{1H} NMR (CDCl3): δ 138.8, 136.4, 135.0, 134.6, 134.3, 132.5, 131.3, 130.4, 128.4, 125.4, 125.1, 123.8, 117.4, 113.4, 101.6, 98.5, 92.2, 87.6, 0.1 (one expected peak not observed). HRESI-MS (pos.): found 1022.3216, calculated for [C66H53N3Si4·Na]+, i.e. [M+Na]+ = 1022.3214 Da.
Tris-alkyne 4SiCN: A solution of 3SiCN (0.37 g, 0.37 mmol) and KF (0.13 g, 2.2 mmol) in 1:1 THF:MeOH (10 mL) was stirred for 2 hours at room temperature under a nitrogen atmosphere. The solvent was removed under reduced pressure, the solid was dissolved in dichloromethane (60 mL) and washed with water (2 × 20 mL) and then brine (20 mL). The organic layer was dried over MgSO4 to give 4SiCN. Yield: 0.27 g (0.34 mmol, 92%).
1H NMR (CDCl3): δ 7.84 (t, J = 1.6 Hz, 3 H), 7.77 (t, J = 1.6 Hz, 3 H), 7.70 (t, J = 1.6 Hz, 3 H), 7.41–7.58 (m, 17 H), 3.22 (s, 3 H). 13C{1H} NMR (CDCl3): δ 138.9, 136.4, 135.1, 134.8, 134.8, 132.5, 131,3, 130.4, 128.4, 125.3, 124.4, 123.7, 117.3, 113.6, 92.4, 87.4, 80.6 (two expected peaks not observed). HRESI-MS (pos.): found 806.2028, calculated for [C57H29N3Si·Na]+, i.e. [M+Na]+ = 806.2028 Da.
Tris-azide 5: 4-Iodophenylazide (0.10 g, 0.42 mmol), Pd(PPh3)4 (14 mg, 0.012 mmol) and CuI (2.3 mg, 0.012 mmol) were dissolved in NEt3 (5 mL) and stirred at 30°C. Tris-alkyne compound 1Si (0.050 g, 0.12 mmol) dissolved in THF (5 mL) was added dropwise and the reaction stirred for 30 minutes under a nitrogen atmosphere. The solution was diluted with DCM (80 mL) and filtered through Celite, washing through with more DCM. The combined filtrates were taken to dryness and then suspended in methanol and sonicated for 5 minutes. The resulting solid was isolated by filtration and then further purified by column chromatography (1:5 DCM:pet. spirits) to obtain 5 as a yellow powder. Yield: 43 mg (0.057 mmol, 48%).
1H NMR (CDCl3): δ 7.39–7.56 (m, 23 H), 7.01 (d, J = 8.6 Hz, 6 H). 13C{1H} NMR (CDCl3): δ 140.6, 136.6, 136.5, 134.3, 133.4, 133.2, 131.2, 130.3, 128.4, 124.9, 119.9, 119.4, 90.3, 89.8. HRESI-MS (pos.): found 782.2215, calculated for [C48H29N9Si·Na]+, i.e. [M+Na]+ = 782.2213 Da.
Synthesis of cages
CCN≡·≡CCN: A Schlenk flask was charged with Cu(OAc)2·H2O (0.48 g, 2.4 mmol) and dried at 70°C for 2 hours under vacuum. CuCl (0.14 g, 1.3 mmol) and dry pyridine (30 mL) were added and the mixture was heated at 70°C under a nitrogen atmosphere. Tris-alkyne 4CCN (30 mg, 0.037 mmol) dissolved in dry pyridine (30 mL) was added dropwise to this mixture, which was stirred at 70°C for 2 hours. The reaction mixture cooled to room temperature and stirred overnight at room temperature and then the solvent was removed under vacuum. The residue was extracted with dichloromethane (80 mL) and washed with aqueous HCl(aq) (1.0 M, 50 mL) and water (2 × 50 mL). The organic extract was dried over MgSO4 and the residue obtained after removal of solvent was purified by preparative TLC (DCM) to give CCN≡·≡CCN as a yellow powder. Yield: 6.5 mg (0.041 mmol, 22%).
