
ABSTRACT
Objective
To develop an updated staging system for long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency (LCHADD) chorioretinopathy based on contemporary multimodal imaging and electrophysiology.
Methods
We evaluated forty cases of patients with genetically confirmed LCHADD or trifunctional protein deficiency (TFPD) enrolled in a prospective natural history study. Wide-field fundus photographs, fundus autofluorescence (FAF), optical coherence tomography (OCT), and full-field electroretinogram (ffERG) were reviewed and graded for severity.
Results
Two independent experts first graded fundus photos and electrophysiology to classify the stage of chorioretinopathy based upon an existing published system. With newer imaging modalities and improved electrophysiology, many patients did not fit cleanly into a single traditional staging group. Therefore, we developed a novel staging system that better delineated the progression of LCHADD retinopathy. We maintained the four previous delineated stages but created substages A and B in stages 2 to 3 to achieve better differentiation.
Discussion
Previous staging systems of LCHADD chorioretinopathy relied on only on the assessment of standard 30 to 45-degree fundus photographs, visual acuity, fluorescein angiography (FA), and ffERG. Advances in recordings of ffERG and multimodal imaging with wider fields of view, allow better assessment of retinal changes. Following these advanced assessments, seven patients did not fit neatly into the original classification system and were therefore recategorized under the new proposed system.
Conclusion
The new proposed staging system improves the classification of LCHADD chorioretinopathy, with the potential to lead to a deeper understanding of the disease’s progression and serve as a more reliable reference point for future therapeutic research.
1. Introduction
Long chain 3-hydroxy-acyl-CoA dehydrogenase deficiency (LCHADD) and trifunctional protein deficiency (TFPD) are rare mitochondrial disorders resulting from the deficiency of enzymes that catalyze the second through fourth steps of the long-chain fatty acid β-oxidation pathway (Citation1). Mitochondrial trifunctional protein (MTP) is a heterotetramer composed of two alpha and two beta subunits containing enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase and 3-ketothiolase activities encoded by the nuclear HADHA (OMIM # 600890) and HADHB (OMIM # 143450) genes (Citation2,Citation3). Mutations in these genes result in dysfunction of one or all of these activities (Citation4). Complete MTP deficiency is often caused by mutations that affect amino acid residues at the interface of the dimerization domains, compromising the integrity of MTP stability resulting in loss of all three enzymatic activities (Citation2). In contrast, some reported mutations in either HADHB or HADHA, typically located in the catalytic sites, can affect only a single enzymatic function (Citation5). In LCHADD, a common pathogenic G to C transition at nucleotide 1528 (1528 G>C) of the HADHA gene results in a glutamic acid to glutamine amino acid change (E510Q) in the LCHAD active site and affects the 3-hydroxyacyl-CoA dehydrogenase enzyme activity, with relatively normal activity in the other two MTP enzymes (Citation6). Affected individuals have life-threatening episodes of hypoketotic hypoglycemia with fasting or physiologic stress, recurrent rhabdomyolysis, polyneuropathy, hepatopathy, myopathy, and cardiomyopathy (Citation7). Pigmentary chorioretinopathy is a common complication of LCHADD, occurring in at least 70% of LCHADD individuals, but is rare in TFPD and all other fatty acid oxidation disorders (Citation4,Citation8,Citation9). Hypoglycemia, rhabdomyolysis, and cardiomyopathy can be mitigated to some extent through the dietary therapy for LCHADD, which includes restriction of dietary long-chain fat, treatment with medium-chain triglycerides (MCT) including triheptanoin, and avoidance of fasting (Citation10–12). However, chorioretinopathy, which can appear as early as the first year of life, and the neuropathy are only marginally improved. Patients identified by newborn screening have slower progression of pigmentary clumping and chorioretinal atrophy compared to those identified symptomatically later in life (Citation13,Citation14).
