Abstract
In order to understand the binding modes of human DNA polymerase α (pol α) inhibitors on a molecular level, a 3D homology model of the active site of the enzyme was proposed based on the application of molecular modelling methods and molecular dynamic simulations using available crystal coordinates of pol α relatives. Docking results for a series of known nucleotide analogue inhibitors were consistent with reported experimental binding data and offered the possibility to elucidate structure-activity relationships via investigations of active site-inhibitor interactions. Furthermore, the study could explain, at least partially, the inhibitory effect of aphidicolin on pol α. In molecular dynamics simulations, aphidicolin occupied the catalytic centre, but acted in a not truly competitive manner with respect to nucleotides. It destabilized the replicating “closed” form of the pol α and transferred the enzyme into the inactive “open” conformation. This result is consistent with recent experiments on the binding mode of aphidicolin.
Introduction
Due to an increased exposure of western society to UV, the number of patients with actinic keratosis and cutaneous squamous cell carcinoma has increased dramatically over the last 30 years. A further increase is to be awaited for future years. Current therapy of actinic keratosis is based on 5-fluorouracil, applied as cream or as a polymeric microsponge preparation, photodynamic therapy using the methyl ester prodrug of the UV sensitizer 5-aminolaevulinic acid or inhibition of cyclooxygenase2 which is over-expressed in these tumors by diclofenac/hyaluronic acid. Another option is imiquimod, which by interacting with toll-like receptors increases the immunogenic response. Complete therapeutic response rates, however, do not exceed 60%, a result which is not satisfactory in cancer treatment. Moreover, treatment is often associated with severe local irritation as well as pain and secondary infections and is not suitable for patients which exhibit extensive numbers of basal cell carcinomas [Citation1]. Thus alternative approaches have to be looked for, which do not only increase cure rates of single lesions but allow topical drug application to larger UV– exposed skin areas, in order to eliminate both obvious skin carcinomas and minimal micro-lesions.
Our investigations suggest that this may become possible by interfering with DNA synthesis. In viral diseases the introduction of DNA polymerase inhibitors has meant a break-through in the treatment of herpes and human immunodeficiency virus infections. We aim at a similar effect in the topical treatment of skin pre-cancerous and cancerous lesions. Polymerase α is an essential enzyme for DNA replication and cell division. Pol α inhibitors could therefore be considered as a group of potentially useful anticancer drugs. Some nucleosides and nucleotide analogues are already used systemically for leukaemia (cytarabine, fludarabine) and pancreatic carcinoma (gemcitabine), whereas others like N2-butylphenyl-dGTP, 2-butylanilino-dATP and aphidicolin are used in vitro for studying the DNA replication system.
Pol α belongs to the DNA polymerase B family which includes the prokaryotic pol II, several eukaryotic and archaeal polymerases, as well as the viral adenovirus, herpes simplex virus-1, bacteriophage RB69, T4 and T6 polymerases. Despite their diverse biological origins, the structural and chemical mechanisms of base incorporation seem to be highly conserved among the polymerases of the B family. Crystal structures of DNA pols indicate that these enzymes are composed of several functionally distinct domains and subdomains. A general feature of these structures, a large cleft comprised of three subdomains, the “fingers”, the “palm”, and the “thumb”, can be characterized as a “right hand” that is capable of holding DNA in its gaps [Citation2]. The palm subdomain at the bottom of the cleft contains the catalytic site, while the finger and thumb subdomains form the walls of the cleft. Three different conformations of RB69 polymerase have been observed in crystal structures: the apo state without DNA, the editing mode forming a binary complex with DNA bound to the exonuclease site, and the replicating mode, a ternary complex with DNA and a dNTP bound in the polymerase catalytic site. In the apo state and editing mode, the enzyme is in the “open” form, showing a rotation of the fingers domain 60° away from the palm. In the “closed” conformation one arginine and two lysines of the fingers domain are involved in hydrogen bonding to the phosphate groups of the incoming dNTP. DNA synthesis is mediated by transfer of a phosphoryl group from the incoming dNTP to the DNA 3′-OH, liberating pyrophosphate and forming a new DNA phosphoester bond. This reaction is catalysed by a mechanism that involves two divalent metal ions, with the participation of two aspartic acid residues, which are structurally conserved among the different polymerase enzymes [Citation3]. Although crystal structures of several type B DNA polymerases are available, the 3D structure of the human pol α has yet to be determined. Since a detailed 3D model of the active site could facilitate the rational design of new inhibitors, we have started to construct a molecular model of the active site of pol α which would permit us to study the structural requirements of pol α inhibition. We report in this paper a model for the tertiary structure of human pol α restricted to the 836–1102 region that contains, by analogy with other B family polymerases of known structures, the fingers, palm and thumb regions of the enzyme.
