
Abstract
More than 35 million people are living with HIV worldwide with approximately 2.3 million new infections per year. Cascade of events (cell entry, virus replication, assembly and release of newly formed virions) is involved in the HIV-1 transmission process. Every single step offers a potential therapeutic strategy to halt this progression and HIV fusion into the human host cell is one such stage. Controlling the initial event of HIV-1 transmission is the best way to control its dissemination especially when prophylaxis is concerned. Action is required either on the HIV’s or host’s cell surface which is logically more rational when compared with other intracellular acting moieties. Aim of this manuscript is to detail the significance and current strategies to halt this initial step, thus blocking the entry of HIV-1 for further infection. Both HIV-1 and the possible host cell’s receptors/co-receptors are under focus while specifying the targets available for inhibiting this fusion. Current and under investigation moieties are categorized based on their versatile mechanisms. Advanced drug delivery and nanotechnology approaches present a key tool to exploit the therapeutic potential in a boosted way. Current drug delivery and the impact of nanotechnology in potentiating this strategy are detailed.
Introduction
HIV infections are considered to be the global threat of present era. A huge percentage of human community is influenced by the direct as well as related impacts of this pandemic. Antiretrovirals are currently the best option for prolonged and maximal viral suppression (Hammer et al., Citation2008). Currently used antiretroviral drug classes include reverse transcriptase inhibitors (RTIs), protease inhibitors (PIs), entry inhibitors (CCR5 antagonists and fusion inhibitors) and integrase inhibitors (das Neves et al., Citation2010a). Since no curative therapy is available, prevention is a cornerstone in the battle against HIV/AIDS, around 30 individual drugs and fixed-dose combinations are available to manage this pandemic. In context to HIV prophylaxis, current research period is able to develop an overall potency of 39% with topical tenofovir (RTI) gel (Galvin & Cohen, Citation2004; Karim & Baxter, Citation2013).
Bulk of efforts has been invested to select an effective and specific target for both preventive and treatment therapeutics. Targeting the entry of enveloped viruses is a very attractive strategy in this context, since the site of action is likely to be extracellular and therefore relatively accessible; this could also limit cell toxicity. All the stages of viral entry are potentially amenable to therapeutic intervention. In this review, we are focusing on inhibition strategies of binding and fusion of HIV-virus at cellular entry step. Discussing the possible sub-targets in the context of recently developed antiviral molecules certainly opens the access to newer targets and inhibitors (Nikolic et al., Citation2007; Teissier et al., Citation2011; Chauhan et al., Citation2013; McGowan, Citation2014; Montgomery, Citation2015).
Factors related to virus
Description about HIV-1 membrane, membrane lipids and membrane glycoproteins is detailed in supplementary data.
Host cell receptors and co-receptors
Host cell receptors
Attachment of the virus to the cell surface, involves recognition and binding to specific cell surface receptors. provides descriptive information about these receptors.
Table 1. Description of host cell receptors involved in HIV-1 transmission.
Host cell co-receptors
CD4 binding causes conformational changes in gp120 that binds it to a second receptor, a co-receptor. HIV-1 strains use the chemokine receptor CCR5 in conjunction with CD4 for virus entry and in absence of CCR5, CXCR4 either in place of or in addition to CCR5. Co-receptor choice is notably the V3loop and, to a lesser degree, the V1/2 region within gp120. This highly conserved region may be the target for neutralizing antibodies (NABs) (Kwong et al., Citation1998). The relatively strong binding to CD4 causes the joining of two β-hairpins to form the bridging sheet domain. gp120 domains rotate and move away from the central stalk, exposing the V1/V2 and V3 loops to the target membrane (Liu et al., Citation2008). Membrane fusion is a cooperative process of four to six CCR5 receptors, multiple CD4 molecules and three to six Env trimers are needed to form a fusion pore (Kuhmann et al., Citation2000). Viruses using CCR5 are largely transmitted and endure throughout infection and those that use CXCR4 often emerge later on in the course of infection and have been associated with more rapid disease progression and CD4 cell decline.
Mode of transmission
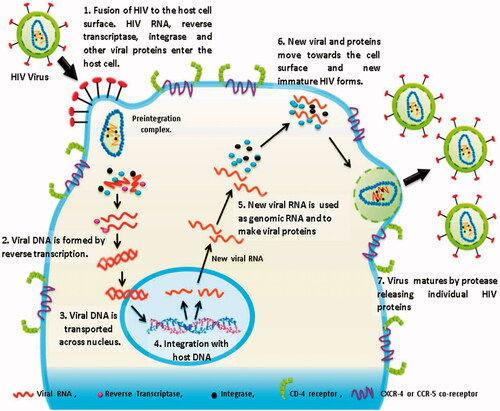
Briefly describing this whole transmission process of HIV, we take this transmission as a triple phase process. First phase (P-1) is HIV invasion; second phase (P-2) is HIV replication and third is budding of newly formed virons from the host cell () (description about the mode of HIV-1 transmission is detailed in supplementary data).
Figure 1. Transmission of HIV-1 virus.
Targets for fusion and binding inhibition
Fusion inhibitors perform extracellularly prior to assault of the host cell. So they are not liable to cellular efflux transporters that lesser the effective intracellular concentrations of additional classes of antiretrovirals. Their absence of intracellular processing may pay to their low toxicity profile and reduces cross-resistance with recognized intracellular agents. With an exclusive mechanism of action they signify a new fourth class of ARVs (Greenberg & Cammack, Citation2004). They block the last step in the three-step viral entry process comprising of attachment, co-receptor binding and fusion (Greenberg & Cammack, Citation2004). Three key steps in viral entry have been embattled for drug development, i.e. inhibition of CD4 binding, inhibition of co-receptor binding and obstruction of the gp41 conformational changes that allow viral fusion. explains these abovementioned targets in a descriptive way. Fusion inhibitor molecules also act as peptide mimics, which block the interactions that are obligatory for HIV-1 viral entry (Wild et al., Citation1994).
Figure 2. Targets for fusion and binding inhibition.
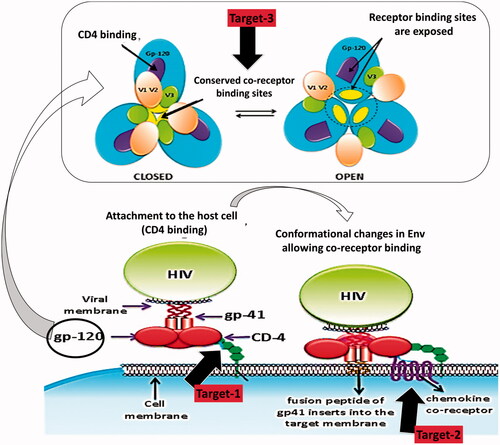
Inhibition of CD4 binding
The CD4 binding site (CD4BS) is at the interface of all three gp120 domains. CD4 binding site is extremely well-preserved and NABs can efficiently block this step. Tetravalent CD4-IgG and Pro542 are some examples that can bind gp120 and restrict its ability to accept the CD4-bound conformation, thus disturbing the entry process. Although CD4 is not involved in the process, CD4BS antibodies exert inhibitory activity on co-receptor binding and envelope glycoprotein mediated syncytium formation. It was also believed (not entirely) that HIV-1 envelope glycoproteins elicit NABs raised against both the variable and conserved regions of the envelope glycoproteins during natural infection (Raja et al., Citation2003).
Inhibition of co-receptor binding
CCR5 is a member of a large family of G-protein-coupled, seven-membrane-spanning receptors (GPCRs). CCR5 has progressed as an attractive target for therapeutic intervention because of its cell type-specific expression and its important role as the major co-receptor for HIV-1. The CCR5 molecule does not appear essential for immune function and survival. So inhibition of CCR5 may not be accompanying with adverse effects on cellular function (Marmor et al., Citation2006). Chemokine receptor CCR5 and to a slighter extent CXCR4, have been under extreme targeting for small molecular weight inhibitors. This includes the approaches like prevention of co-receptor binding to gp120 through steric or allosteric hindrance, second by inducing receptor down-regulation thereby limiting the number of HIV entry points and third by the development of inhibitors for blocking the co-receptor binding site on gp120 (Kuhmann et al., Citation2000). Antibodies against the CCR5 N-segments also block entry with variable degrees of activity, but usually are less potent than those against the extracellular region (Lee et al., Citation1999). Only one potent anti-CXCR4 antibody (12G5) that inhibits HIV-1 entry has been well characterized. The 12G5 murine monoclonal recognizes an epitope in extracellular loops of CXCR4 (Phogat et al., Citation2007). The chemokine variants also competitively block interaction of gp120 with the chemokine receptors (Matos et al., Citation2010). Chemokines bind to their receptors at two positions, one in the NT and the other within the TM domain. The binding sites on CXCR4 have been restricted to the extracellular domain (Donzella et al., Citation1998).
gp41 targeting
A prominent pocket on the surface of a central trimeric coiled coil within gp41 is a potential target for inhibiting HIV-1 entry. In gp41, a central three stranded coiled coil formed by the N-terminal regions of gp41 is surrounded by helices derived from the C-terminal end of the gp41 ectodomains (Eckert et al., Citation1999). Peptides corresponding to these regions of gp41 are known as N-peptides and C-peptides (Kahle et al., Citation2009). The principal targets for gp41 inhibitors are two heptad repeat (HR) fragments in the N- and C-terminal regions of the gp41 ectodomain symbolized N-HR and C-HR, respectively (Lu et al., Citation1995). Synthetic C-peptides act in a dominant-negative manner by binding to the transiently exposed coiled-coil N-peptide region in the prehairpin intermediate. Therapeutic agent that goals this gp41 pocket would likely be relatively elusive to the emergence of resistant viral strains (Chan et al., Citation1998). However, C-peptides not containing pocket-binding residues, such as T-20, are more vulnerable to the emergence of resistant virus than C-peptides containing pocket-binding residues, such as T649 (Rimsky et al., Citation1998).