The original staging system for retinal disease proposed four stages for LCHADD chorioretinopathy (Citation9). In stage 1, the fundus appears normal or pale, similar to the fundus of the newborn. In stage 2, pigment accumulation is most abundant in the macula with or without retinal pigment epithelium (RPE) hypopigmentation. In stage 3, the posterior pole demonstrates progressive chorioretinal atrophy sparing the central fovea and peripheral fundus and posterior staphyloma may be observed. In stage 4, there is significant chorioretinal atrophy extending to the central fovea and progressing peripherally. An update to this system was proposed in 2012, dividing stage 2 into subcategories by an additional grading of pigmentary deposits into 3 severity levels (P1–P3) and dividing RPE atrophy into 3 categories (A1–A3) (Citation15).
Since the development of the original staging system, advances in electrophysiology and wide field imaging have identified new functional markers not included in the current grading system. For example, improvements in electrodes or amplifiers used for ERG recording have improved the signal-to-noise ratio that is achievable thus allowing for the recording of preserved signals that would have been categorized as unrecordable in the past (Citation16–20). The introduction of widefield imaging allows routine imaging of the peripheral retina up to the vortex veins. Finally, the widespread adaptation of OCT and optical coherence tomography angiography (OCTA) now permits near cellular resolution of the retina, RPE, choriocapillaris, and large choroidal vessels. We have employed these imaging technologies in our current prospective natural history study of LCHADD and TFPD. Based on our findings, we propose a refined staging classification for LCHADD that builds off the original, but updates criteria by utilizing newer diagnostic techniques and improved electrophysiology.
2. Subjects and methods
2.1. Eligibility criteria
Forty patients were part of an observational prospective LCHADD Natural History Study. Inclusion criteria included patients aged 2–60 years diagnosed with TFPD or LCHADD. They were examined at the ophthalmic genetics’ clinic at the Casey Eye Institute of Oregon Health & Science University or the University of Pittsburgh from January 2020 through December 2022. Diagnosis of LCHADD or TFPD was confirmed by obtaining medical records, results of acylcarnitine profiles, fatty acid oxidation probe studies in cultured fibroblasts, and/or genetic testing. The patients, 10 who were homozygous and 27 who were compound heterozygous for the common HADHA 1528 G>C variant and 3 with other pathogenic mutations, underwent an ocular examination, including measurement of best-corrected visual acuity, slit-lamp biomicroscopy, and a dilated fundus exam.
2.2. Retinal imaging
Thirty-degree fundus photographs were obtained with a FF450 camera (Carl Zeiss Meditec, Dublin, CA) and ultra-widefield fundus photography and fundus autofluorescence (FAF) were obtained with an Optos 200Tx confocal scanning laser ophthalmoscope (Optos PLC, Marlborough, MA). Structural macular assessment was performed by a Spectralis spectral-domain OCT2 (Heidelberg Engineering, Heidelberg, Germany), OCTA by Avanti RTVue XR (OptoVue, Inc., Freemont, CA) or Solix (OptoVue, Inc., Freemont, CA) and functional assessment of cones and rods were obtained by full-field electroretinogram (ffERG)(Diagnosys LLC; Lowell, MA) using DTL electrodes in most patients, and either skin electrodes(n = 4) or Burian-Allen electrodes (Hansen Ophthalmic Development Lab, Inc., Bellingham, WA)(n = 2) in younger participants who were sedated.
