Abstract
Acetylcholinesterase (AChE) is a widely spread enzyme playing a very important role in nerve signal transmission. As AChE controls key processes, its inhibition leads to the very fast death of an organism, including humans. However, when this feature is to be used for killing of unwanted organisms (i.e. mosquitoes), one is faced with the question - how much do AChEs differ between species and what are the differences? Here, a theoretical point of view was utilized to identify the structural basis for such differences. The various primary and tertiary alignments show that AChEs are very evolutionary conserved enzymes and this fact could lead to difficulties, for example, in the search for inhibitors specific for a particular species.
Introduction
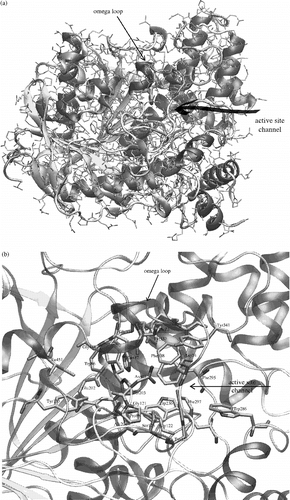
Acetylcholinesterase (AChE, EC 3.1.1.7) is one of the most important enzymes in many living organisms, including humans and vertebrates, and is located in the nervous system and in muscles [Citation1]. AChE is responsible for regulation of acetylcholine concentration during nerve signal transmission. In the central and peripheral nervous systems, the nerve signal is usually transmitted by the neurotransmitter acetylcholine. The transmission is terminated by the cleavage of acetylcholine by AChE directly in the synaptic gap [Citation2]. The AChE is a very efficient catalyst. It catalyses the hydrolysis of acetylcholine with a turnover number of 108s− 1 [Citation3]. This high turnover implies that Nature has created a perfect enzyme working on rates very close to diffusion limits. AChE has been studied for a long time Citation1-7 and many important and interesting observations have been achieved. The first crystallographic structure [Citation6] was solved in 1991 (pdb code: 1ACJ). AChE is an alpha/beta protein with a large twisted beta sheet spanning through the whole molecule (see ).
Figure 1 a) AChE crystal structure (1N5M) with highlighted secondary structure elements and active site entrance. Beta sheets are yellow, alpha helical structures are violet and unordered structures are grey. Catalytic triad is represented by bold tubes. b) A detailed view of the AChE active site, which is located under the omega loop. Secondary structures are highlighted and active site residues are represented by bold tubes. The numbering used is valid for human and mouse structures of AChE. (Please see colour online)
Nowadays in the PDB database, there are 69 structures of AChE's from various organism sources including Homo sapiens (1B41, 1F8U), Mus musculus (1N5M, 1N5R, 1JO6, 1JO7), Torpedo californica (1EA5), Electrophorus electricus (1C2O) and Drosophila melanogaster (1QO9). These structures are very important for grasping the mode of action of AChE. Because the crystal structures are known, the active site of AChE is quite well described, although the precise function of some of the active site residues remains unclear.
There are several subsites, which can be distinguished in AChE's active site: esteratic subsite, also called the catalytic triad (Ser203, His447, Glu334), oxyanion hole (Gly121, Gly122, Ala204), anionic subsite (Trp86, Tyr133, Glu202, Gly448, Ile451), acyl binding pocket (Trp236, Phe295, Phe297, Phe338), peripheral anionic subsite (Asp74, Tyr124, Ser125, Trp286, Tyr337, Tyr341) and other residues of the omega loop (Thr83, Asn87, Pro88) (see ). The omega loop is a disulphide-linked loop (Cys69 - Cys96) that covers the active site of AChE, which is buried at the bottom of a 20A deep gorge approximately in the centre of the molecule (see and 1b).
Knowledge gained about AChE is utilized, for example, to control its inhibition, which is then used in the development of medicinal agents for the treatment of Alzheimer disease, and in agriculture to design pesticides especially against insects and other arthropod vertebrates [Citation7]. In regard to pesticides, an inhibitor specific for each species would be an ideal situation. Unfortunately, in the terms of AChE inhibitory specificity, it is only possible to distinguish between mammals and insects, due to the very high functional and structural similarity of AChE's of different species that are evolutionary more similar than the above mentioned mammals and insects. This fact is firmly based on the primary and tertiary structure of AChE's of various species and the aim of this article is to elucidate these differences and similarities using an alignment method.