The peptide inhibitor efforts to bind in the trimeric coiled loop of gp41, performs a dominant-negative mechanism (Weissenhorn et al., Citation1997). C peptides perform at this intermediate phase, subsequently the native state has undertaken a conformational transition but before establishment of the hairpin structure since, once this gp41 core is accumulated, it is extremely stable and the melting temperature of the gp41 core is in surplus of 90 °C which is unlikely to be interrupted by exogenous peptides (Matos et al., Citation2010). Consistent using this vision, the C peptide DP178 binds to gp41 only after collaboration of the envelope complex with cellular receptors (Furuta et al., Citation1998).
Structurally mimicking engineered proteins of all or part of the N-HR coiled coil can prevent HIV-1 entry by binding the gp41 C-HR regions (Eckert & Kim, Citation2001; Louis et al., Citation2001). A well-considered example is the 5-helix protein that contains all three N-HR sections but only two C-HR segments; when suitably folded, 5-helix exposes a single C peptide binding spot that strongly interacts with gp41 C-HR fragment (Root et al., Citation2001). 5-Helix and C-peptides do not interact with the native state of Env earlier to gp120/CD4 interaction (Melikyan et al., Citation2000).
Effects of first and second generation peptide inhibitors in inhibiting conformational changes necessary for viral fusion provide a proof of principle that the entry process is a viable target for therapeutic intervention. Molecules that block TOH (trimers of hairpins) formation can effectively inhibit HIV-1 membrane fusion both in vitro and in vivo (Kahle et al., Citation2009). Inhibitors bind the N-HR or C-HR section prior to bundle creation and prevent folding of gp41 into its TOH conformation (Root & Steger, Citation2004). The broad inhibitory activity of C peptides against diverse HIV isolates is explained by the highly conserved nature of the hydrophobic groove to which these peptides bind (Chan et al., Citation1997). C-peptides goal the N-HR in its coiled-coil form, binding the same hydrophobic indentations that would normally interact with gp41 CHR divisions (Kilgore et al., Citation2003). Several peptides that mimic the sequence of the C- and N-helical regions of HIV-1 have been found to inhibit fusion either by blocking the interaction between the N- and C-helical regions or by promoting the dissociation of gp41 trimers into monomers and thus preventing the formation of the 6-helix bundle fusogenic state of gp41 (Weissenhorn et al., Citation1999).
Present therapeutics and prophylaxis
CD4–gp120 attachment inhibitors
Attachment inhibitors like BMS-043 and BMS-806, PRO 2000, TMB-355 or Ibalizumab and PRO 542, are designed to inhibit the binding of gp120 to the CD4 receptor and are in pre-clinical or clinical development phase. These act extracellular prior to viral cell invasion but fewer like TMB-355 targets the essential host CD4 receptor instead of a specific viral target, which may result in undesirable side effects.
PRO-542 (CD4–IgG2) is a tetravalent soluble recombinant antibody-like fusion protein that includes four replicas of the virus binding CD4 domain (Allaway et al., Citation1995) and mimics the CD4 receptor. It is one of the gp120–CD4 binding inhibitors in more progressive phases of clinical development. Phase I studies determined that PRO-542 was well tolerated without apparent dose-limiting toxicities (Jacobson et al., Citation2000). Besides, pre-clinical studies uniting PRO-542 and enfuvirtide have recommended synergy in the inhibition of HIV replication (Nagashima et al., Citation2001). TMB-355 is a non-immunosuppressive monoclonal antibody focused against the CD4 receptor to compete with HIV gp120 for CD4 binding. Therefore, TMB-355 might inhibit post-viral binding conformational changes which are essential for the successful entry of HIV into the cell. cyclotriazadisulfonamide (CADA) compounds, down-regulate the cellular receptor, CD4. Some synthesized analogs of CADA proved to be highly effective in decreasing cellular CD4 and in acting as HIV entry inhibitors (Vermeire et al., Citation2002). CADA shows specific inhibition of the CD4–gp120 binding by assuming down-regulation of the CD4 receptor expression at the post-translational level (Vermeire et al., Citation2003). BMS-378806 (a prototype small molecule attachment inhibitor) is a newly exposed molecule with high affinity to prevent the conformational changes induced in gp120 after CD4 binding. Binding of BMS-806 to HIV gp120 is highly specific, reversible and co-receptor independent. This compound exhibits potent inhibitory activity against a panel of R5, X4 and R5/X4 HIV-1 (Lin et al., Citation2003). The study data shows that BMS-378806 is a representative of a new class of HIV inhibitors having the potential to become a valued addition to our current antiretroviral drugs (Lin et al., Citation2003). BMS-663068 has revealed effective antiviral activity in early phase studies and phase 2b trials are at present in progress (Henrich & Kuritzkes, Citation2013).
Synthetic compound 2-aminothiazolones evolved as novel anti-HIV agents. It acts at the very early stage of the HIV-1 entry process through inhibition of the gp120–CD4 protein–protein interaction (Tiberi et al., Citation2014). Synthetic compound pyrimidinediones signify high efficacy and potency against HIV-1 with a dual mechanism of antiviral action including both virus entry and reverse transcription (Watson Buckheit et al., Citation2011). Cyanovirin-N (CV-N) is a cyanobacterial protein having potent activity against HIV (Dey et al., Citation2000). CV-N directly goals HIV spike glycoproteins, preventing attachment and consequent viral fusion, and successfully renders the virus non-infectious (Shenoy et al., Citation2001). This comprises several binding to high-mannose oligosaccharides mainly in the C2–C4 region of the gp120 protein (Hu et al., Citation2007). Then it blocks the conformational changes essential for virus–target cell attachment and subsequent fusion (Buffa et al., Citation2009). CV-N has broad-spectrum antiviral activity, both for numerous steps in the HIV entry mechanism (Dey et al., Citation2000). CV-N has high binding affinity and nonexistence of toxicity (Balzarini et al., Citation2006). The green tea flavonoid, epigallocatechingallate (EGCG), anticipated having an anti-HIV-1 effect also high affinity to CD4 molecule for blocking the binding of gp120 to the CD4 molecule on T cells (Williamson et al., Citation2006).
CCR5 and CXCR4 antagonists
CCR5 and CXCR4 antagonists are divided into groups depending on their size. Large molecules, such as PRO-140, or molecules with a medium size, as Met-RANTES and AOP-RANTES, which are modified CCR5 natural ligands, make CCR5 inaccessible. Finally, several small-molecule inhibitors directed against CCR5 (TAK-779, SCH-C, SCH-D, UK427, 857 and GW-873140) or CXCR4 (KRH1636) are in several stages of clinical development phase. Most CCR5 antagonists are small molecules which block the gp120–CCR5 interaction after binding to the co-receptor. These also cause undesirable side effects, since these are target essential host molecules, rather than specific viral targets (Greenberg & Cammack, Citation2004). Maraviroc (MRV) (UK-427, 857) is a CCR5 antagonist. It attached to the transmembrane co-receptor cavity, within the 2, 3, 6 and 7 helix, that is different from the region targeted by TAK-779. The drug is presently in innovative steps of clinical development and is anticipated to be approved in the near future (Briz et al., Citation2006; van Lelyveld et al., Citation2015). Studies implemented with Aplaviroc (GW-873140) and other CCR5 antagonists (SCH-C and TAK-779) showed that they exhibit potent activity against R5 viruses. Aplaviroc act directly with second extracellular loops (ECL2) and not with the transmembrane cavity (Maeda et al., Citation2004).