Molecular dynamics simulations and docking of a series of known inhibitors, including aphidicolin (molecular structures see ) have been used to prove our active site model. In a forthcoming study (Part 2) we have established an assay to detect if pol α inhibition may be of value in skin cancer, investigating the influence of aphidicolin on keratinocyte proliferation and cell death. In addition, we have used the homology model to develop strategies for designing new pol α inhibitors.
Table I. Molecular structure and inhibition data of ligands used for active site docking.
Methods and materials
The sequences of human pol α, bacteriophage RB69 DNA pol, and T9N7 pol in the palm, fingers and thumb regions have been aligned by using multiple sequence and pairwise alignment programmes CLUSTALW [Citation4] and BLAST [Citation5]. Secondary structure predictions were performed with the programme PSIPRED [Citation6]. All of the initial modelling, including loop building and side chain placement, was performed with the programme SYBYL 7.0 [Citation7]. Assignment of the coordinates of homologous regions has been achieved using the crystallographic RB69 pol coordinates, Protein Data Bank accession code 1IG9 [Citation8] in the replicating mode for template structure. The backbone coordinates were transferred from RB69 pol to pol α. Side chains conformations have been assigned using existing side chain conformations for identical or closely similar residues or manual selection from a rotamer library. Structurally variable regions (loops) have been generated by scanning the Protein Data Bank for similar sequences. The resulting 3D protein model was subjected to energy minimization and finally validated with PROCHECK [Citation9]. A detailed description of the protein modelling procedures used has been given elsewhere [Citation10,Citation11]. Molecular structures of primer and template DNA as well as the incoming thymidine triphosphate (dTTP) and three Ca2 + (of which only two are relevant to coordinate the triphosphate) ions were taken from the RB69 pol crystal coordinates and merged into the active site model. The resulting complex was solvated in a cubic box filled with water molecules and submitted to molecular dynamics simulations.
Molecular dynamics simulations have been carried out with the GROMACS simulation package [Citation12]. Hydrogen bond lengths have been constrained, allowing an integration step of 2 fs. A system with periodic boundary conditions was used where electrostatic interactions were calculated with the Particle-Mesh Ewald method. Temperature and pressure were kept constant. The GROMACS united atom force field has been applied. The total simulation length was 2.4 ns. During the first 200 ps the water molecules were allowed to equilibrate with positional restraints on all the other atoms. Subsequently a 2.2 ns run with positional restraints on DNA, dTTP and the ions only has been performed in order to obtain a relaxed protein structure.
For the docking studies the dTTP ligand from the model complex was changed against a set of known nucleoside triphosphate inhibitors [Citation13,Citation14] and the non-nucleoside inhibitor aphidicolin [Citation15]. The inhibitor molecules (all nucleosides contained a thymine base in order to get a correct Watson-Crick base pairing with the given template) were docked manually into the active site by superimposing the structures with the natural substrate in a conformation close to the dTTP template. The resulting interaction complexes were minimized with steepest descent and conjugate gradient methods.
