Abstract
Novel mercapto-1,3,4-oxadiazole and -1,2,4-triazole derivatives were synthesized by various pathways starting from 4-(4-halogeno-phenylsulfonyl)benzoic acid hydrazides which were reacted with carbon disulfide or isothiocyanates. The heterocyclic mercaptans prepared in this way were assayed as inhibitors of three physiologically relevant isoforms of the zinc enzyme carbonic anhydrase (CA, EC 4.2.1.1), i.e., the cytosolic CA I and II, and the tumor-associated, transmembrane isozyme CA IX. Interesting biological activity was detected for some of the new mercaptans, with inhibition constants in the low micromolar range.
Introduction
The carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous metallo-enzymes, present in prokaryotes and eukaryotes, being encoded by four distinct, evolutionarily unrelated gene families: the α-CAs (present in vertebrates, Bacteria, algae and cytoplasm of green plants), the β-CAs (predominantly in Bacteria, algae and chloroplasts of both mono- as well as dicotyledons) the γ-CAs (mainly in Archaea and some Bacteria), and the δ-CAs, present in some marine diatoms, respectively Citation1-8. In mammals, 16 different α-CA isozymes or CA-related proteins (CARP) have been described, with very different subcellular localization and tissue distribution Citation1-8. Basically, there are several cytosolic forms (I–III, and VII), five membrane-bound isozymes (IV, IX, XII, XIV and XV), two mitochondrial forms (VA and VB), as well as a secreted isozyme in saliva and milk, CA VI. Among the membrane-bound CAs, isoforms CA IV and XV are anchored to membranes by means of GPI (glycosylphosphatidylinositol) tails, whereas isozymes IX, XII and XIV are transmembrane proteins possessing just one transmembrane domain Citation1-8. However, all these five isozymes have their active site outside the cell, being commonly termed as extracellular CAs Citation1-8. These enzymes catalyze a very simple physiological reaction, the interconversion between carbon dioxide and the bicarbonate ion, and are thus involved in crucial physiological processes connected with respiration and transport of CO2/bicarbonate between metabolizing tissues and lungs, pH and CO2 homeostasis, electrolyte secretion in a variety of tissues/organs, biosynthetic reactions (such as gluconeogenesis, lipogenesis and ureagenesis), bone resorption, calcification, tumorigenicity, and many other physiological or pathological processes Citation1-8. Many of these isozymes are important targets for the design of inhibitors or activators with clinical applications Citation1-8.
A wide variety of heterocycles, including 1,3,4-thiadiazole, 1,2,4-triazole and 1,3,4-oxadiazole derivatives have been described for their possible applications as antimicrobials, inhibitors of glycosidase, urease or carbonic anhydrase (CA) enzymes Citation9-13. Among such compounds we have also investigated the interaction of a series of heterocyclic mercaptans incorporating 1,3,4-thiadiazole- 1,2,4-triazole moieties, of types A–C, with CA isozymes I, II, IV and IX, finding several potent inhitors, potentially useful as lead compounds for obtaining isozyme-selective derivatives [Citation11,Citation12].
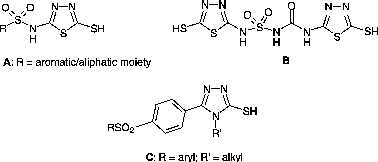
Based on the interesting CA inhibitory activity of some heterocyclic mercaptans A–C investigated earlier by us, and continuing our research in this field, we report here the synthesis of some new mercapto-1,3,4-oxadiazoles/-1,2,4-triazole Mannich bases and investigated their CA inhibition activities against three physiologically relevant isoforms, the cytosolic CA I and II, and the tumor-associated, transmembrane isozyme CA IX. Our main interest was as mentioned above, the detection of enzyme inhibitors with an inhibition profile leading to isozyme-selectivity in view of the fact that the presently used CA inhibitors indiscriminately inhibit most of the 15 isoforms widely distributed in mammals Citation1-8. As a consequence, clinically used CA inhibitors showed many undesired side effects Citation1-8.