1H NMR (CDCl3): δ 7.89 (s, 6 H), 7.75 (s, 6 H), 7.69 (s, 6 H), 7.45 (d, J = 8.6 Hz, 12 H), 7.16–7.23 (m, 15 H), 6.88 (d, J = 8.8 Hz, 4 H), 3.84 (s, 6 H). 13C{1H} NMR (CDCl3): δ 158.3, 147.8, 140.4, 136.5, 134.7, 134.1, 132.1, 131.4, 131.1, 125.7, 123.5, 120.0, 117.1, 113.9, 113.5, 92.8, 86.3, 79.8, 76.1, 64.5, 55.4. HRESI-MS (pos.): found 1611.4377, calculated for [C118H56N6O2·Na]+, i.e. [M+Na]+ = 1611.4362 Da.
SiCN≡·≡SiCN: A Schlenk flask was charged with Cu(OAc)2·H2O (0.66 g, 3.3 mmol) and dried at 70°C for 2 hours under vacuum. CuCl (0.18 g, 1.8 mmol) and dry pyridine (30 mL) were added and the mixture was heated at 70°C under a nitrogen atmosphere. Tris-alkyne 4SiCN (40 mg, 0.051 mmol) dissolved in dry pyridine (30 mL) was added dropwise to this mixture and stirred at 70°C for 2 hours. The reaction mixture cooled to room temperature and stirred overnight at room temperature and then the solvent was removed under vacuum. The dark green oil was extracted with dichloromethane (2 × 80 mL) and washed with HCl(aq) (1.0 M, 50 mL) and water (2 × 50 mL). The organic extracts were combined, dried over MgSO4, and the residue obtained after removal of solvent was purified by column chromatography (1:2 DCM:pet. spirits) to give cage SiCN≡·≡SiCN as a yellow powder. Yield: 11 mg (7.0 μmol, 28%).
1H NMR (CDCl3): δ 7.91 (s, 6 H), 7.78 (s, 6 H), 7.71 (s, 6 H), 7.68 (d, 4 H, J = 8.0 Hz), 7.47–7.55 (m, 30 H).
13C{1H} NMR (CDCl3): δ 140.3, 136.5, 136.4, 135.5, 134.9, 134.3, 131.5, 131.3, 130.7, 128.6, 125.6, 123.5, 123.5, 117.1, 114.0, 92.8, 87,1, 79.8, 76.2.HRESI-MS (pos.): found 1583.3690, calculated for [C114H52N6Si2·Na]+, i.e. [M+Na]+ = 1583.3690 Da.
SiH≡SiH: A suspension of Pd(PPh3)4 (8.0 mg, 0.0060 mmol) in DIPEA (8 mL) was warmed to 80°C under a nitrogen atmosphere. A solution of tris-alkyne 4SiH (7.0 mg, 0.010 mmol) and tris-iodide 2SiH (13 mg, 0.013 mmol) in DMF (4 mL) was added dropwise to the preheated catalyst mixture and the temperature was raised to 100°C. The reaction was stirred overnight and then cooled to room temperature. The solvent was removed under reduced pressure and the resulting solid washed with MeOH (10 mL). The resulting solid was dissolved in dichloromethane (50 mL) and washed with water (2 × 20 mL). The organic layer was concentrated and purified by preparative TLC (2:1 pet. spirits:DCM) to give SiH≡SiH as a sticky white powder. Yield: 1.3 mg (0.97 μmol, 10%).
1H NMR (CDCl3): δ 7.77 (s, 6 H), 7.71 (d, 4 H), 7.43–7.61 (m, 40 H), 7.29–7.39 (m, 8 H). 13C{1H} NMR (CDCl3): δ 136.6, 136.4, 136.1, 134.6, 132.3, 131.2, 131.0, 131.0, 130.4, 128.7, 128.4, 124.7, 123.7, 12 3.6, 90.0, 89.8, 89.5. HRESI-MS (pos.): found 1361.3942, calculated for [C102H58Si2·Na]+, i.e. [M+Na]+ = 1361.3975 Da.