Methods
The alignment method attempts to arrange the sequences in such a way, that the most similar regions are superposed. The amino acids in these regions can be either identical (identity) or similar (similarity). The similarity is based on amino acid properties such as acidity, basicity, polarity, aromaticity etc. The mathematical expression of these properties is called a weight (scoring) matrix. So each pair of amino acids is assigned a score. Furthermore the sequences can be moved against one another, or a gap can be inserted. However the insertion of the gap and the length of the gap contribute negatively to the whole score. The alignment of two sequences (pair-wise alignment) is solved by algorithms of dynamic programming [Citation9]. The alignment of more sequences (multiple alignment) computes firstly pair-wise alignments, constructs phylogenetic tree and then proceeds by adding sequences according to the phylogenetic tree branching order to finally construct the multiple alignment. This algorithm is called progressive multiple sequence alignment [Citation10]. Clustal W [Citation11] and Clustal X [Citation12] utilised in this work use a modified progressive approach.
Firstly, an alignment of the primary sequences of known crystallographic AChE structures was carried out. The structures were taken from the PDB database [Citation13]. The human (1B41, Homo sapiens, [Citation14]), mouse (1N5M, Mus musculus, [Citation15]), torpedo (1EA5, Torpedo californica, [Citation16]) and drosophila (1QO9, Drosophila melanogaster, [Citation17]) AChE's were taken into account in this first step. Then the Swiss-Prot database, version 50.1 released 13-Jun-2006 [Citation18], was used to extract more AChE sequences (Rattus norvegicus, Felis silvestris catus, Oryctolagus cuniculus and Bos taurus). These are sequences of AChE from species that could serve as testing animals. Multiple alignments of these sequences with human AChE were also performed.
If the structure and function of a protein are tightly tied together, then proteins with similar structures are likely to have similar functions. Three dimensional alignment can give useful information in this type of study. The 3-D alignment is a method that attempts to fit two structures in such a way that the RMSD (root-mean-square deviation) between atoms of two structures is as small as possible. There are many algorithms that are able to do this task. We have used the “Best fit procedure” for backbone atoms implemented in the Swiss-Pdb Viewer [Citation19,Citation20], because the crystal structures of compared AChE's do not have the same number of atoms or even residues.
Results and discussion
As mentioned earlier, AChE's from different species are generally similar or different depending on how evolutionary similar the species are. The question is how to measure their similarities or differences. One branch of reliable sources to measure this is certainly enzyme kinetics and the specificity of a chosen set of inhibitors and AChE reactivators [Citation21]. The second branch, utilized in this paper, is based on primary and/or tertiary structure of the AChE molecules.
First we performed an alignment between the aminoacid sequences of four crystallized AChE's (see ). The identity in this case is 27.68%.
Figure 2 Alignment of primary structures of AChE sequences from Homo sapiens (HUMAN), Mus musculus (MOUSE), Torpedo californica (TORCA) and Drosophila melanogaster (DROSO). Positions of secondary structures depicted under alignment are taken from mouse 1N5M AChE structure. The red arrows point to active site residues. The grey row of bars under each row of alignment is a graphical representation of the alignment score for each column of residues. The score is also marked by signs (star, colon, full stop, space) above the alignment rows. Aminoacid residues are highlighted by colours: acidic (D,E) are red, basic (K,R,H) are blue, polar (N,Q,S,C,T,W,Y) are yellow, non-polar (A,V,L,I,M,F) are green and the others (P,G) stayed white. The far right column shows the number of the last residue in the row. Colour online version. (Please see colour online)
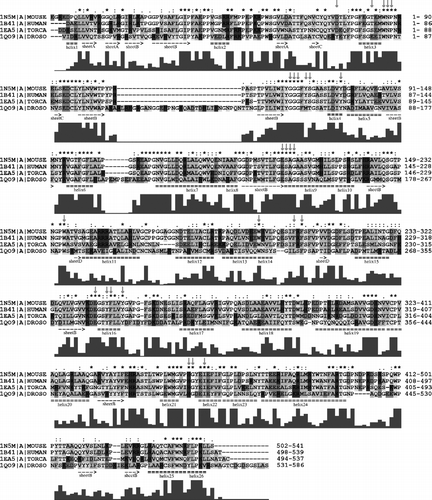
We then excluded sequence 1QO9 (the drosophila sequence). The identity significantly increased to 56.14% (). In , the identity of the Swiss-prot-extracted sequences is 80.13%. The identity of the active site residues is high in all cases. The values in the three previously mentioned alignments (, and ) are 76%, 92% and 100%, respectively. It can be seen that despite the low overall identity between human and drosophila sequences, the identity of active site residues is still high. When the drosophila sequence was excluded from the alignment, the identity significantly increased and when the torpedo sequence was excluded, the overall identity increased once again and the identity of active site residues reached even 100%.