Results and discussion
The pol α model: in the absence of experimental data, model-building on the basis of the known 3D structure of a homologous protein is at present the only reliable method to obtain structural information. For accurate structure predictions, at least 30% sequence identity of the target protein with templates of known structures is required. According to alignment results, the palm, fingers and thumb domains of human pol α share 34% identical residues and 49% similar residues with T9N7 pol, but only 20% identical and 28% similar with RB69 pol. Thus, T9N7 pol is the closer homologue, and should be a better choice for homology modelling. However, crystal coordinates were available only in the “apo” (and therefore inactive) state, a conformation not useful for creating an active site model. Comparisons of the tertiary structures of homologous proteins have shown that 3D structures have been much better conserved during evolution than protein primary structures. A close inspection of the crystal structures of the apo forms of T9N7 pol and RB69 pol indicated a good match of the active sites, despite their low sequence identity of about 17%. On the basis of these data, we concluded, that RB69 pol seems to be a quite suitable template for modelling the pol α active site.
The resulting architecture of our model is very similar to the crystal structure of RB69 pol, as can be seen from . The protein model was found to be stable, since restraints have not been necessary and the ternary complex with primer, template, dTTP and ions has remained fully intact during the molecular dynamics simulations. The dTTP binding site () shows similar binding characteristics as seen in the crystal structure of RB69 pol: three conserved, positively charged residues, Arg922, Lys926, and Lys950 form salt bridges with the α and γ-phosphate oxygens of the dTTP. The metal ions (Ca2 + ions were taken according to the RB69 pol crystal structure) are ligated by Asp860 and Asp1004 and further complexed with the phosphate groups. Between the aromatic ring of Tyr865 and the ribose of dTTP, a stacking interaction is formed accompanied by a hydrogen bond between the amide hydrogen of Tyr865 and the oxygen of the 3′-OH group of the ribose. Docking of the inhibitors to the nucleotide binding pocket of the pol α model shows interesting correlations with the reported biological data (Table 1).
Figure 1 Human DNA polymerase α model. a) Superposition of the protein backbones of the template (α-helices = red, β-sheets = yellow, loops = cyan) and the pol α model. The model contains the nucleotide binding domain only, consisting of finger, palm and thumb region (α-helices = blue, β-sheets = green, loops = grey). b) Close-up view of the dTTP binding complex. Hydrogen bonds are shown as black dashed lines. The ligand is presented in ball-and-stick- mode. Colour code: carbon = white, hydrogen = cyan, oxygen = red, nitrogen = blue, phosphorus = orange, calcium = green). (Please see colour online).
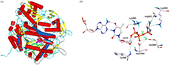
Adefovir-DP
Phosphonylmethoxyethyl derivatives like adefovir, belong to a class of acyclic nucleotide analogues containing a metabolically stable P-C bond in a phosphonylmethoxyethyl group (see molecular structure in ). The active diphosphate forms function as chain terminators after their incorporation into DNA due to the lack of a 3′-OH moiety in the molecule. Adefovir-DP fits well in our model in terms of molecular interactions. The flexible side chain easily adopts a conformation required for interaction with the active site of pol α. Even though stacking interaction and hydrogen bonding like in dTTP cannot occur with its acyclic side chain, adefovir-DP is a good substrate due to its conformational flexibility and good positional adaptability.
Tenofovir-DP
Tenofovir is a phosphonylmethoxypropyl derivative with significantly lower inhibitory effects on pol α compared to adefovir-DP. Our docking studies indicate that the additional methyl group of this inhibitor reduces the probability of a good fit due to steric hindrance. Especially, the proximity of the methyl group to the phenyl ring atoms of Tyr865 is unfavourable and may lower the stability of the replicative complex.