CH≡SiH: In a heavy-walled tube, tris-iodide 2CH (41 mg, 0.040 mmol), tris-alkyne 4SiH (28 mg, 0.040 mmol) and Pd(PPh3)4 (28 mg, 0.024 mmol) were dissolved in a 2:1 DMF:DIPEA (30 mL) mixture and this was deoxygenated with bubbling nitrogen for 10 min. The solution was then introduced into a preheated oil bath at 80°C and the temperature was gradually raised to 100°C. The solution was stirred for 48 hours and was then allowed to cool down to room temperature. The solvent was removed under reduced pressure and the resulting solid washed with MeOH (10 mL). The residue was dissolved in dichloromethane (50 mL) and washed with water (2 × 20 mL). The organic layer was dried over MgSO4 and the residue obtained after removal of solvent was purified by preparative TLC (2:1 pet. spirits:DCM) to give CH≡SiH as a white powder. Yield: 6.0 mg (0.0049 mmol, 12%)
1H NMR (CDCl3): δ 7.71–7.78 (m, 7 H), 7.44–7.59 (m, 32 H), 7.31–7.36 (m, 7 H), 7.07–7.21 (m, 9 H), 6.87 (d, J = 8.8 Hz, 2 H), 3.84 (s, 3 H). 13C{1H} NMR (CDCl3): δ 147.1, 136.5, 136.4, 136.2, 136.1, 134.6, 132.3, 131.2, 131.2, 131.1, 130.9, 130.9, 128.7, 128.4, 124.6, 123.8, 123.7, 123.6, 123.6, 121.0, 113.3, 90.0, 89.9, 89.5, 89.5, 88.9, 64.4, 55.4 (eight expected signals not observed, which we attribute to a low amount of sample and the large number of very similar chemical environments in the molecule). HRESI-MS (pos.): found 1375.4304, calculated for [C104H60OSi·Na]+, i.e. [M+Na]+ = 1375.4311 Da.
SiH≡SiCN: A round bottom flask was charged with Pd(PPh3)4 (29 mg, 0.025 mmol), DMF (4 mL) and DIPEA (4 mL) and warmed to 80°C under a nitrogen atmosphere. The tris-alkyne 4SiH (30 mg, 0.042 mmol) and tris-iodide 2SiCN (42 mg, 0.039 mmol) dissolved in DMF (4 mL) were added dropwise to the preheated solution and the temperature was raised to 100°C. The solution was stirred overnight and was then allowed to cool down to room temperature. The solvent was removed under reduced pressure and the resulting solid was washed with MeOH (10 mL). The residue was dissolved in dichloromethane (30 mL) and washed with water (2 × 20 mL). The organic layer was dried over MgSO4 and the residue obtained after removal of solvent was purified by preparatory thin layer chromatography (1:1 DCM:pet. spirits) to give SiH≡SiCN as a pale yellow powder. Yield: 1.7 mg, 0.0012 mmol, 3%).
1H NMR (CDCl3): δ 7.92 (s, 3 H), 7.69–7.76 (m, 12 H), 7.46–7.58 (m, 36 H), 7.35–7.39 (d, J = 7.8 Hz, 4 H). HRESI-MS (pos.): found 1414.3958, calculated for [C105H56N3Si2], i.e. [M∙H]+ = 1414.4013 Da.
Siamid≡·≡Siamid∙6Cl: SiCN≡·≡SiCN (20 mg, 13 μmol) was added to a Schlenk flask and the flask degassed and backfilled with nitrogen three times. Dry THF (5.0.mL) was added under a nitrogen atmosphere and the solution was cooled to −78°C. LiHMDS (1.0 M in THF, 0.16 mL, 0.16 mmol) was added causing the solution to turn purple and then brown upon warming to room temperature. After stirring the solution overnight at room temperature, the reaction was cooled to 0°C and quenched using ethanolic HCl (prepared by cautiously adding 0.50 mL acetyl chloride to 5 mL ethanol). The solvent was removed under reduced pressure and the resultant solid was washed with water (7 mL) to remove inorganic salts. The remaining dark brown solid was dried in vacuo to afford Siamid≡·≡Siamid∙6Cl. Yield: 18 mg (9.0 μmol, 67%).
1H NMR (d6-DMSO): δ 9.58 (br. s, 12 H), 9.36 (br. s, 12 H), 8.20 (s, 6 H), 8.13 (s, 6 H), 8.11 (s, 6 H), 7.54–7.71 (m, 34 H). 13C{1H} NMR (d6-DMSO): δ 164.0, 140.3, 136.1, 134.9, 131.3, 129.9, 128.7, 123.6, 123.1, 121.5, 91.6, 88.0, 80.4, 74.9 (five expected peaks not observed). HRESI-MS (pos.): 1663.5413, calculated for [C114H71N12Si2]+, i.e. loss of 6 Cl– and 5 H+ = 1663.5463 Da.
Acknowledgments
We thank the Australian Research Council for financial support (FT210100495), Prof. Christian Doonan (University of Adelaide) for helpful discussions, Anitha Jeyasingham (Australian National University) for assistance with mass spectrometry and Dr Michael Gardiner (Australian National University) for assistance with synchrotron X-ray crystallography. Parts of this work were conducted using beamline MX2 at the Australian Synchrotron [52], part of ANSTO, and made use of the Australian Cancer Research Foundation detector.