The TAK-779 is a small molecule which acts at the membrane fusion stage by obstructing the interaction of gp120 with co-receptor CCR5. The binding site for TAK-779 is on ECL of CCR5. TAK-779 also avoids the binding of the ordinary CCR5 ligand, RANTES. It is not in clinical development phase but TAK-652 is under clinical development. PRO-140 is a monoclonal antibody focused against the CCR5 co-receptor, to inhibit the binding of HIV gp120. Cenicriviroc is a small-molecule CCR5 antagonist arrived in phase 2b studies. The potent antiviral activity of AMD3100 against X4 strains has been established in different in vitro and in vivo studies (but it is not further developed for HIV therapy because of its poor bioavailability severe side effects) (Donzella et al., Citation1998; Flomenberg et al., Citation2005). KRH-1636 is another CXCR4 antagonist, with antiviral activity similar to that of AMD3100. KRH-2731 is a novel CXCR4 antagonist (Ichiyama et al., Citation2003). Phase II trials are currently continuing. The binding site for CXCR4 antagonists is positioned in the ECL2 of the CXCR4 co-receptor (Murakami et al., Citation1997; Labrosse et al., Citation1998). Due to high negative charge on the surface of CXCR4, the interactions with the HIV gp120 V3 loop are electrostatic. HIV strains using CCR5 co-receptors are inhibited by the CCR5 ligands RANTES, MIP-1a and MIP-1b; correspondingly strains using CXCR4 co-receptor are inhibited by the CXCR4 ligand SDF (stromal-derived factor). Small molecule inhibitors alike ALX40-4C and T22 prevent entry of HIV strains that make use of CXCR4, thereby blocking its interaction with gp120 (Matos et al., Citation2010).
Fusion inhibitors
Enfuvirtide is the first in this category to reach market approval. Enfuvirtide has a unique mechanism of action, high viral target specificity, high efficacy and low toxicity. Enfuvirtide is a peptide mimetic (Greenberg & Cammack, Citation2004). It is homologous to a segment of the HR2 region of gp41 corresponding to amino acids 643–678. Enfuvirtide obstructs the HR2 binding site on the gp41 trimeric coiled coil assembly, thereby inhibiting the development of the hairpin structure and subsequently, the fusion (Matthews et al., Citation2004). Enfuvirtide is previously known as T-20 and DP-178 would drag to the opposite NHR (Matos et al., Citation2010). T20 (enfuvirtide) C-peptide, efficiently suppress HIV-1 infection in human (Kahle et al., Citation2009). Second generation of peptide inhibitors symbolizes T-1249, it fixes to gp41 and avoids its fusogenic conformation, hindering viral entry into host cells (Fatkenheuer et al., Citation2005). This agent is a synthetic compound utilizing non-native sequences corresponding to the HR2 region and has been shown to inhibit enfuvirtide-resistant virus. Development of drug resistance in first generation (T20) and drug formulation problem in second generation (T-1249) halted their further clinical development. Third generation fusion inhibitors in the form of sifuvirtide are as initially designed on the basis of three-dimensional structural information of gp41. It has been assessed by the phase I clinical trials and is presently in phase II clinical studies. It was recently demonstrated that the M-T hook structure can be used to design a short CHR peptide that specifically targets the conserved gp41 pocket rather than the T20-resistant sites. Recently an attempt was made to develop more potent HIV-1 fusion inhibitors using multiple biophysical and functional approaches. HP23 (a 23-residue peptide), following M-T hook structure and pocket-binding sequence, had significantly improved anti HIV-1 activity (including on T20 and MT-SC22EK resistant HIV-1 mutants). It was considered as the most potent inhibitor of both M-T hook-modified and unmodified control peptides.
5-Helix binds the C-peptide region of gp41 and acts by sequestering one of the helices necessary for the formation of the six-helix bundle protein. Peptide mimetics fusion inhibitors T-649 having homology with the HR2 region of gp41 inhibit hairpin structure formation in a parallel way to enfuvirtide. C34 peptide has homology to a region which creates the six-helix bundle and blocks its formation. A cyclic molecule D-peptide (DP-107) is homologous to HR1 that binds HR2 (a pocket region within the six-helix structure) (Root et al., Citation2001; Koshiba & Chan, Citation2003). “RPR103611” a non-peptide triterpene compound disrupts the association of gp120–gp41 in CXCR4-tropic viruses by targeting the loop region of the gp41 leucine zipper (Greenberg & Cammack, Citation2004).
C-helical peptides such as C34 blocks fusion through binding to the hydrophobic grooves that line the interior N-terminal trimeric coiled coil core of the gp41 ectodomain (Lu et al., Citation1995). N-helical-derived peptides such as N36 are much less operative inhibitors. They goal the N-helical region of the prehairpin intermediate by making fusion-incompetent heterotrimers (Weissenhorn et al., Citation1999). Binding of HIV-1 envelope glycoprotein (Env) to its cellular receptors provokes a variety of signaling events, comprising the activation of selected tyrosine kinases (Stantchev et al., Citation2007). Genistein prevents infection of macrophages made by Env glycoprotein. This inhibitory effect of genistein implied prevention of the virus entry process (Tobiume et al., Citation2003; Cavrois et al., Citation2004).
Lectins
Several lectins having strong-mannose carbohydrates binding site on the surface of virus envelopes have been found to have antiviral activity. Some specific algal lectins such as Cyanovirin-N, Griffithsin, Microcystisviridislectin, Scytovirin, Oscillatoriaagardhii agglutinin, demonstrate high anti-HIV activity (Lin et al., Citation2003). Lectin actinohivin (AH), exhibits potent in vitro anti-HIV activity by interacting to high-mannose type glycans (HMTGs) of highly glycosylated gp120 of HIV. Griffithsin (GRFT), a 12.7 kDa carbohydrate-binding protein, is highly potent inhibitor of both CXCR4 and CCR5-tropic viruses of HIV-1. GRFT also blocks cell fusion and cell-to-cell transmission of HIV (Doms & Trono, Citation2000). Griffthsin with tenofovir, MRV and enfuvirtide gives synergistic activity profile. GRFT inhibits viral replication at primary levels (Emau et al., Citation2007). GRFT efficiently prevents DC-SIGN-mediated transfer of HIV-1 to CD4+ T-lymphocytes and capable of expelling gp120 from the gp120–DC-SIGN complex.
Drug delivery
Disappointing results of the current approaches, demand smart drug delivery approaches with better delivery, superior safety profiles and potential for improved patient compliance. Challenge to design an efficient delivery system is a smart task. This designing contest requires a need based approach where different delivery routes have their specific requirement. Localized drug delivery is very much explored in this case, as it offers several additional advantages when compared to other routes. Now, the dosage from designing approach must be entirely different in every category. Specifically for topical purpose; gels, creams, tablets suppositories are among the conventional formulations and vaginal rings are most common for long term controlled delivery (Goyal et al., Citation2015; Gaurav et al., Citation2015b; D'Cruz & Uckun, Citation2014). In this specific case bioavailability achieved is affected by numerous physiological factors, and the ability of the formulation to effectively deliver drug molecule may vary with the menstrual cycle, pH variations and the presence of co-pathogens. There is a special emphasis on mucoadhesive gel systems for the delivery of intravaginal microbicide. The remarkable elastic character and the improved rheological properties of the mucoadhesive gel prolong the residence time at the application site. Selection of correct viscosity of the formulation is important in order to provide adequate retention and distribution in the vagina. A list of selected polymers is mentioned in .
Table 2. List of polymers used for the formulation of mucoadhesive vaginal gel formulation system.
Nanotechnology has its own role to play when considered for this task. The small size of the nanocarriers allows them to be transported more easily across the mucosal barriers by passive or active transport and possess the host targeting ability after entering the blood stream (Mallipeddi & Rohan, Citation2010b; Date & Destache, Citation2013). Essential properties of different nanosystems such as size, particle shape and surface charge can modify its bioavailability and targeting. In case of active targeting, a suitable surface modification “most frequently by surface attachment of specific ligands” is capable of identifying target cells or sites (das Neves et al., Citation2010b; Gaurav et al., Citation2015b). Enfuvirtide and MRV therapies in a conventional solid dosage form (tablet and capsules) have been tried and still under surveillance. Poor in vivo performance after a successful in vitro screening points towards the poorly managed drug delivery. T20 and sifuvirtide are also delivered by subcutaneous injection but a huge difference in half-life was observed for both these candidates. One important limitation of many current antiretroviral drugs is their unavailability to circumvent efflux pumps (particularly P-glycoprotein) that are present, in the membrane of several HIV-target cells. An effective delivery of such drugs requires an ideal platform and nanotechnology based approaches are a good area to watch out.