Materials and methods
Chemistry
Melting points were determined with a Boetius apparatus and are uncorrected. The IR spectra were recorded on a FTS-135 BIO-RAD instrument in KBr pellets. The UV spectra were recorded on a SPECORD 40 Analytik Jena instrument using 2 × 10− 5 M methanolic solutions. The NMR spectra were recorded on a VARIAN GEMINI 300 BB instrument at 300 MHz for 1H and at 75 MHz for 13C and using TMS as internal standard.
General procedure for preparation of mannich bases 4(a–c), 5(a–c)
A mixture of 1,3,4-oxadiazole 2(a–c) (0.01 mole) and secondary cyclic amine (0.01 mole) was refluxed in ethanol (50 mL) with 37% formaldehyde (0,02 mole) for 3 h. The resulting solid was filtered, dried and recrystallised from absolute ethanol.
5-(4-phenylsulfonyl)phenyl-3-(piperidin-1-yl-methyl)-1,3,4-oxadiazole-2(3H)-thione (4a)
m.p. 134–135°C; 92% yield. Found: C: 57.92; H: 4.95; N: 10.18. Calcd. for C20H21N3O3S2 (415.54 g/mol): C: 57.81; H: 5.09; N: 10.11%; UV spectrum (methanol; λ max nm; ϵ max): 247 (44054); 347 (26913); IR-; 1H-NMR and 13C-NMR spectra–See Tables and
Table I. Selected IR data for some of the new compounds 4(a–c) and 5(a–c).
Table II. NMR data for compounds 4(a–c); 5(a–c)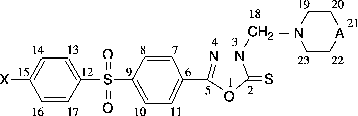
5-[4-(4-chloro-phenylsulfonyl)phenyl]-3-(piperidin-1-yl-methyl)-1,3,4-oxadiazole-2(3H)-thione (4b)
m.p. 239–241°C; 91% yield. Found: C: 53,43; H: 4.42; N: 9.41. Calcd. for C20H20ClN3O3S2 (449.98 g/mol): C: 53.39; H: 4.48; N: 9.34%; UV spectrum (methanol; λ max nm; ϵ max): 251 (43804); 350 (26528); IR-; 1H-NMR and 13C-NMR spectra–See Tables and
5-[4-(4-bromo-phenylsulfonyl)phenyl]-3-(piperidin-1-yl-methyl)-1,3,4-oxadiazole-2(3H)-thione (4c)
m.p. 241–243°C; 94% yield. Found: C: 48.59; H: 4.02; N: 8.57. Calcd. for C20H20BrN3O3S2 (494.43 g/mol): C: 48.64; H: 4.08; N: 8.50%; UV spectrum (methanol; λ max nm; ϵ max): 252 (44910); 350 (26162); IR-; 1H-NMR and 13C-NMR spectra–See Tables and
5-(4-phenylsulfonyl)phenyl-3-(morpholin-4-yl-methyl)-1,3,4-oxadiazole-2(3H)-thione (5a)
m.p. 148–150°C; 94% yield. Found: C: 54.71; H: 4.50; N: 10.12. Calcd. for C19H19N3O4S2 (417.51 g/mol): C: 54.66; H: 4.59; N: 10.06%; UV spectrum (methanol; λ max nm; ϵ max): 246 (40067); 349 (26265); IR-; 1H-NMR and 13C-NMR spectra–See Tables and
5-[4-(4-chloro-phenylsulfonyl)phenyl]-3-(morpholin-4-yl-methyl)-1,3,4-oxadiazole-2(3H)-thione (5b)
m.p. 250–252°C; 92% yield. Found: C: 50.55; H: 3.97; N: 9.38. Calcd. for C19H18ClN3O4S2 (451.95 g/mol): C: 50.49; H: 4.01; N: 9.30%; UV spectrum (methanol; λ max nm; ϵ max): 250 (42521); 350 (25607); IR-; 1H-NMR and 13C-NMR spectra–See Tables and
5-[4-(4-bromo-phenylsulfonyl)phenyl]-3-(morpholin-4-yl-methyl)-1,3,4-oxadiazole-2(3H)-thione (5c)
m.p. 251–252°C; 95% yield. Found: C: 46.02; H: 3.59; N: 8.52. Calcd. for C19H18BrN3O4S2 (496.41 g/mol): C: 45.97; H: 3.65; N: 8.46%; UV spectrum (methanol; λ max nm; ϵ max): 253 (47884); 350 (28321); IR-; 1H-NMR and 13C-NMR spectra–See Tables and
CA inhibition assay
An Applied Photophysics stopped-flow instrument was used for assaying the CA-catalysed CO2 hydration activity [Citation20]. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5) as buffer, 0.1 M Na2SO4 (for maintaining constant the ionic strength), and following the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7–17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction was used for determining the initial velocity. The uncatalyzed rates were determined in a same manner and subtracted from the total observed rates. Stock solutions of inhibitor (1 mM) were prepared in distilled-deionized water with 10–20% (v/v) DMSO (which is not inhibitory at these concentrations) and dilutions up to 0.01 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, from Lineweaver-Burk plots, as reported earlier, and represent the mean from at least three different determinations [Citation12].