3. Results
Participants were 2 to 36 years of age at enrollment. All retinal imaging was reviewed and staged based on the Tyni system independently by two ophthalmologists familiar with LCHADD (MEP and NW; ). Disagreement on the staging system was settled by open adjudication. During adjudication, it became clear that many patients were not easily categorized for several reasons. For example, in patients who would have previously been defined as Stage 2, we found that 3 out of 13 exhibited early evidence of photoreceptor degeneration in the form of a disruption of the ellipsoid zone. OCT revealed that the accumulation of pigment in the fovea correlated with a thickening of the RPE-Bruch complex on OCT and subretinal fibrotic scars were more visible, suggesting past choroidal neovascularization. As chorioretinal atrophy progressed, outer retinal tubulations were observed. Based on widefield photos and FAF imaging, it was apparent that patients could be divided into those who had speckled autofluorescence changes mostly within the vascular arcades and those with changes anterior to the arcades (9 with stage 2A and 4 with stage 2B). By prior definition, Stage 2 patients have a progressively low ERG, but we found 9 patients who were consistent with Stage 2 but had ERGs that fell within the normal range. To differentiate between these different stage 2 patients, we propose dividing this group into Stage 2A and Stage 2B as defined in and illustrated in . For the three out of forty patients who were diagnosed with TFP deficiency (defined as mutations other than G1528C), two were stage 1 and the other was stage 2A. All three subjects presented with a milder form of retinopathy, with visual acuity 20/25 or better and normal ERGs.
Figure 1. First row; fundus images representing from stage 1 to 4. Second row representing fundus autofluorescence start from speckle hypoautofluorescence mostly within vascular arcades in stage 2A progressing to nearly total dense hypoautofluorescence towards periphery in stage 4 (red arrow) third row demonstrates cross-sectional OCT scan of central macular area comparing each stages, severe attenuation of choroid was observed by OCT in stage 3B.(yellow arrow) fourth and fifth rows showing scotopic ERG and photopic ERG response respectively, amplitude of b wave gradually decline in more advance stages.
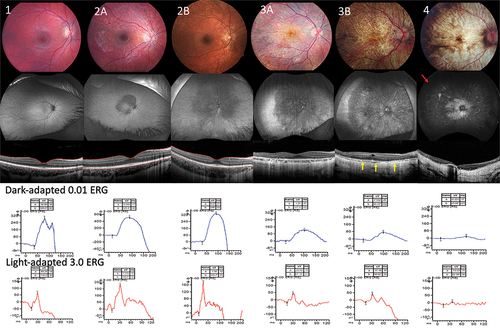
Table 1. Clinical staging based on multimodality ophthalmological findings of patients with LCHADD chorioretinopathy.
Table 2. A comparison of patient staging between the two systems of grading.
In stage 3, chorioretinal atrophy starts at the peripapillary margin and within the paracentral macula and eventually spreads from vascular arcades to the peripheral fundus. Structural OCT or OCTA allows a more accurate assessment of chorioretinal atrophy than previously possible with fundus photos. For example, it is possible to observe areas of RPE hypopigmentation and atrophy as well as photoreceptor layer thinning without frank choroidal atrophy. To better separate Stage 3, we have defined Stage 3A to be where RPE/Photoreceptor atrophy can be present, but the choroid is still relatively preserved. We designate Stage 3B as the point at which localized areas of complete or nearly complete choroidal atrophy are visible on OCTA (defined in and illustrated in ).
In the original Tyni system, stage 4 is defined by total atrophy of the posterior pole, posterior staphyloma, sparing of the peripheral fundus, and an unrecordable ERG. Interestingly, we were able to detect recordable ERGs in several cases of Stage 4 patients, albeit severely reduced (3 out of 8). We also observed 1 out of 8 Stage 4 patients with complete atrophy of the periphery. Visual acuity of stage 4 patients usually declines due to photoreceptor loss, however, we observed 38% (6 out of 16 eyes) with a visual acuity of 20/50 or better. Posterior staphyloma was observed in 8 patients in stage 4 (100%) and 2 patients in stage 3B (50%). None of patients in stage 1, 2, and 3A have posterior staphyloma. Advancing age and low visual acuity were also related to the advance stage of the disease. A complete comparison of patient staging between the two systems of grading is presented in . Additional findings such as epiretinal membranes (ERM) and cystoid macular edema (CME) can also frequently be observed in most structural OCT scans of stage 3 and stage 4 patients. The frequency of observed abnormal retinal findings by the new proposed staging system is listed in supplemental .