Nanotechnology approaches
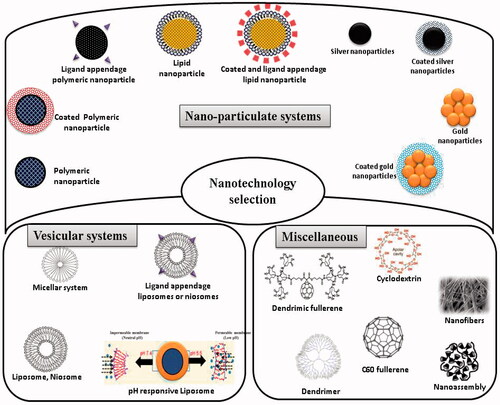
General properties of nanosystems that favor their use in antiretroviral drug delivery are well known and include versatility (virtually drugs may be encapsulated), good toxicity profile, possibility of drug-release modulation, high drug payloads, relative low cost, easiness to produce and possible scale-up to mass production scale (Vyas et al., Citation2006; Mallipeddi & Rohan, Citation2010a; Gaurav et al., Citation2015b,c). Nanocarriers provide an option for the optimal and controlled drug delivery at the target sites in the body. The small size of the nanocarriers enables them to penetrate through cells and deliver drugs intracellular without risking extracellular degradation (Mallipeddi & Rohan, Citation2010b). Conversion of pure drug to nano-scale particles leads to dramatic increase in the surface area and dissolution kinetics leading to increase in bioavailability and reduction in pharmacokinetic variability (Patravale & Kulkarni, Citation2004; Desai et al., Citation2012). Moreover, their ability to incorporate, protect and/or promote the absorption of non-orally administrable anti-HIV drugs, namely mono- or oligonucleotides is of importance to improve the bioavailability of several molecules (Leroux et al., Citation1996; De Jaeghere et al., Citation2000; Gaurav et al., Citation2015c). Some properties of these nanotechnology-based systems, e.g. prolonged release of active agents and ability to penetrate epithelial linings, are important advantages that may favor their utilization in the field of microbicides. mentions a compilation of the nanosystems that are currently under research or can be used in future.
Figure 3. Nanotechnology tools that can be exploited for the rational delivery of fusion inhibitors.
Vesicular systems
This delivery system can be explained as a highly ordered association of one or more concentric lipid bilayers for delivery of drug to the site of infection, preventing drug degradation with no adverse effects (Kumar et al., Citation2011; Chen et al., Citation2015; Goyal et al., Citation2015). The encapsulation of the drug in the vesicular structures prolongs the survival of the drug in systemic circulation, thus reduces the toxicity. Bioavailability of poorly soluble drug increases thereby decreasing the cost of therapy by encapsulating the drug in vesicular delivery system. They can incorporate both lipophilic as well as hydrophilic drugs. Vesicular systems increase the bioavailability of the poorly available drugs. Drug selectively reaches to the site of infection and remains in the systemic circulation for longer time to produce effective results without causing toxicity. The elimination time of rapidly metabolizable drugs increases by its encapsulation in vesicular system thereby causing sustained release effect. Various novel approaches have been developed and are under development focusing different routes of administration to attain sustained, controlled and targeted drug delivery (das Neves et al., Citation2016). A liposomal system Novasomes® was used to deliver 2 RANTES (a truncated fragment of RANTES presenting high specificity for CCR5) binding via vaginal route. Novasomes® 7474 are 200–700 nm non-phospholipidic liposomes comprising monoesters of PEO fatty acids, cholesterol and free fatty acids (Kish-Catalone et al., Citation2007). Native activity of −2 RANTES was effectively restored in this liposomal formulation where only mild signs of inflammation were seen during in vivo studies. This liposomal formulation also showed an efficient pre-exposure prophylaxis when applied (−2 RANTES/liposome complexes in PBS) 30 min before challenge in cynomolgus macaques. Cationic liposomes were successfully developed for the delivery of fusion inhibitor sifuvirtide. Strong electrostatic interactions were studied between sifuvirtide with cationic phosphoplipids providing a great platform for the delivery of anionic peptide sifuvirtide with improved stability and efficacy (Franquelim et al., Citation2011). Earlier in an attempt to develop safe, effective and stable intravaginal microbicides a large multilamellar liposomal formulation (in ointment base) was developed. These liposomes comprising cardiolipin (an anti-HIV lipid) significantly inhibited HIV infections, where reports suggest that cardiolipin composition affect the rate and extent of HIV-1 fusion (Malavia et al., Citation2011).
Polymeric and lipid based nanoparticles
Polymeric nanoparticles range between 10 and 1000 nm, catch great attention while delivering various anti-HIV moieties via different routes (Corsi et al., Citation2016; Narasimhan et al., Citation2016). Owing to several properties possessed by these systems such as easy to synthesize, biocompatible, biodegradable, non-immunogenic, inexpensive, nontoxic, etc., they are considered as versatile nanosystems to study. Different polymers are such as poly(lactic-co-glycolic acid) (PLGA), chitosan, poly(actic acid) (PLA), polyethylene glycol (PEG), poly(γ-glutamic acid) (γ-PGA) and polylysine (PLL). Polymeric nanoparticles can (1) protect encapsulated active molecule from in local biological degradation, (2) yield sustained and enhanced cross-presentation of the active moiety to the viral as well as host cell, (3) undergo endolysosomal escape after uptake by immune cells; and (4) can be tailored by various methods to target specific site (either viral or host cell receptor) (Peek et al., Citation2008; Hamdy et al., Citation2011; Qu et al., Citation2016).
Lipid nanoparticles are particles of nanosize range 50–500 nm made up of lipids that are solid at body temperature. They have potential for sustained/controlled and targeted drug delivery so preferred for pharmaceutical application. Types of lipid nanoparticles were considered for medicinal use. Solid lipid nanoparticles (SLN) have ability to incorporate a wide variety of drugs. These are the spherical shaped particles made up of biodegradable solid lipids and emulsifiers that protects chemically labile ingredients against decomposition (Zhang et al., Citation2007; Elsabahy & Wooley, Citation2012). Nanolipid carriers (NLC) consist of solid lipid matrix with a liquid lipid phase content that enhances greater drug encapsulation and drug loading due to long term stability. SLN and NLCs because of its inherent properties can accommodate both hydrophilic and lipophilic drugs. Further the presence of amphiphilic lipids and emulsifiers makes lipid nanoparticles a suitable alternative to delivery of BCS II and IV (Müllertz et al., Citation2010) drugs.
SLNs are composed of low cost and biodegradable solid lipid (Pardeike et al., Citation2009). Sustained drug release and site specificity for drug delivery can be achieved by altering the properties of lipid based nanocarriers, such as their lipid composition, size and surface charge. The presence of liquid lipid in the NLC confers long-term colloidal stability and greater drug encapsulation and loading unlike SLN phospholipids such as cardiolipin has shown ability to inhibit HIV-1 in vitro.
Polymeric nanoparticles are amongst the trial candidates gaining focus for their potential delivery of fusion inhibitors. Poly(lactic-co-glycolic acid), PLGA nanoparticles encapsulating PSC-RANTES revealed significant anti-HIV activity in cell cultures when compared with unformulated PSC RANTES (Ham et al., Citation2009; Fumakia et al., Citation2016). An attempt was made for the co-delivery of HIV-1 entry inhibitor and nonnucleoside RTI shuttled by nanoparticles. Biodegradable polymeric nanoparticles were synthesized to encapsulate nonnucleoside RTI (NNRTI) DAAN-14f (14f), surface-conjugated with HIV-1 fusion inhibitor T1144, designated T1144-NP-DAAN-14f (T1144-NP-14f) and aiming to achieve enhanced cellular uptake, improved antiviral activity and prolonged blood circulation time (Li et al., Citation2016).
Metal nanoparticles
Metallic nanoparticles are interesting options owing to their intrinsic therapeutic potential and drug carrying properties. Exceptional surface property and smaller size (2–40 nm) provides ample opportunity to functionalize their surface and attach a selective therapeutic moiety. Inorganic metals such as antimony, iron, platinum, calcium, gold and silver have long history as therapeutic agents. Infection inhibition could be enhanced by altering the nanoparticle diameter and/or physical properties and the density of conjugates on the nanoparticle surface (Gaurav et al., Citation2015b).