Results and discussion
Chemistry
In the present work 4-(4-X-phenylsulfonyl)benzoic acid hydrazides 1(a–c), (X = H, Cl, Br) were used as key intermediates for the synthesis of compounds investigated here as enzyme inhibitors. Nucleophilic addition of 4-(4-X-phenylsulfonyl)benzoic acid hydrazides 1(a–c), (X = H, Cl, Br,) to different isothiocyanate led to N4-R-N1-[4-(4-X-phenylsulfonyl)benzoyl-thiosemicarbazides 6(a–c), 7(a–c) (X = H, Cl, Br), R = –CH(CH3)2, -2-methoxyphenyl. By ring closure of these compounds in alkaline medium the 5-[4-(4-X-phenylsulfonyl)-phenyl]-4-R-4H-1,2,4-triazole-3-thiols [Citation14, Citation15] 8(a–c), 9(a–c) (X = H, Cl, Br), R = –CH(CH3)2, -2-methoxyphenyl were produced (Scheme ).
Scheme 1 Synthesis of the heterocycles 2–9
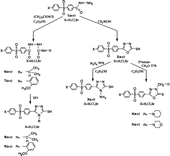
When compounds 1(a–c) were treated with carbon disulphide and potassium hydroxide in ethanolic medium, 5-[4-(4-X-phenylsulfonyl)phenyl]-1,3,4-oxadiazole-2-thiols 2(a–c), (X = H, Cl, Br) were obtained, which have been reported earlier [Citation16] (Scheme ). Diphenylsulfones incorporated into the triazole moiety were then synthesized by the reaction of compounds 2(a–c) with hydrazine hydrate 99% in absolute ethanol when 4-amino-5-[4-(4-X-phenylsulfonyl)phenyl]-4H-1,2,4-triazole-3-thiols 3(a–c) were obtained (X = H, Cl, Br) [Citation16] (Scheme ). Compounds 2(a–c) were allowed to undergo the Mannich reaction with various secondary amines, such as morpholine or piperidine, in the presence of 37% formaldehyde (in absolute ethanol) for the synthesis of compounds 4(a–c) and 5(a–c), respectively (Scheme ).
Analytical and physical data of the new compounds 4(a–c) and 5(a–c) are provided in the Experimental Section and Tables and . The structure of the Mannich bases 4(a–c) and 5(a–c) is supported by the IR spectral data (). Thus in these spectra a new band characteristic of the–CH2–N–CH2– group around 1186–1223 cm− 1 appears simultaneously with the disappearance of the band around 3100–3300 cm− 1 characteristic for the–NH– group of 1,3,4-oxadiazole-2-thione. In the NMR spectra () a characteristic signal due to the–N–CH2–N– protons appeared at 5.06–5.07 ppm as a singlet, whereas the signal due to–N–CH2–N– carbon appeared at 70.72–71.82 ppm. On the other hand, the 1H-NMR spectra exhibited features and characteristics for the diarylsulfone moiety and for the remaining functional side-chains, and presented significant similarities with the related heterocyclic thiols possessing a different ring system and a diphenylsulfone moiety previously reported in the literature Citation17-19.