DdNTP
2′,3′-Dideoxynucleotides (like ddATP) are poor inhibitors of pol α. The only structural difference between dNTP and ddNTP is the lack of the 3′-OH group. However, because of their close molecular similarity to dNTPs, ddNTP derivatives fit well in the active site. Its decreased inhibition rates have been ascribed to the loss of an intramolecular hydrogen bond between the 3′-OH of the deoxyribose and a phosphate oxygen (which is found in the case of dNTP, see Figure 1b), possibly affecting correct conformational alignment of the reactive phosphate groups [Citation16]. Furthermore, ddNTP derivatives are not capable of forming a hydrogen bond with the Tyr865 backbone [Citation17]. However, it seems somewhat paradoxical that in the case of adefovir-DP none of these hydrogen bonds exist and yet adefovir-DP is favoured over the ddNTP analogues. To investigate the differences in the conformational properties, we have calculated force field energies for the appropriate dTTP, ddTTP and adefovir-DP conformations. In accordance with the cited postulations and the inhibition data, dTTP exhibits the energetically most favourable conformation, due to its intramolecular hydrogen bond, followed by adefovir-DP, which inhibits much better torsional energies than the more rigid ddTTP (data not presented here).
Stavudine-TP
The structure-activity relationships, which have been used to explain the inhibition properties of ddNTP, can be applied just as well to stavudine. The still lower inhibition rate of stavudine-TP may be due to an increased rigidity caused by the double bond in the dideoxyribose. We have performed a Monte Carlo conformational search method to compare the molecular flexibility of stavudine-TP and a ddNTP analogue. From these calculations, stavudine-TP exhibited a distinctly reduced number of energetically favourable conformations compared to ddNTP.
Lamivudine-TP and zidovudine-TP
Docking of lamivudine-TP clearly shows that its poor inhibitory effects are due to a sterical conflict with the phenyl ring of Tyr865 caused by the large sulphur atom in the dideoxyribose, as can be seen from . The same is true for zidovudine-TP, a molecule bearing a large and rigid azido group. In contrast to the HIV-1 reverse transcriptase, which is able to accommodate such bulky molecules, the pol α binding pocket seems to be much tighter and thus more discriminating.
Figure 2 Inhibitor docking results. a) Lamivudine-TP/polymerase α complex. The sulphur atom of the lamivudine thioribose produces a steric clash with tyrosine 865 of the active site. Amino acids are shown in orange coloured lines, Tyr 865 (orange) and lamivudine-TP are shown as space filled models (carbon = green, hydrogen = white, nitrogen = blue, oxygen = red, phosphorus = magenta, sulphur = yellow). b) BuAdATP bound in the active site pocket of the polymerase α model. BuAdATP is presented in ball-and-stick mode. Hydrogen bonds are shown as black dashed lines (colour code see Figure 1).
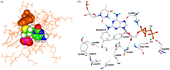
Buadatp
The nucleotide analogue 2-butylanilino-dATP (see Table I) is a potent and highly selective inhibitor of mammalian pol α. Since BuAdATP, in contrast to the other investigated inhibitors, contains a bulky butylphenyl moiety attached to the base, manual docking generated some bad van der Waals’ contacts. Thus a molecular dynamics simulation was performed to reach an energetically favourable interaction complex. shows an extract of the resulting complex. The butylphenyl moiety of BuAdATP occupies a lipophilic pocket, formed by the residues Leu960, Leu972, Val976, Ile869, Tyr865 and Tyr957. These lipophilic interactions between the butylphenyl moiety and the pol α active site are likely to be responsible for the good inhibition efficiency of the butylanilino derivatives.