Gold nanoparticles are effective HIV-1 fusion inhibitors. Gold nanoparticles stabilized with PEG were shown to inhibit M-tropic, T-tropic, dual tropic and resistant isolates of HIV-1. They prevent the viral entry by binding with gp120 and inhibit CD4 attachment (Vijayakumar & Ganesan, Citation2012). Gold nanoparticles layered with numerous copies of an amphiphilic sulfate-ended ligand are able to bind the HIV envelope gp120 to prevent HIV infection of T-cells (Di Gianvincenzo et al., Citation2010). Gold-based compounds have shown favorable activity against HIV-1. A typical example is auranofin which resulted in an elevated CD4+ T-cell count in an HIV patient being treated for psoriatic arthritis (Fonteh et al., Citation2010).
Silver nanoparticles (AgNPs) and NABs target both envelope gp120 and gp41. The addition of AgNPs to NABs exerts an additive effect and highly neutralizing potency to prevent cell-associated HIV-1 infection (Lara et al., Citation2011). Silver nanoparticles show anti-HIV activity at primary stage of viral replication, as a virucidal agent or as an inhibitor of viral entry. They get attached to gp120 and prevent CD4-dependent virion binding, fusion and infectivity. Also both silver sulfadiazine (AgSD) and silver nitrate (AgNO) salts, show anti-HIV activity. Silver nanoparticles interact with the two disulfide bonds situated in the carboxyl half of the HIV-1 gp120 glycoprotein (Lekutis et al., Citation1992) and modify this viral protein by denaturing its disulfide-bonded domain (Lara et al., Citation2010). PVP-coated nanoparticles synthesized by consuming glycerin as both reducing agent and solvent have shown favorable activity against HIV (Elechiguerra et al., Citation2005).
Dendrimers
The word dendrimers itself defines the class of macromolecules as novel, highly branched having 3D architecture differentiating it from linear polymers (Poxon et al., Citation1996). Dendrimers word can also be described as the combo of two words, “Dendron” means Tree and “mer” meaning branching. Thus it is a tree like structure characterized by multiple layers of interior branching from central core, which forms repetitive addition of branched units to its central core molecule by step-growth polymerization process. Dendrimers are very effective against viral infection caused by especially HIV and Herpes virus, thus having good antiviral/antibacterial properties. The key factors behind its antiviral/antibacterial properties are surface charge property, its 3D structure and the effective size of dendrimer. The conformational changes occur during branching of the dendrimer molecule which directly affects the viscosity of the molecule. The generation number of dendrimers relates viscosity parameter of dendrimers. As generation number increases, the viscosity increases to a maximum and then decreases at higher molecular weight causing change in conformation at maximum viscosity and exerts a bell-shaped viscosity curve. Thus higher molecular weight and high generation dendrimers are not very viscous thus easy to handle as compared to other linear polymers.
These are synthetic branched polymer which act as drugs in their own right against HIV (McCarthy et al., Citation2005). Dendrimers act both as therapeutic agents and non-viral vectors of chemical agents for HIV treatment. Some dendrimers with functional end groups associate with the gp120 of HIV and CD4 molecule of host cell to inhibit the attachment of HIV to the host cell, where some of the dendrimers are capable of interrupting into the cell and restrict with the later stages of HIV replication as well (Peng et al., Citation2013). Study on polyanionic carbosilane dendrimers G3-S16 and G2-NF16 with sulfated and naphthylsulfonated showed blocking the entry of different X4 and R5 HIV-1. The combinations of G2-STE16 with other carbosilane dendrimers showed a synergistic profile with 100% inhibition against different HIV-1 isolates (Sepulveda-Crespo et al., Citation2014). Combinations of carbosilane dendrimers/MRV against HIV-1 strains showed synergistic profile (Córdoba et al., Citation2013). SPL7013 is a dendrimer with broad spectrum activity against both X4 and R5 HIV-1 strains. It prevents viral entry by blocking viral attachment and entry. The polyamidoamine (PAMAM) dendrimer targets the gp120–CD4 complex at two stages: it deteriorates the complex and also modifies its dissociation pathway, potentially inhibiting HIV-1 entry. PAMAM disrupted salt bridges and hydrogen bonds across the gp120–CD4 interface and altered the hydration pattern of the hydrophobic cavity in the interface (Nandy et al., Citation2013).
Nanofibers
Various properties of nanofibers make them useful in different fields including its enormous surface area, high porosity, small pore size and the diameter of fibers (Rath et al., Citation2015, 2016b). Different techniques are available for the synthesis of nanofiber including electrospinning, self-assembly and phase separation (Rath et al., Citation2016a). Jiang and coworkers prepared pH-responsive drug delivery system by coating the mussel-inspired protein, polydopamine using poly (ɛ-caprolactone) polymer and mediated surface functionalization of electrospun nanofibers. Nanofibers were also used for the treatment of HIV by introducing anti-HIV drugs into vagina. Electrospun fibers of cellulose acetate phthalate (CAP) were prepared for semen induced anti-HIV vaginal drug delivery. Fibers prepared by electrospinning process were incorporated with anti-viral drugs in CAP fibers for pH dependent release of anti-viral drug from fibers in presence of human semen. Cellulose acetate phthalate nanofibers were well tolerated by vaginal epithelial cells and vaginal microflora. Due to pH-sensitive nature of CAP, nanofibers maintained integrity in acidic pH (vaginal environment). However, addition of semen to nanofibers led to immediate dissolution of CAP. CAP nanofibers retained the ability to prevent HIV-1 entry. Incorporation of tenofovir in these nanofibers significantly improved its antiviral activity. Polymers like polyvinylpyrrolidone, poly-l-lactide, PLGA, PCL were also used for the fabrication and incorporating different antiviral drugs either individually or as composite.
Others nanosystems (cyclodextrins, polymeric micelles) ()
Cyclodextrins are cyclic oligosaccharides that form hydrophilic inclusion complexes with small or large compounds or can form cyclodextrin drug conjugates (Gaurav et al., Citation2015a). They can be used in pharmaceutical applications for multifunctional purposes due to its biocompatible nature. Cyclodextrins exhibit property of complexation with different substances which are useful for the enhancement of different properties including increased efficacy, stability and solubility of poorly soluble drugs. Different anti HIV-1 molecules including some nano-metalo herbal complexes were also delivered after forming inclusion complex with different cyclodextrin derivatives (Gaurav et al., Citation2014, 2015a). Reports also suggest that a derivative cyclodextrin sulfate can block attachment of HIV virions in a nonspecific sense.
Figure 4. Cyclodextrins and polymeric micelle as nanodrug delivery systems.
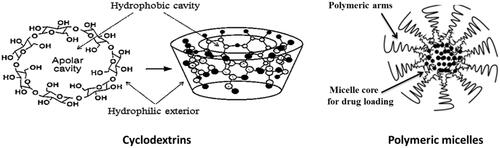
Polymeric micelles are a type of nanodelivery systems that hold the poorly aqueous soluble drug in the hydrophobic interior of the micelles and cause drug delivery. They are nanocarriers of nanoscopic shell/core structures formed through amphiphilic block copolymers. Targeted drug delivery, controlled drug release and drug solubilization are their properties that make them suitable for drug delivery purposes. The polymeric micelles showed better results as vaginal microbicide therapy rather than other routes. NanoViricides Inc. is a trademark for polymeric chemical chain which is covalently attached to ligands on virus particles to engulf or coat it for specific virus target. This leads to neutralize the virus infectivity, destabilize and may dismantle it. Thus nano-microbicides are potentially advanced techniques (Aliabadi & Lavasanifar, 2006). Croy et al. reviewed on polymeric micelles for drug delivery to reduce toxicities, delivery to targeted sites and to enhance the therapeutic efficacy of active pharmaceutical ingredients. Polymeric micelles composed of block copolymers have been utilized for improving aqueous solubility, membrane permeability and site-specific delivery of several drug moieties. They have a vesicular or core shell structure similar to surfactant-based micelles, but self-associate at much lower concentrations, typically in the 0.1–1 μM range compared with 0.1–1 mM for surfactant-based micelles. The core of the micelles is usually derived from polymers such as propylene oxide, aspartic acid, l-lysine and caprolactone which constitute the hydrophobic block.
Conclusion
An efficient anti-HIV treatment will surely have a great impact on the world’s population specifically in the microbicide arena where topical prophylaxis is the key. The widespread requirement of the era where vaccination is not available critically requires a dependable strategy. New class of ARVs and their combination therapeutics provided a positive reflection but no real curable success. Need is there to reassess the potential of the targets we are following till date. Fusion and binding inhibition seems to possess that potential, with the only target which control the viral bioload before it get host entry. Basics of the anatomical and physiological factors related to HIV and its host cells, ease us to understand various available as well as hidden targets. Selection of the delivery system is the key to exploit these targets and the outcomes will be more positive if rational use of nanotechnological approaches would be made.