4. Discussion
Tyni et al. described 4 stages of LCHADD based on fundus photography, FA, acuity and electrophysiology. We propose a revised clinical staging system based on three patients with TFP and 37 patients with LCHADD who were grouped according to the fundus changes, OCT characteristics, FAF, and full-field ERG findings.
Fundus photographs in the previous staging system rely on multiple standard 30 to 45-degree color images that do not easily allow imaging of the far peripheral retina. Early areas of chorioretinal atrophy in stage 3 can be more difficult to distinguish with color photos as compared to hypofluorescence area in FAF and thinning area in OCT. In addition, OCT and or OCTA can detect concurrent ocular findings including choroidal neovascularization (CNV), epiretinal membrane (ERM), and cystoid macular edema (CME) that were difficult to observe in retinal examination or in fundus photographs (Citation21). We submitted our recent analysis using OCTA in LCHADD patients, demonstrating that the prevalence of CNV is 21% higher than previously thought and occurs at an early stage of LCHADD retinopathy (Citation22).
The pathophysiology of chorioretinopathy in LCHADD is still not completely understood. One leading hypothesis is that the accumulation of hydroxy acyl-carnitines following periods of hypoglycemia or stress may have a toxic effect on the retina. There has been debate as to the cell layer that is initially affected in LCHAAD retinopathy, but several lines of evidence suggest that the RPE might be the first cells affected. Immunohistochemistry demonstrates high expression of enzymes related to beta-oxidation antibodies in the RPE (Citation23,Citation24). Our study shows early presence of hypo/hyper autofluorescence speckling in the posterior pole of stage 2A, prior to detection of photoreceptor loss by OCT, which supports that RPE may be affected prior to photoreceptor degeneration. Further prospective longitudinal studies are needed to confirm these findings. The presence of outer retinal tubulations (ORT) also suggests the loss of RPE prior to the loss of photoreceptors. Although ORTs have been reported in numerous conditions, they are more commonly observed in chorioretinal degenerations such as Bietti crystalline dystrophy and choroideremia (Citation25,Citation26).
We show that multi-modal imaging techniques including wide-field fundus photographs, FAF, OCT, and OCTA provide superior imaging quality and spatial localization, sufficient for grading disease severity. In this study, FA was not necessary for clinical characterizing, especially with the use of OCTA. Moreover, FA is time-consuming, invasive, and carries a small but real risk of anaphylaxis. We also show that visual acuity can have high variability among stages. Some patients maintained good visual acuity in the late stage of the disease depending on the presence of residual photoreceptors and RPE in the central fovea. Additionally, CNV which can occur anytime from Stage 2 to Stage 4 can cause a disconnect between visual acuity and stage. Thus, we propose that visual acuity and FA can be excluded from the grading criteria for LCHADD retinopathy.
Tyni et al. staging described severely decreased ERG in stage 2 and unrecordable ERG at stages 3 and 4. Historically, the signal-to-noise ratio and inter-test variability can differ greatly among ERG labs. However, the availability of high-quality commercially-available ERG equipment has improved tremendously in the past twenty years, resulting in an increased standardization in the field towards these more robust ERG systems. Using one of these commercial systems, we found that reliable responses could still be recorded from most LCHADD patients in our cohort and previous studies (Citation8,Citation27,Citation28). We therefore have modified the ERG staging criteria to better reflect improvements in ERG systems.
5. Conclusions
Advanced technologies in retinal imaging facilitate a better understanding of the disease course and progression of LCHAD deficiency retinopathy. The new staging system better explains fundus changes in both central and peripheral retina, provides more detail of the retinal layers affected, and more accurately represents functional changes over time. Early diagnosis and treatment of LCHAD deficiency are associated with a slower progression of retinopathy and improved staging will be important in assessing the efficacy of future therapeutic trials.
Supplemental Material
Download PDF (142.8 KB)Acknowledgments
We would like to thank Wayne Tschetter, Assistant Staff Scientist, Casey Eye Institute, Department of Ophthalmology, Oregon Health & Science University for kindly providing the ffERGs illustration in .