Carbonic anhydrase inhibition
Heterocyclic mercaptans have been investigated earlier as CA inhibitors against isoforms I, II, IV and IX Citation11-13, and some interesting data were obtained. Thus, we decided to investigate other classes of such derivatives for their interaction with some isoforms. Inhibition data against three physiologically relevant CA isoforms, i.e., the cytosolic human hCA I and hCA II, and the tumor-associated, transmembrane isozyme hCA IX, were obtained by means of a stopped-flow assay [Citation20] and the results are provided in .
Table III. Inhibition data for derivatives 2-9 investigated here and the standard sulfonamide CAI (acetazolamide AAZ), against isozymes hCA I, II and IX.
The following SAR can be drawn from the data of : (i) against the slow [Citation1,Citation2] cytosolic isozyme hCA I, the heterocycles 2–5 showed good inhibitory activity, with inhibition constants in the range 5.1–9.3 μM, being on the other hand weaker inhibitors as compared to the classical sulfonamide inhibitor acetazolamide (5-acetamido-1,3,4-thiadiazole-2-sulfonamide), which had a KI of 0.25 μM. Thus, all types of substitution patterns in derivatives 2–5 lead to approximately the same inhibitory pattern, whether in the 1,3,4-oxadiazole or 1,2,4-triazole series of derivatives. Thus, substitution of the nitrogen atom close to the thiol (thione) moiety (in derivatives 4 and 5) with the rather bulky piperidine-methyl or morpholine-methyl groups was not detrimental to the hCA I inhibitory activity of these compounds as compared to the corresponding parent, unsubstituted compounds 2. The 4-X-phenyl moiety in all these derivatives was also not very important for their inhibitory power, since both compounds with X = H or X = halogens, showed similar activity. For the triazoles 3, the presence of the amino moiety in the 4-position also did not negatively influence the inhibitory power. However, for the compounds incorporating the much bulkier iso-propyl and 2-methoxy-phenyl moieties in this position, i.e., derivatives 8a–8c, and 9a–c, the hCA I inhibitory activity was very much diminished as compared to the corresponding derivatives 3a–c (possessing the more compact amino group in this position), with KIs in the range 340–460 μM. Probably this is due to the steric hindrance produced by these bulky moieties, which interferes with the binding of the mercaptide to the zinc ion within the enzyme active site, as observed earlier for other heterocyclic mercaptans possessing bulky groups in the neighborhood of the zinc-binding group Citation11-13; (ii) against the physiologically most important cytosolic isoform, hCA II, the new compounds investigated here showed good inhibitory activity, with KIs in the range 0.63–31 μM, being again less efficient inhibitors as compared to the sulfonamide acetazolamide (KI of 12 nM). The oxadiazoles 2, the aminotriazoles 3 and the Mannich bases 4 and 5 showed a similar level of activity. So, similarly to what mentioned above for isozyme I, the substitution pattern of these compounds does not greatly influence their CA II inhibitory activity. The only much less active derivatives were again the iso-propyl/2-methoxy-phenyl substituted triazoles 8 and 9 which were less inhibitory probably for the same reason mentioned above, i.e., steric impairment due to the bulky moiety substituting the N-4 atom. Unexpectedly however, the best inhibitor was the Mannich base incorporating the thiadiazole ring 5a, which has a rather bulky group at the nitrogen atom close to the zinc-binding function; (iii) against the tumor-associated isoform hCA IX, compounds 2–5 showed a moderate - weak inhibitory activity, with KIs in the range of 1.25–59 μM, whereas the bulky triazoles 8 and 9 were quite weak inhibitors, with KIs in the range 129–476 μM. The SAR is again quite similar to that mentioned above for isozymes I and II.
In conclusion, we synthesized and assayed as CA inhibitors a series of heterocyclic mercaptans incorporating oxadiazole and triazole rings, and various other substituted-diphenylsulfone side chains. Some of the new compounds proved to be moderately active inhibitors of isozymes hCA I, II and IX, with inhibition constants in the low micromolar range.