Reduced bio-load is always beneficial to tackle any pandemic’s progression and especially in case of HIV where host involvement is the key for its spread. Among the versatile therapeutic strategies to sequester its spread or provide prophylaxis from the existing modes, inhibiting the access of virus inside the host cell seems to be most interesting. attempts to analyze a very simple concept of viral load and the site where an efficient inhibition can be attained. HIV-1 binding and fusion (entry) inhibitors seem to be the fascinating option in this contrast. A number of candidates had gone through the screening with their possible targets of viral attachment, co-receptor binding and fusion. Structural exploration of the receptors and co-receptors is continuously aiding the research units to seek newer targets. Especially for topical pre-exposure prophylaxis (PrEP) where sexual transmission via vaginal and rectal route is under scan, this strategy responds quite early (usually within few minutes to couple of hours). After surfactants and viral disrupting agents which act on the spot of viral exposures this is fastest acting control, while RTI, protease and integrase inhibitors generally take hours to days. Presently, the clinical stake of binding and fusion inhibitors is not up to the mark expressing the negligence of such key target. Moreover the selection of the drug delivery approaches is not need based.
Figure 5. Logical assessment of the best inhibition target with respect to HIV viral load.
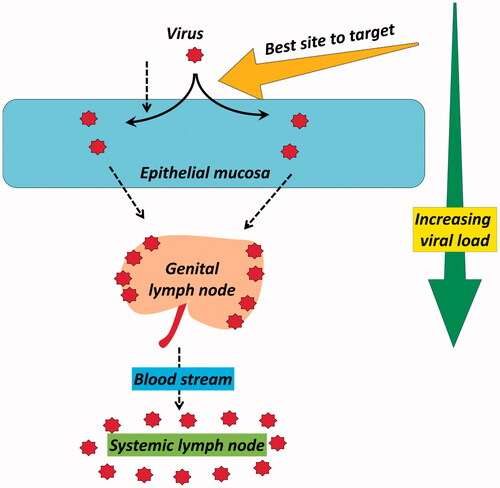
Focusing on the anatomical factors of both HIV and host cells is the key to exploit this fusion inhibition. First challenge in this context is the structural exploration of all the associated receptors and co-receptors. Pharmacoinformatic persons are working in this field to detail the structures and to optimize the pharmacophores of new entities having the capacity to bind these sites. Kinetics and specificity of binding of a new entity with a receptor or co-receptor will determine its potential in real world. Failure of immunization trials literally ends the investment in the area of anti-HIV vaccine development. New vaccination strategies are required aiming the targets responsible for fusion and binding of HIV.
As mentioned in the introduction, lipid composition of the virus is one of the decisive reasons behind the smartness of HIV. Till date, only receptors and co-receptors are targeted for this kind of inhibition. But it is important to understand that it is the flexibility of the viral envelop which regulates most of viral fusion events. Nanotechnology approaches aiming this lipid such as nanocarbon and cyclodextrins which can carry drug too, will be a great solution to strengthen this inhibition approach. Rational selection of drug delivery platform and exploiting the nanosystems can surely improve this approach.
Declaration of interest
The authors declare no competing financial interest.
Supplementary materials available online.
Supplementary_information.docx
Download MS Word (40 KB)Acknowledgements
The authors acknowledge Department of Biotechnology (DBT) India, Punjab Technical University, Jalandhar and Punjab State Council of Science and Technology (PSCST), India for the support.
References
- Ackerman AL, Cresswell P. (2004). Cellular mechanisms governing cross-presentation of exogenous antigens. Nat Immunol 5:678–84
- Allaway GP, Davis-Bruno KL, Beaudry GA, et al. (1995). Expression and characterization of CD4-IgG2, a novel heterotetramer that neutralizes primary HIV type 1 isolates. AIDS Res Hum Retroviruses 11:533–9
- Balzarini J, Van Laethem K, Peumans WJ, et al. (2006). Mutational pathways, resistance profile, and side effects of cyanovirin relative to human immunodeficiency virus type 1 strains with N-glycan deletions in their gp120 envelopes. J Virol 80:8411–21
- Briz V, Poveda E, Soriano V. (2006). HIV entry inhibitors: mechanisms of action and resistance pathways. J Antimicrob Chemother 57:619–27
- Buffa V, Stieh D, Mamhood N, et al. (2009). Cyanovirin-N potently inhibits human immunodeficiency virus type 1 infection in cellular and cervical explant models. J Gen Virol 90:234–43
- Cavrois M, Neidleman J, Yonemoto W, et al. (2004). HIV-1 virion fusion assay: uncoating not required and no effect of Nef on fusion. Virology 328:36–44
- Chan DC, Chutkowski CT, Kim PS. (1998). Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci 95:15613–7
- Chan DC, Fass D, Berger JM, Kim PS. (1997). Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–73
- Chang JJ, Altfeld M. (2009). TLR-mediated immune activation in HIV. Blood 113:269–70
- Chauhan G, Rath G, Goyal AK. (2013). In-vitro anti-viral screening and cytotoxicity evaluation of copper–curcumin complex. Artif Cells Nanomed Biotechnol 41:276–81
- Chen J, Lin A, Peng P, et al. (2015). Lipid composition has significant effect on targeted drug delivery properties of NGR-modified liposomes. Drug Deliv 23:1426–33
- Córdoba EV, Arnáiz E, Francisco J, et al. (2013). Synergistic activity of carbosilane dendrimers in combination with maraviroc against HIV in vitro. AIDS 27:2053–8
- Corsi F, Fiandra L, Rizzardini G. (2016). New perspectives on nanotechnology and antiretroviral drugs: a ‘small’ solution for a big promise in HIV treatment? AIDS 30:963–4
- Das Neves J, Amiji MM, Bahia MF, Sarmento B. (2010a). Nanotechnology-based systems for the treatment and prevention of HIV/AIDS. Adv Drug Deliv Rev 62:458–77
- Das Neves J, Amiji MM, Bahia MF, Sarmento B. (2010b). Nanotechnology-based systems for the treatment and prevention of HIV/AIDS. Adv Drug Deliv Rev 62:458–77
- Das Neves J, Nunes R, Rodrigues F, Sarmento B. (2016). Nanomedicine in the development of anti-HIV microbicides. Adv Drug Deliv Rev 103:57–75
- Date AA, Destache CJ. (2013). A review of nanotechnological approaches for the prophylaxis of HIV/AIDS. Biomaterials 34:6202–28
- D'cruz OJ, Uckun FM. (2014). Vaginal microbicides and their delivery platforms. Expert Opin Drug Deliv 11:723–40
- De Jaeghere F, Allemann E, Kubel F, et al. (2000). Oral bioavailability of a poorly water soluble HIV-1 protease inhibitor incorporated into pH-sensitive particles: effect of the particle size and nutritional state. J Control Release 68:291–8
- Delézay O, Yahi N, Fantini J. (1996). Detection of functional galactosylceramide (GalCer) receptors on CD4-negative HIV-1 target cells. Perspect Drug Discov Des 5:192–202
- Desai PP, Date AA, Patravale VB. (2012). Overcoming poor oral bioavailability using nanoparticle formulations – opportunities and limitations. Drug Discov Today: Technol 9:e87–95
- Dey B, Lerner DL, Lusso P, et al. (2000). Multiple antiviral activities of cyanovirin-N: blocking of human immunodeficiency virus type 1 gp120 interaction with CD4 and coreceptor and inhibition of diverse enveloped viruses. J Virol 74:4562–9
- Di Gianvincenzo P, Marradi M, Martinez-Avila OM, et al. (2010). Gold nanoparticles capped with sulfate-ended ligands as anti-HIV agents. Bioorg Med Chem Lett 20:2718–21
- Doms RW, Trono D. (2000). The plasma membrane as a combat zone in the HIV battlefield. Genes Dev 14:2677–88
- Donzella GA, Schols D, Lin SW, et al. (1998). AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med 4:72–7
- Eckert DM, Kim PS. (2001). Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc Natl Acad Sci 98:11187–92
- Eckert DM, Malashkevich VN, Hong LH, et al. (1999). Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell 99:103–15
- Elechiguerra JL, Burt JL, Morones JR, et al. (2005). Interaction of silver nanoparticles with HIV-1. J Nanobiotechnol 3:1–10
- Elsabahy M, Wooley KL. (2012). Design of polymeric nanoparticles for biomedical delivery applications. Chem Soc Rev 41:2545–61