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/13816810.2024.2303682.
Additional information
Funding
References
- Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol. 2002;64(1):477–502. doi:10.1146/annurev.physiol.64.082201.154705.
- Xia C, Fu Z, Battaile KP, Kim JP. Crystal structure of human mitochondrial trifunctional protein, a fatty acid β-oxidation metabolon. Proc Natl Acad Sci USA. 2019;116(13):6069–74. doi:10.1073/pnas.1816317116.
- Liang K, Li N, Wang X, Dai J, Liu P, Wang C, Chen XW, Gao N, Xiao J. Cryo-EM structure of human mitochondrial trifunctional protein. Proc Natl Acad Sci USA. 2018;115(27):7039–7044. doi:10.1073/pnas.1801252115.
- Fletcher AL, Pennesi ME, Harding CO, Weleber RG, Gillingham MB. Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. Mol Genet Metab. 2012;106(1):18–24. doi:10.1016/j.ymgme.2012.02.015.
- Boutron A, Acquaviva C, Vianey-Saban C, de Lonlay P, de Baulny HO, Guffon N, Dobbelaere D, Feillet F, Labarthe F, Lamireau D, et al. Comprehensive cDNA study and quantitative analysis of mutant HADHA and HADHB transcripts in a French cohort of 52 patients with mitochondrial trifunctional protein deficiency. Mol Genet Metab. 2011;103(4):341–348. doi:10.1016/j.ymgme.2011.04.006.
- IJ L, Ruiter JP, Hoovers JM, Jakobs ME, Wanders RJ. Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest. 1996;98(4):1028–33. doi:10.1172/JCI118863.
- Vockley J, Bennett MJ, Gillingham MB. Mitochondrial fatty acid oxidation disorders. In: Valle D, Antonarakis S, Ballabio A, Beaudet A, and Mitchell G, editors. The online metabolic and molecular bases of inherited disease. New York, NY: McGraw-Hill Education; 2019. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=247995158.
- Fahnehjelm KT, Liu Y, Olsson D, Amrén U, Haglind CB, Holmström G, Halldin M, Andreasson S, Nordenström A. Most patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency develop pathological or subnormal retinal function. Acta Paediatr. 2016;105(12):1451–1460. doi:10.1111/apa.13536.
- Tyni T, Kivelä T, Lappi M, Summanen P, Nikoskelainen E, Pihko H. Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: a new type of hereditary metabolic chorioretinopathy. Ophthalmology. 1998;105(5):810–824. doi:10.1016/S0161-6420(98)95019-9.
- Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33(5):527–532. doi:10.1007/s10545-010-9090-x.
- Tyni T, Pihko H. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr. 1999;88(3):237–245. doi:10.1111/j.1651-2227.1999.tb01089.x.
- Gillingham M, Van Calcar S, Ney D, Wolff J, Harding C. Dietary management of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). A case report and survey. J Inherit Metab Dis. 1999;22(2):123–131. doi:10.1023/A:1005437616934.
- Schrijver-Wieling I, van Rens GHMB, Wittebol-Post D, Smeitink JAM, de Jager JP, de Klerk HBC, van Lith GHM. Retinal dystrophy in long chain 3-hydroxy-acyl-coA dehydrogenase deficiency. Br J Ophthalmol. 1997;81(4):291. doi:10.1136/bjo.81.4.291.
- Lim CC, Vockley J, Ujah O, Kirby RS, Edick MJ, Berry SA, Arnold GL. Outcomes and genotype correlations in patients with mitochondrial trifunctional protein or isolated long chain 3-hydroxyacyl-CoA dehydrogenase deficiency enrolled in the IBEM-IS database. Mol Genet Metab Rep. 2022;32:100884. doi:10.1016/j.ymgmr.2022.100884.
- Tyni T, Immonen T, Lindahl P, Majander A, Kivelä T. Refined staging for chorioretinopathy in long-chain 3-hydroxyacyl coenzyme a dehydrogenase deficiency. Ophthalmic Res. 2012;48(2):75–81. doi:10.1159/000334874.