Disclosure statement
NGW is an associate editor of Supramolecular Chemistry. He was blinded to all aspects of the peer review process of this manuscript.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Yang X, Ullah Z, Stoddart JF, et al. Porous organic cages. Chem Rev. 2023;123(8):4602–4634. doi: 10.1021/acs.chemrev.2c00667
- Yoshizawa M, Klosterman JK, Fujita M. Functional molecular flasks: new properties and reactions within discrete, self-assembled hosts. Angew Chem Int Ed. 2009;48(19):3418–3438. doi: 10.1002/anie.200805340
- Cook TR, Stang PJ. Recent developments in the preparation and chemistry of Metallacycles and metallacages via coordination. Chem Rev. 2015;115(15):7001–7045. doi: 10.1021/cr5005666
- Hasell T, Cooper AI. Porous organic cages: soluble, modular and molecular pores. Nat Rev Mater. 2016;1(9):16053. doi: 10.1038/natrevmats.2016.53
- Sudik AC, Millward AR, Ockwig NW, et al. Design, synthesis, structure, and gas (N 2, ar, CO 2, CH 4, and H 2) sorption properties of porous metal-organic tetrahedral and Heterocuboidal Polyhedra. J Am Chem Soc. 2005;127(19):7110–7118. doi: 10.1021/ja042802q
- Tozawa T, Jones JTA, Swamy SI, et al. Porous organic cages. Nat Mater. 2009;8(12):973. doi: 10.1038/nmat2545
- Zhang G, Presly O, White F, et al. A permanent mesoporous organic cage with an exceptionally high surface area. Angew Chem Int Ed. 2014;53(6):1516–1520. doi: 10.1002/anie.201308924
- Jones JTA, Hasell T, Wu X, et al. Modular and predictable assembly of porous organic molecular crystals. Nature. 2011;474(7351):367–371. doi: 10.1038/nature10125
- Evans JD, Huang DM, Hill MR, et al. Molecular design of amorphous porous organic cages for enhanced gas storage. J Phys Chem C. 2015;119(14):7746–7754. doi: 10.1021/jp512944r
- Taggart GA, Antonio AM, Lorzing GR, et al. Tuning the porosity, solubility, and gas-storage properties of cuboctahedral coordination cages via Amide or Ester Functionalization. ACS Appl Mater Interfaces. 2020;12(22):24913–24919. doi: 10.1021/acsami.0c06434
- Gosselin AJ, Decker GE, Antonio AM, et al. A charged coordination cage-based porous salt. J Am Chem Soc. 2020;142(21):9594–9598. doi: 10.1021/jacs.0c02806
- Smulders MMJ, Riddell IA, Browne C, et al. Building on architectural principles for three-dimensional metallosupramolecular construction. Chem Soc Rev. 2013;42(4):1728–1754. doi: 10.1039/C2CS35254K
- Liu X, Liu Y, Li G, et al. One-pot, 18-component synthesis of an octahedral nanocontainer molecule. Angew Chem Int Ed. 2006;45(6):901–904. doi: 10.1002/anie.200504049
- Francesconi O, Ienco A, Moneti G, et al. A self-assembled pyrrolic cage receptor specifically recognizes β-Glucopyranosides. Angew Chem Int Ed. 2006;45(40):6693–6696. doi: 10.1002/anie.200602412
- Skowronek P, Gawronski J. Chiral iminospherand of a tetrahedral symmetry spontaneously assembled in a [6 + 4] cyclocondensation. Org Lett. 2008;10(21):4755–4758. doi: 10.1021/ol801702j
- Nishimura N, Kobayashi K. Self-assembly of a cavitand-based capsule by dynamic boronic ester formation. Angew Chem Int Ed. 2008;47(33):6255–6258. doi: 10.1002/anie.200802293
- Christinat N, Scopelliti R, Severin K. Multicomponent assembly of boronic acid based macrocycles and cages. Angew Chem Int Ed. 2008;47(10):1848–1852. doi: 10.1002/anie.200705272
- Mastalerz M, Schneider MW, Oppel IM, et al. A salicylbisimine cage compound with high surface area and selective CO 2 /CH 4 adsorption. Angew Chem Int Ed. 2011;50(5):1046–1051. doi: 10.1002/anie.201005301
- Zhang G, Mastalerz M. Organic cage compounds – from shape-persistency to function. Chem Soc Rev. 2014;43(6):1934–1947. doi: 10.1039/C3CS60358J
- We note that recent advances in alkyne metathesis chemistry and hydrazone chemistry may offer a method to prepare robust cages under thermodynamic control, although the ability to form low symmetry species using these approaches is still limited. See e.g. Lee S, Yang A, Moneypenny TP II, et al. Kinetically trapped tetrahedral cages via Alkyne Metathesis. J Am Chem Soc. 2016;138(7):2182–2185. doi: 10.1021/jacs.6b00468 and Jiao T, Wu G, Zhang Y, et al. Self-assembly in water with N-substituted imines. Angew Chem Int Ed. 2020;59(42):18350-18367. doi: 10.1002/anie.201910739