- Emau P, Tian B, O’keefe B, et al. (2007). Griffithsin, a potent HIV entry inhibitor, is an excellent candidate for anti‐HIV microbicide. J Med Primatol 36:244–53
- Fatkenheuer G, Pozniak AL, Johnson MA, et al. (2005). Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med 11:1170–2
- Flomenberg N, Devine SM, Dipersio JF, et al. (2005). The use of AMD3100 plus G-CSF for autologous hematopoietic progenitor cell mobilization is superior to G-CSF alone. Blood 106:1867–74
- Fonteh PN, Keter FK, Meyer D. (2010). HIV therapeutic possibilities of gold compounds. Biometals 23:185–96
- Franquelim HG, De-Sousa FF, Veiga AS, et al. (2011). Cationic liposomes are possible drug-delivery systems for HIV fusion inhibitor sifuvirtide. Soft Matter 7:11089–92
- Fumakia M, Yang S, Gu J, Ho EA. (2016). Protein/peptide‐based entry/fusion inhibitors as anti‐HIV therapies: challenges and future direction. Rev Med Virol 26:4–20
- Furuta RA, Wild CT, Weng Y, Weiss CD. (1998). Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Mol Biol 5:276–9
- Galvin SR, Cohen MS. (2004). The role of sexually transmitted diseases in HIV transmission. Nat Rev Microbiol 2:33–42
- Gaurav C, Goutam R, Rohan KN, et al. (2014). (Copper–curcumin) β-cyclodextrin vaginal gel: delivering a novel metal–herbal approach for the development of topical contraception prophylaxis. Eur J Pharm Sci 65:183–91
- Gaurav C, Goutam R, Rohan KN, et al. (2015a). In situ stabilized AgNPs and (Cu-Cur) CD dispersed gel, a topical contraceptive antiretroviral (ARV) microbicide. RSC Adv 5:83013–28
- Gaurav C, Nikhil G, Deepti S, et al. (2015b). Albumin stabilized silver nanoparticles – clotrimazole β-cyclodextrin hybrid nanocomposite for enriched anti-fungal activity in normal and drug resistant Candida cells. RSC Adv 5:71190–202
- Gaurav C, Saurav B, Goutam R, K Goyal A. (2015c). Nano-systems for advanced therapeutics and diagnosis of atherosclerosis. Curr Pharm Des 21:4498–508
- Geijtenbeek TB, Kwon DS, Torensma R, et al. (2000). DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100:587–97
- Goyal G, Garg T, Malik B, et al. (2015). Development and characterization of niosomal gel for topical delivery of benzoyl peroxide. Drug Deliv 22:1027–42
- Greenberg ML, Cammack N. (2004). Resistance to enfuvirtide, the first HIV fusion inhibitor. J Antimicrob Chemother 54:333–40
- Groot F, Welsch S, Sattentau QJ. (2008). Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 111:4660–3
- Ham AS, Cost MR, Sassi AB, et al. (2009). Targeted delivery of PSC-RANTES for HIV-1 prevention using biodegradable nanoparticles. Pharm Res 26:502–11
- Hamdy S, Haddadi A, Hung RW, Lavasanifar A. (2011). Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations. Adv Drug Deliv Rev 63:943–55
- Hammer SM, Eron JJ, Reiss P, et al. (2008). Antiretroviral treatment of adult HIV infection: 2008 recommendations of the International AIDS Society – USA panel. JAMA 300:555–70
- Henrich TJ, Kuritzkes DR. (2013). HIV-1 entry inhibitors: recent development and clinical use. Curr Opin Virol 3:51–7
- Hladik F, Sakchalathorn P, Ballweber L, et al. (2007). Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity 26:257–70
- Hu Q, Mahmood N, Shattock RJ. (2007). High-mannose-specific deglycosylation of HIV-1 gp120 induced by resistance to cyanovirin-N and the impact on antibody neutralization. Virology 368:145–54
- Ichiyama K, Yokoyama-Kumakura S, Tanaka Y, et al. (2003). A duodenally absorbable CXC chemokine receptor 4 antagonist, KRH-1636, exhibits a potent and selective anti-HIV-1 activity. Proc Natl Acad Sci USA 100:4185–90
- Jacobson JM, Lowy I, Fletcher CV, et al. (2000). Single-dose safety, pharmacology, and antiviral activity of the human immunodeficiency virus (HIV) type 1 entry inhibitor PRO 542 in HIV-infected adults. J Infect Dis 182:326–9
- Kahle KM, Steger HK, Root MJ. (2009). Asymmetric deactivation of HIV-1 gp41 following fusion inhibitor binding. PLoS Pathog 5:e1000674
- Karim QA, Baxter C. (2013). Microbicides for the prevention of sexually transmitted HIV infection. Expert Rev Anti-Infect Ther 11:12–23
- Kilgore NR, Salzwedel K, Reddick M, et al. (2003). Direct evidence that C-peptide inhibitors of human immunodeficiency virus type 1 entry bind to the gp41 N-helical domain in receptor-activated viral envelope. J Virol 77:7669–72
- Kish-Catalone T, Pal R, Parrish J, et al. (2007). Evaluation of-2 RANTES vaginal microbicide formulations in a nonhuman primate simian/human immunodeficiency virus (SHIV) challenge model. AIDS Res Hum Retrovirus 23:33–42
- Koshiba T, Chan DC. (2003). The prefusogenic intermediate of HIV-1 gp41 contains exposed C-peptide regions. J Biol Chem 278:7573–9
- Kuhmann SE, Platt EJ, Kozak SL, Kabat D. (2000). Cooperation of multiple CCR5 coreceptors is required for infections by human immunodeficiency virus type 1. J Virol 74:7005–15
- Kumar R, Kumar S, Jha SS, Jha AK. (2011). Vesicular system-carrier for drug delivery. Der Pharm Sin 4:192–202
- Kwong PD, Wyatt R, Robinson J, et al. (1998). Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–59
- Labrosse B, Brelot A, Heveker N, et al. (1998). Determinants for sensitivity of human immunodeficiency virus coreceptor CXCR4 to the bicyclam AMD3100. J Virol 72:6381–8
- Lara HH, Ayala-Nuñez NV, Ixtepan-Turrent L, Rodriguez-Padilla C. (2010). Mode of antiviral action of silver nanoparticles against HIV-1. J Nanobiotechnol 8:1
- Lara HH, Ixtepan-Turrent L, Treviño ENG, Singh DK. (2011). Use of silver nanoparticles increased inhibition of cell-associated HIV-1 infection by neutralizing antibodies developed against HIV-1 envelope proteins. J Nanobiotechnol 9:38
- Lee B, Sharron M, Blanpain C, et al. (1999). Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J Biol Chem 274:9617–26
- Lekutis C, Olshevsky U, Furman C, et al. (1992). Contribution of disulfide bonds in the carboxyl terminus of the human immunodeficiency virus type I gp120 glycoprotein to CD4 binding. JAIDS: J Acquir Immune Defic Syndr 5:78–81
- Leroux JC, Cozens RM, Roesel JL, et al. (1996). pH-sensitive nanoparticles: an effective means to improve the oral delivery of HIV-1 protease inhibitors in dogs. Pharm Res 13:485–7
- Li W, Yu F, Wang Q, et al. (2016). Co-delivery of HIV-1 entry inhibitor and nonnucleoside reverse transcriptase inhibitor shuttled by nanoparticles: cocktail therapeutic strategy for antiviral therapy. AIDS 30:827–38
- Lin PF, Blair W, Wang T, et al. (2003). A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci 100:11013–18
- Lindahl U. (2007). Heparan sulfate-protein interactions – a concept for drug design? Thromb Haemost-Stuttgart 98:109
- Liu J, Bartesaghi A, Borgnia MJ, et al. (2008). Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–13
- Louis JM, Bewley CA, Clore GM. (2001). Design and properties of NCCG-gp41, a chimeric gp41 molecule with nanomolar HIV fusion inhibitory activity. J Biol Chem 276:29485–9
- Lu M, Blacklow SC, Kim PS. (1995). A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Mol Biol 2:1075–82
- Maeda K, Nakata H, Koh Y, et al. (2004). Spirodiketopiperazine-based CCR5 inhibitor which preserves CC-chemokine/CCR5 interactions and exerts potent activity against R5 human immunodeficiency virus type 1 in vitro. J Virol 78:8654–62
- Malavia NK, Zurakowski D, Schroeder A, et al. (2011). Liposomes for HIV prophylaxis. Biomaterials 32:8663–8
- Mallipeddi R, Rohan LC. (2010a). Nanoparticle-based vaginal drug delivery systems for HIV prevention. Expert Opin Drug Deliv 7:37–48
- Mallipeddi R, Rohan LC. (2010b). Progress in antiretroviral drug delivery using nanotechnology. Int J Nanomed 5:533
- Marmor M, Hertzmark K, Thomas SM, et al. (2006). Resistance to HIV infection. J Urban Health 83:5–17