Figure 3 Alignment of primary structures of AChE sequences from Homo sapiens (HUMAN), Mus musculus (MOUSE), Torpedo californica (TORCA). For further description, see Figure 2. For more details see the text. (Please see colour online)

Figure 4 Alignment of primary structures of AChE sequences from Homo sapiens (HUMAN), Mus musculus (MOUSE), Rattus norvegicus (RAT), Felis silvestris catus (FELCA), Oryctolagus cuniculus (RABIT) and Bos taurus (BOVIN). For further description, see Figure 2. For more details see the text.

It is accepted in protein homology modelling that a homology model is likely to be accurate when the sequence identity of the model and template(s) is more than 40%. A protein sequence that has at least 40% identity to a known structure can be modelled automatically with an accuracy approaching that of a low resolution X-ray structure or a medium resolution NMR structure [Citation22]. Once the overall sequence identity of drosophila and human sequences is below 40% (28% in this case), these two proteins might not have similar 3D structures. The sequence of the Drosophila fly could have a different tertiary structure and it might not even have an AChE fold (with assumption that the human sequence has an AChE fold). However, this finding is inconsistent with the very high identity of active site residues. According to the threshold value of 40%, the torpedo structure should have the same fold as mouse or human structures, and the tertiary structure should also be similar. Another interesting feature, which can be seen directly from the alignments, is that the insertions and deletions in drosophila and torpedo sequences appear solely in the unordered structure or in the loops, when the mouse secondary structures are used as reference. This fact together with the conservation of active site residues indicates a large structural and functional conservation of AChE's from different species.
When one considers the 3-D structure of the active sites, and , it can be seen, that although the primary structures of drosophila AChE and human AChE are very different, the active sites are similar. On the other hand there are several mutations, especially in the peripheral anionic subsite, which change the properties of the active site. The most important mutations, in our opinion, are the mutations of Asp74 in human to Tyr71 in Drosophila and Tyr124 to Met153, which influence both the steric and electrostatic properties of the upper part of the active site gorge. The negative charge, located in the human structure at the entrance of the gorge, disappears by Asp74 to Tyr71 mutation and appears on the opposite side of the gorge entrance due to mutation of Gly342 to Asp375. The active site of torpedo AChE is nearly identical to the mouse and human AChEs. The only minor difference is the mutation of Tyr337 in human to Phe330 in the torpedo structure, which should not cause any significant differences in both the electrostatic and steric properties of the active site gorge.
Figure 5 3-D alignment of human 1B41 and drosophila 1QO9 structures. Mutated residues and flipped Trp86(83) are coloured by red for human and orange for the drosophila structure. The first residue number always belongs to the human AChE and “/” character means a mutation, where the first aminoacid is from the human structure. Colour online version.
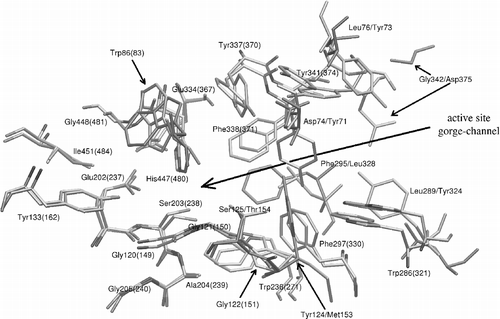
Figure 6 3-D alignment of human 1B41, mouse 1N5M and torpedo 1EA5 structures. Residues which show the biggest deviations are coloured by red for human, blue for mouse and yellow for the torpedo structure. Torpedo californica has also one mutation of Tyr337 (in the case of human and mouse) to Phe330 (torpedo). The first residue number belongs to the human or mouse AChE. Colour online version.
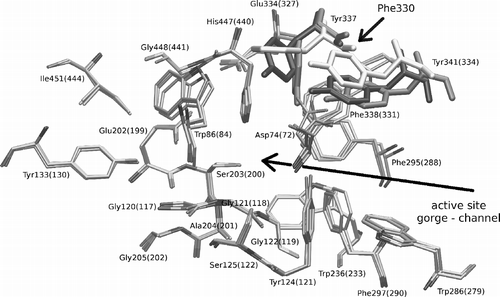
The trends seen in the 3-D alignment of active site structures are also reflected by the RMSD values of backbone atoms of drosophila, torpedo and mouse structures from the human one; the values are 1.20A, 0.87A, and 0.57A, respectively. The values were measured on the maximal number of residues common in both structures; about 500 residues were utilized in average to compute this backbone 3-D alignment. According to RMSD values we can see that all aligned AChE structures have the same fold and specific differences are in an exact position, and also it can be seen that the type of side chain retrospectively influences the backbone position. For this reason the drosophila structure, which has the lowest primary structure identity, has the largest RMSD. But on the contrary to the results from primary structure alignments, which show low overall identity between drosophila and human sequence, the drosophila tertiary structure and fold is very similar to the human tertiary structure and fold. Based on this observation, we predict that the tertiary structures of AChEs from Rattus norvegicus, Felis silvestris catus, Oryctolagus cuniculus and Bos taurus will be very similar to the human structure, because the identity of the alignment of their primary sequences with the human sequence is very high.