References
- Carbonic anhydrase–its inhibitors and activators, CT Supuran, A Scozzafava, J Conway. CRC Press, Boca Raton (FL), USA 2004; 1–363, And references cited therein
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors and their therapeutic potential. Expert Opin Ther Pat 2000; 10: 575–600
- Supuran CT, Scozzafava A. Applications of carbonic anhydrase inhibitors and activators in therapy. Expert Opin Ther Pat 2002; 12: 217–242
- Scozzafava A, Mastrolorenzo A, Supuran CT. Modulation of carbonic anhydrase activity and its applications in therapy. Expert Opin Ther Pat 2004; 14: 667–702
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003; 23: 146–189
- Supuran CT, Vullo D, Manole G, Casini A, Scozzafava A. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem–Cardiovasc Hematol Agents 2004; 2: 49–68
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects. J Enz Inhib Med Chem 2004; 19: 199–229
- Supuran CT. Carbonic anhydrase inhibitors in the treatment and prophylaxis of obesity. Expert Opin Ther Pat 2003; 13: 1545–1550
- Rossi LL, Basu A. Glycosidase inhibition by 1-glycosyl-4-phenyl triazoles. Bioorg Med Chem Lett 2005; 15: 3596–3599
- Amtul Z, Rasheed M, Choudhray MI, Rosanna S, Khan KM, Rahman A. Kinetics of novel competitive inhibitors of urease enzymes by a focused library of oxadiazoles/thiadiazoles and triazolesoxadiazoles/thiadiazoles and triazoles. Biochem Biophys Res Commun 2004; 319: 1053–1063
- Supuran CT, Scozzafava A, Saramet I, Banciu MD. Carbonic anhydrase inhibitors. Inhibition of isozymes I, II and IV with heterocyclic mercaptans, sulfenamides, sulfonamides and their metal complexes. J Enz Inhib 1998; 13: 177–194
- Almajan GL, Innocenti A, Puccetti L, Manole G, Barbuceanu S, Saramet I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the cytosolic and tumor-associated carbonic anhydrase isozymes I, II and IX with a series of 1,3,4-thiadiazole- and 1,2,4-triazole-thiols. Bioorg Med Chem Lett 2005; 15: 2347–2352
- Zareef M, Innocenti A, Iqbal R, Zaidi JH, Arfan M, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of human tumor-associated isozymes IX and cytosolic isozymes I and II with some 1,3,4-oxadiazole-thiols. J Enz Inhib Med Chem 2006; 21: 351–359
- Barbuceanu S, Almajan L, Saramet I, Draghici B. Noi compusi din clasa 1,2,4-triazolilor si 1,3,4-tiadiazolilor obtinuţi prin ciclizarea unor aroiltiosemicarbazide. Rev Chim(Buc.) 2005; 56(12)1270–1274
- Barbuceanu SF, Almajan GL, Saramet I, Draghici B. Noi tiosemicarbazide obtinute prin aditia nucleofila a hidrazidei acidului 4-(4-X-fenilsulfonil)benzoic la izotiocianatul de 2-metoxifenil si ciclizarea acestora la heterociclii din clasa 1,2,4-triazolilor si 1,3,4-tiadiazolilor. Rev Chim(Buc.) 2006, in press
- Almajan GL, Barbuceanu SF, Saramet I, Draghici C. Noi compusi heterociclici obtinuti din hidrazidele acizilor fenilsulfonilbenzoici-4-substituiti. Rev Chim(Buc.) 2005; 56(11)1182–1187
- Saramet I. Synthesis and physico-chimical characterization of some thiosemicarbazides and mercaptotriazoles with phenazinic moieties. Rev Roum Chim 2000; 45(7)643–651
- Saramet I, Draghici C, Barcutean C, Radulescu V, Loloiu T, Banciu M. Synthesis of new 4-substituted-1-aroyl-thiosemicarbazides and their cyclization to mercaptotriazoles and aminothidiazoles. Heterocycl Comm 2001; 7(4)369–377
- Saramet I, Almajan GL, Barbuceanu S, Draghici C, Banciu MD. Synthesis of some substituted aroylthiosemicarbazides,-mercaptotriazoles and aminothiadiazoles. Rev RoumChim 2005; 50(1)19–27
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C.J. Biol Chem 1971; 246: 2561–2573