- Schneider MW, Oppel IM, Griffin A, et al. Post-modification of the interior of porous shape-persistent organic cage compounds. Angew Chem Int Ed. 2013;52(13):3611–3615. doi: 10.1002/anie.201208156
- Bhat AS, Elbert SM, Zhang W-S, et al. Transformation of a [4+6] salicylbisimine cage to chemically robust amide cages. Angew Chem Int Ed. 2019;58(26):8819–8823. doi: 10.1002/anie.201903631
- Alexandre P-E, Zhang W-S, Rominger F, et al. A robust porous quinoline cage: transformation of a [4+6] salicylimine cage by Povarov Cyclization. Angew Chem Int Ed. 2020;59(44):19675–19679. doi: 10.1002/anie.202007048
- Andrews KG, Christensen KE. Access to amide-linked organic cages by in situ trapping of Metastable Imine Assemblies: solution phase bisamine recognition. Chem: Eur J. 2023;29(26):e202300063. doi: 10.1002/chem.202300063
- Bloch WM, Clever GH. Integrative self-sorting of coordination cages based on ‘naked’ metal ions. Chem Commun. 2017;53(61):8506–8516. doi: 10.1039/C7CC03379F
- Lewis JEM, Tarzia A, White AJP, et al. Conformational control of Pd 2 L 4 assemblies with unsymmetrical ligands. Chem Sci. 2020;11(3):677–683. doi: 10.1039/C9SC05534G
- Tian C-B, Sun Q-F. Combinatorial coordination self-assembly for organopalladium cages with fine-tuned structure and function. Chem: Eur J. 2023;29(28):e202300195. doi: 10.1002/chem.202300195
- Wu K, Benchimol E, Baksi A, et al. ChemRxiv. doi:10.26434/chemrxiv-2023-5gb4g.
- Preston D, Evans JD. A lantern-shaped Pd(II) cage constructed from four different low-symmetry ligands with positional and orientational control: an ancillary pairings approach. Chem Int Ed. 2023;62(49):e202314378. doi: 10.1002/anie.202314378
- Zhang C, Chen C-F. Synthesis and structure of a Triptycene-based nanosized molecular cage. J Org Chem. 2007;72(24):9339–9341. doi: 10.1021/jo7017526
- Avellaneda A, Valente P, Burgun A, et al. Kinetically controlled porosity in a robust organic cage material. Angew Chem Int Ed. 2013;52(13):3746–3749. doi: 10.1002/anie.201209922
- Kitchin M, Konstas K, Sumby CJ, et al. Continuous flow synthesis of a carbon-based molecular cage macrocycle via a three-fold homocoupling reaction. Chem Commun. 2015;51(75):14231–14234. doi: 10.1039/C5CC05181A
- Burgun A, Valente P, Evans JD, et al. Endohedrally functionalised porous organic cages. Chem Commun. 2016;52(57):8850–8853. doi: 10.1039/C6CC04423A
- Tominaga M, Ohara K, Yamaguchi K, et al. Hollow sphere formation from a three-dimensional structure composed of an Adamantane-Based Cage. J Org Chem. 2014;79(14):6738–6742. doi: 10.1021/jo500989c
- Ma H, Zhai T-L, Wang Z, et al. Switching porosity of stable triptycene-based cage via solution-state assembly processes. RSC Adv. 2020;10(15):9088–9092. doi: 10.1039/D0RA00128G
- Zhang Z-Q, Ren Q-X, Tian W-F, et al. Synthesis of enantiopure hydrocarbon cages based on an optically resolved C 3 -symmetric triaminotribenzotriquinacene. Org Lett. 2021;23(4):1478–1483. doi: 10.1021/acs.orglett.1c00176
- Thomas CM, Liang W, Preston D, et al. Post-synthetic modification of a porous hydrocarbon cage to give a Discrete Co 24 organometallic Complex. Chem: Eur J. 2022;28(51):e202200958. doi: 10.1002/chem.202200958
- We note that generally the term Eglinton coupling is used to refer to the Cu(II)-mediated coupling of two terminal alkynes to give a di-alkyne and that our reactions, which are based on those developed by Sumby and Doonan use both a Cu(I) and Cu(II) mediator (reference 31). Other copper-mediated coupling reactions were also investigated (see Supporting Information). In the interests of simplicity, we refer to these reactions as Eglinton reactions throughout.