Conclusions
The 3-D structure of AChE is very evolutionary conserved, despite the lower conservation of the aminoacid sequence. The folding were similar when the structures of AChE from Homo sapiens and Drosophila melanogaster were compared. In the structure of Drosophila melanogaster AChE, various mutations of the active site residues occur leading to differences in both steric and electrostatic properties of the active site. Most of the mutations were seen at the peripheral anionic site. We predict that the structures of AChEs from Rattus norvegicus, Felis silvestris catus, Oryctolagus cuniculus and Bos taurus should be very similar to the human structure and should have identical properties of the active site.
Acknowledgement
This work was supported by the Ministry of Defence of the Czech Republic (KK, DJ), Grant No. FVZ0000604, Ministry of Education of the Czech Republic, Grants No. MSM0021622413 (ZK) and LC06030 (JK), and Grant Agency of the Czech Republic, Grant No. 204/03/H016 (JW).
References
- Massoulié J, Pezzementi L, Bon S, Krejci E, Vallette FM. Molecular and cellular biology of cholinesterases. Progr Neurobiol 1993; 41: 31–91
- Patočka J, Kuča K, Jun D. Acetylcholinesterase and butyrylcholinesterase — important enzymes in human body. Acta Medica 2004; 47(4)215–228
- Quinn DM. Acetylcholinesterase - enzyme structure, reaction dynamics, and virtual transition-states. Chem Rev 1987; 87(5)955–979
- Nolte HJ, Rosenberry TL, Neumann E. Effective charge on acetylcholinesterase active-sited determined from the ionic-strength dependence of association rate constants with cationic ligands. Biochemistry 1980; 19(16)3705–3711
- Ripoll DR, Faerman CH, Axelsen PH, Silman I, Sussman JL. An electrostatic mechanism for substrate guidance down the aromatic gorge of acetylcholinesterase. Proc Natl Acad Sci USA 1993; 90: 5128–5132
- Sussman J, Harel M, Frolow F, Oefner C, Goldman A, Toker R, Silman I. Atomic structure of acetylcholinesterase from torpedo califonica — a prototypic acetylcholine-binding protein. Science 1991; 253(5022)872–879
- Hougton PJ, Ren Y, Howes MJ. Acetylcholinesterase inhibitors from plants and fungi. Nat Prod Rep 2006; 23: 181–199
- Singh AK. Molecular properties and inhibition kinetics of acetylcholinesterase obtained from rat brain and cockroach ganglion. Toxicol Ind Health 1990; 6(6)551–570
- Needleman SB, Wunch ChD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 1970; 48: 443–453
- Feng DF, Doolittle FR. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol Evol 1987; 25: 351–360
- Thompson JD, Higgins DG, Gibson TJ. Clustal-W - Improving the sensitivity of progressive multiple sequence alignment through sequence weighting position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997; 25(24)4876–4882
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res 2000; 28(1)235–242
- Kryger G, Harel M, Giles K, Toker L, Velan B, Lazar A, Kronman C, Barak D, Ariel N, Shafferman A, Silman I, Sussman JL. Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr 2000; 56(D)1385–1394
- Bourne Y, Taylor P, Radic Z, Marchot P. Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. EMBO J 2003; 22(1)1–12
- Harel M, Weik M, Silman I, Sussman JL. Native Acetylcholinesterase (E.C. 3.1.1.7) from torpedo californica at 1.8A resolution. To be Published.
- Harel M, Kryger G, Rosenberry TL, Mallender WD, Lewis T, Fletcher RJ, Guss JM, Silman I, Sussman JL. Three-dimensional structures of Drosophila melanogaster acetylcholinesterase and of its complexes with two potent inhibitors. Protein Sci 2000; 2(6)1063–1072
- Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS. The universal protein resource (UniProt). Nucleic Acids Res 2005; 33: D154–D159
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997; 18(15)2714–2723
- Swiss-Pdb Viewer URL: http://www.expasy.org/spdbv/.
- Kuca K, Cabal J, Kassa J. In vitro reactivation of sarin-inhibited brain acetylcholinesterase from different species by various oximes. J Enz Inhib Med Chem 2005; 20(3)227–232
- Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol 1993; 234(3)779–815