- Matos PM, Castanho MA, Santos NC. (2010). HIV-1 fusion inhibitor peptides enfuvirtide and T-1249 interact with erythrocyte and lymphocyte membranes. PLoS One 5:e9830
- Matthews T, Salgo M, Greenberg M, et al. (2004). Enfuvirtide: the first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov 3:215–25
- Mccarthy TD, Karellas P, Henderson SA, et al. (2005). Dendrimers as drugs: discovery and preclinical and clinical development of dendrimer-based microbicides for HIV and STI prevention. Mol Pharm 2:312–18
- Mcgowan I. (2014). The development of rectal microbicides for HIV prevention. Expert Opin Drug Deliv 11:69–82
- Melikyan GB, Markosyan RM, Hemmati H, et al. (2000). Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol 151:413–23
- Montgomery CM. (2015). ‘HIV has a woman's face’: vaginal microbicides and a case of ambiguous failure. Anthropol Med 22:250–62
- Müllertz A, Ogbonna A, Ren S, Rades T. (2010). New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol 62:1622–36
- Murakami T, Nakajima T, Koyanagi Y, et al. (1997). A small molecule CXCR4 inhibitor that blocks T cell line-tropic HIV-1 infection. J Exp Med 186:1389–93
- Nagashima KA, Thompson DA, Rosenfield SI, et al. (2001). Human immunodeficiency virus type 1 entry inhibitors PRO 542 and T-20 are potently synergistic in blocking virus-cell and cell-cell fusion. J Infect Dis 183:1121–5
- Nandy B, Bindu DH, Dixit NM, Maiti PK. (2013). Simulations reveal that the HIV-1 gp120-CD4 complex dissociates via complex pathways and is a potential target of the polyamidoamine (PAMAM) dendrimer. J Chem Phys 139:024905
- Narasimhan B, Goodman JT, Vela Ramirez JE. (2016). Rational design of targeted next-generation carriers for drug and vaccine delivery. Annu Rev Biomed Eng 18:25–49
- Nikolic DS, Garcia E, Piguet V. (2007). Microbicides and other topical agents in the prevention of HIV and sexually transmitted infections. Expert Rev Anti-Infect Ther 5:77–88
- Nishibu A, Ward BR, Jester JV, et al. (2006). Behavioral responses of epidermal Langerhans cells in situ to local pathological stimuli. J Invest Dermatol 126:787–96
- Nittayananta W, Weinberg A, Malamud D, et al. (2016). Innate immunity in HIV‐1 infection: epithelial and non‐specific host factors of mucosal immunity – a workshop report. Oral Dis 22:171–80
- Pardeike J, Hommoss A, Müller RH. (2009). Lipid nanoparticles (SLN, NLC) in cosmetic and pharmaceutical dermal products. Int J Pharm 366:170–84
- Patravale V, Kulkarni R. (2004). Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol 56:827–40
- Peek LJ, Middaugh CR, Berkland C. (2008). Nanotechnology in vaccine delivery. Adv Drug Deliv Rev 60:915–28
- Peng J, Wu Z, Qi X, et al. (2013). Dendrimers as potential therapeutic tools in HIV inhibition. Molecules 18:7912–29
- Phogat S, Wyatt R, Karlsson Hedestam G. (2007). Inhibition of HIV‐1 entry by antibodies: potential viral and cellular targets. J Intern Med 262:26–43
- Poxon S, Mitchell P, Liang E, Hughes J. (1996). Dendrimer delivery of oligonucleotides. Drug Deliv 3:255–61
- Qu J, Zhang L, Chen Z, et al. (2016). Nanostructured lipid carriers, solid lipid nanoparticles, and polymeric nanoparticles: which kind of drug delivery system is better for glioblastoma chemotherapy? Drug Deliv 1–28
- Raja A, Venturi M, Kwong P, Sodroski J. (2003). CD4 binding site antibodies inhibit human immunodeficiency virus gp120 envelope glycoprotein interaction with CCR5. J Virol 77:713–18
- Rath G, Hussain T, Chauhan G, et al. (2015). Fabrication and characterization of cefazolin-loaded nanofibrous mats for the recovery of post-surgical wound. Artif Cells Nanomed Biotechnol 1–10
- Rath G, Hussain T, Chauhan G, et al. (2016a). Collagen nanofiber containing silver nanoparticles for improved wound-healing applications. J Drug Target 24:520–9
- Rath G, Hussain T, Chauhan G, et al. (2016b). Development and characterization of cefazolin loaded zinc oxide nanoparticles composite gelatin nanofiber mats for postoperative surgical wounds. Mater Sci Eng: C 58:242–53
- Rimsky LT, Shugars DC, Matthews TJ. (1998). Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J Virol 72:986–93
- Root MJ, Kay MS, Kim PS. (2001). Protein design of an HIV-1 entry inhibitor. Science 291:884–8
- Root MJ, Steger HK. (2004). HIV-1 gp41 as a target for viral entry inhibition. Curr Pharm Des 10:1805–25
- Rusnati M, Vicenzi E, Donalisio M, et al. (2009). Sulfated K5 Escherichia coli polysaccharide derivatives: a novel class of candidate antiviral microbicides. Pharmacol Ther 123:310–22
- Sedwick C. (2012). How HIV sneaks aboard mature dendritic cells. PLoS Biol 10:e1001454
- Sepulveda-Crespo D, Lorente R, Leal M, et al. (2014). Synergistic activity profile of carbosilane dendrimer G2-STE16 in combination with other dendrimers and antiretrovirals as topical anti-HIV-1 microbicide. Nanomedicine 10:609–18
- Shenoy SR, O'keefe BR, Bolmstedt AJ, et al. (2001). Selective interactions of the human immunodeficiency virus-inactivating protein cyanovirin-N with high-mannose oligosaccharides on gp120 and other glycoproteins. J Pharmacol Exp Ther 297:704–10
- Stantchev TS, Markovic I, Telford WG, et al. (2007). The tyrosine kinase inhibitor genistein blocks HIV-1 infection in primary human macrophages. Virus Res 123:178–89
- Teissier E, Penin F, Pecheur EI. (2011). Targeting cell entry of enveloped viruses as an antiviral strategy. Molecules 16:221–50
- Tiberi M, Tintori C, Ceresola ER, et al. (2014). 2-aminothiazolones as anti-HIV agents which act as gp120-CD4 Inhibitors. Antimicrob Agents Chemother 58:3043–52
- Tobiume M, Lineberger JE, Lundquist CA, et al. (2003). Nef does not affect the efficiency of human immunodeficiency virus type 1 fusion with target cells. J Virol 77:10645–50
- Van Lelyveld SF, Drylewicz J, Krikke M, et al. (2015). Maraviroc intensification of cART in patients with suboptimal immunological recovery: a 48-week, placebo-controlled randomized trial. PLoS One 10:e0132430
- Veazey RS, Marx PA, Lackner AA. (2003). Vaginal CD4+ T cells express high levels of CCR5 and are rapidly depleted in simian immunodeficiency virus infection. J Infect Dis 187:769–76
- Vermeire K, Bell TW, Choi HJ, et al. (2003). The anti-HIV potency of cyclotriazadisulfonamide analogs is directly correlated with their ability to down-modulate the CD4 receptor. Mol Pharmacol 63:203–10
- Vermeire K, Zhang Y, Princen K, et al. (2002). CADA inhibits human immunodeficiency virus and human herpesvirus 7 replication by down-modulation of the cellular CD4 receptor. Virology 302:342–53
- Vijayakumar S, Ganesan S. (2012). Gold nanoparticles as an HIV entry inhibitor. Curr HIV Res 10:643–6
- Vyas TK, Shah L, Amiji MM. (2006). Nanoparticulate drug carriers for delivery of HIV/AIDS therapy to viral reservoir sites Expert Opin Drug Deliv 3:613–28
- Watson Buckheit K, Yang L, Buckheit RW, Jr. (2011). Development of dual-acting pyrimidinediones as novel and highly potent topical anti-HIV microbicides. Antimicrob Agents Chemother 55:5243–54
- Weissenhorn W, Dessen A, Calder LJ, et al. (1999). Structural basis for membrane fusion by enveloped viruses. Mol Membr Biol 16:3
- Weissenhorn W, Dessen A, Harrison SC, et al. (1997). Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–30
- Wild CT, Shugars DC, Greenwell TK, et al. (1994). Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci 91:9770–4
- Williamson MP, Mccormick TG, Nance CL, Shearer WT. (2006). Epigallocatechin gallate, the main polyphenol in green tea, binds to the T-cell receptor, CD4: potential for HIV-1 therapy. J Allergy Clin Immunol 118:1369–74
- Zhang J, Liu J, Li X, Jasti B. (2007). Preparation and characterization of solid lipid nanoparticles containing silibinin. Drug Deliv 14:381–7