- Hoskins BF, Robson R. Infinite polymeric frameworks consisting of three dimensionally linked rod-like segments. J Am Chem Soc. 1989;111(15):5962–5964. doi: 10.1021/ja00197a079
- Robson R. Design and its limitations in the construction of bi- and poly-nuclear coordination complexes and coordination polymers (aka MOFs): a personal view. Dalton Trans. 2008;38:5113–5131. doi: 10.1039/b805617j
- Yaghi OM, O’Keeffe M, Ockwig NW, et al. Reticular synthesis and the design of new materials. Nature. 2003;423(6941):705–714. doi: 10.1038/nature01650
- Morshedi M, Thomas M, Tarzia A, et al. Supramolecular anion recognition in water: synthesis of hydrogen-bonded supramolecular frameworks. Chem Sci. 2017;8(4):3019–3025. doi: 10.1039/C7SC00201G
- Xing G, Yan T, Das S, et al. Synthesis of crystalline porous organic salts with high proton conductivity. Angew Chem Int Ed. 2018;57(19):5345–5349. doi: 10.1002/anie.201800423
- Bassanetti I, Bracco S, Comotti A, et al. Flexible porous molecular materials responsive to CO 2 , CH 4 and Xe stimuli. J Mater Chem A. 2018;6(29):14231–14239. doi: 10.1039/C8TA02211A
- White NG. Amidinium⋯carboxylate frameworks: predictable, robust, water-stable hydrogen bonded materials. Chem Commun. 2021;57(84):10998–11008. doi: 10.1039/D1CC04782E
- Spek AL. PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. 2015;C71:9–8.
- Bruno IJ, Cole JC, Edgington PR, et al. New software for searching the Cambridge structural database and visualizing crystal structures. Acta Crystallogr. 2002;B58(3):389–397. doi: 10.1107/S0108768102003324
- Boer SA, Morshedi M, Tarzia A, et al. Molecular tectonics: a node-and-linker building block approach to a family of hydrogen-bonded frameworks. Chem: Eur J. 2019;25(42):10006–10012. doi: 10.1002/chem.201902117
- Kwon Y-D, Son J, Chun J-H. Catalyst-Free Aromatic Radiofluorination via Oxidized Iodoarene Precursors. Org Lett. 2018;20(24):7902–7906. doi: 10.1021/acs.orglett.8b03450
- Alagille D, DaCosta H, Chen Y, et al. Potent mGlur5 antagonists: pyridyl and thiazolyl-ethynyl-3,5-disubstituted-phenyl series. Bioorg Med Chem Lett. 2011;21(11):3243–3247. doi: 10.1016/j.bmcl.2011.04.047
- Endres A, Maas G. Dirhodium(II) tetrakis(perfluoroalkylbenzoates) as partially recyclable catalysts for carbene transfer reactions with diazoacetates. Tetrahedron. 2002;58(20):3999–4005. doi: 10.1016/S0040-4020(02)00249-1
- Aragao D, Aishima J, Cherukuvada H, et al. MX2: a high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian synchrotron. J Synchrotron Rad. 2018;25(3):885–891. doi: 10.1107/S1600577518003120
- Kabsch W. XDS. Acta Crystallogr. 2010;66(2):125–132. doi: 10.1107/S0907444909047337
- Palatinus L, Chapuis G. SUPERFLIP – a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J Appl Crystallogr. 2007;40(4):786–790. doi: 10.1107/S0021889807029238
- Betteridge PW, Carruthers JR, Cooper RI, et al. CRYSTALS version 12: software for guided crystal structure analysis. J Appl Crystallogr. 2003;36(6):1487. doi: 10.1107/S0021889803021800