- Fricker SJ, Sanders JJ. Ergs and noise: detection probability, time and amplitude errors. In: Dodt E Pearlman J, editors. Xith ISCERG symposium. Dordrecht: Springer; 1974. pp. 395–408.
- Rispoli E, Iannaccone A, Vingolo EM. Low-noise electroretinogram recording techniques in retinitis pigmentosa. Doc Ophthalmol. 1994;88(1):27–37. doi:10.1007/BF01203699.
- Yamashita T, Miki A, Tabuchi A, Funada H, Kondo M. A novel method to reduce noise in electroretinography using skin electrodes: a study of noise level, inter-session variability, and reproducibility. Int Ophthalmol. 2017;37(2):317–324. doi:10.1007/s10792-016-0240-5.
- Man TTC, Yip YWY, Cheung FKF, Lee WS, Pang CP, Brelén ME. Evaluation of electrical performance and properties of electroretinography electrodes. Transl Vis Sci Technol. 2020;9(7):45. doi:10.1167/tvst.9.7.45.
- Yip YWY, Man TC, Pang CP, Brelén ME. Improving the quality of electroretinogram recordings using active electrodes. Exp Eye Res. 2018;176:46–52. doi:10.1016/j.exer.2018.06.007.
- Sacconi R, Bandello F, Querques G. Choroidal neovascularization associated with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Retin Cases Brief Rep. 2022;16(1):99–101. doi:10.1097/ICB.0000000000000923.
- Wongchaisuwat N, Wang J, Yang P, Everett L, Gregor A, Sahel JA, Nischal KK, Pennesi ME, Gillingham MB, Jia Y. Optical coherence tomography angiography of choroidal neovascularization in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). Am J Ophthalmol Case Rep. 2023;32:101958. doi:10.1016/j.ajoc.2023.101958.
- Tyni T, Paetau A, Strauss AW, Middleton B, Kivelä T. Mitochondrial fatty acid beta-oxidation in the human eye and brain: implications for the retinopathy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr Res. 2004;56(5):744–50. doi:10.1203/01.PDR.0000141967.52759.83.
- Polinati PP, Ilmarinen T, Trokovic R, Hyotylainen T, Otonkoski T, Suomalainen A, Skottman H, Tyni T. Patient-specific induced pluripotent stem cell–derived RPE cells: understanding the pathogenesis of retinopathy in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Invest Ophthalmol Visual Sci. 2015;56(5):3371–3382. doi:10.1167/iovs.14-14007.
- Iriyama A, Aihara Y, Yanagi Y. Outer retinal tubulation in inherited retinal degenerative disease. Retina. 2013;33(7):1462–1465. doi:10.1097/IAE.0b013e31828221ae.
- Sun LW, Johnson RD, Williams V, Summerfelt P, Dubra A, Weinberg DV, Stepien KE, Fishman GA, Carroll J, Wen R. Multimodal Imaging of Photoreceptor Structure in Choroideremia. PloS One. 2016;11(12):e0167526. doi:10.1371/journal.pone.0167526.
- Fahnehjelm KT, Holmström G, Ying L, Haglind CB, Nordenström A, Halldin M, Alm J, Nemeth A, Von Döbeln U. Ocular characteristics in 10 children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: a cross-sectional study with long-term follow-up. Acta Ophthalmol. 2008;86(3):329–337. doi:10.1111/j.1755-3768.2007.01121.x.
- Gillingham MB, Weleber RG, Neuringer M, Connor WE, Mills M, Van Calcar S, Ver Hoeve J, Wolff J, Harding CO. Effect of optimal dietary therapy upon visual function in children with long-chain 3-hydroxyacyl CoA dehydrogenase and trifunctional protein deficiency. Mol Genet Metab. 2005;86(1–2):124–133. doi:10.1016/j.ymgme.2005.06.001.