Aphidicolin, a tetracyclic diterpene with no structural similarities to nucleotides (), is a potent inhibitor of eukaryotic polymerases α and δ. The molecular details of the inhibition of pol α activity by aphidicolin have not been determined yet. A competitive inhibition mechanism was proposed with dCTP and perhaps dTTP and non-competitive ones with the other dNTP substrates [17 and references therein]. Studies have been attempted [Citation18] to identify the protein residues which participate in aphidicolin interactions by characterizing mutations within the aphidicolin-sensitive DNA pol of herpes simplex virus type, which alter the substrate specificity of this enzyme. The interaction model which emerged from these studies suggested that the binding sites for aphidicolin and dNTPs are in close proximity and overlap substantially. From mutational studies on human pol α, it has been suggested [Citation19] that aphidicolin forms hydrogen bonds with two of its four hydroxyl groups to the side chains of Asp1002 and Thr1003, blocking any incoming dNTP-metal complex. The competitive inhibitory effect against dCTP (and dTTP) has been postulated to occur via hydrogen bonding of the other two hydroxyl groups to the template purines. We have used this concept as a starting point to investigate the interaction properties of aphidicolin with the active site of the pol α model. shows the starting structure wherein aphidicolin has been placed manually instead of dTTP. The starting position was chosen according to the proposal of Copeland and Wang [Citation19] wherein aphidicolin binds with its two hydroxyl groups of ring A to Asp1002 and Thr1003 and with the ring C hydroxyl groups to a base of the DNA template (this is one possible position; due to its somehow symmetrical character concerning the hydroxyl groups and due to enough space in the catalytic centre, aphidicolin could also bind in the opposite orientation). Figure 3b shows the resulting structure after an 1.5 ns molecular dynamics simulation. In principle, aphidicolin does not change its overall position during the molecular dynamics run. It forms a stable interaction complex via hydrogen bonds between its OH-groups, the template purine and the side chain of Asp1004 (instead of the proposed Asp1002) and Asn954 (instead of Thr1003, which is not close enough for a stable hydrogen bond). However, as can be seen from the Figure, the protein structure undergoes a large conformational change, from the “closed” to the “open” form. This conformational change is due to the loss of the ionic interactions between the substrate and the positively charged residues Arg922, Lys926 and Lys950. In addition, aphidicolin does not form chelates with the metal ions, thus lacking a very important property for stabilizing the “closed” conformation. During the simulation, the Ca2 + ions move away from the catalytic centre. In the active replication mode these divalent cations are coordinated by two aspartic acids and by the triphosphate tail of the incoming nucleotide. Aphidicolin is not able to coordinate the Ca2 + ions. In addition, it interrupts the interaction of the metal ions with the aspartic acids, thus the ions start to move out of the active site, a result that is in accordance with polymerase crystal structures. Only in the presence of a nucleotide triphosphate are the two metal ions found in the active site.
Figure 3 The polymerase α/aphidicolin complex before (a) and after (b) a 1.5 ns molecular dynamics simulation. The protein changes from the closed form to the inactive open form and the Ca2 + ions move away from the catalytic centre. Colour code: α-helices = red, β-sheets = yellow, loops = green, calcium ions = magenta, aphidicolin = cyan.
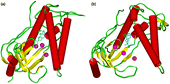
From these results we conclude that aphidicolin may be able to block the active site of pol α; however, it is not able to do this with the replicating “closed” conformation of the enzyme. Compared to the strong and specific contacts of the dNTPs with the polymerase active site, aphidicolin forms only significantly weaker and relatively unspecific hydrogen bonds. Thus the modelling studies do no reflect a true competitive inhibition mechanism for aphidicolin, a result that is in good accordance with the experimental data.
However, one has to keep in mind that all domains of the polymerase may contribute to processing and strand transfer, thus the model of the central core domain presents a detailed, but truncated snap-shot of the enzyme/substrate complex formation and the replication mechanism. Thus, although Hall et al. [Citation18] as well as Copeland and Wang [Citation19] have derived from their experiments a binding position for aphidicolin close to the nucleotide binding site, it is possible that other domains of pol α may be required for the inhibitory effects of that molecule. Despite its four OH-groups, aphidicolin is mainly of hydrophobic nature. It has substantial similarity in its overall structure to non-nucleoside inhibitors of HIV reverse transcriptase, which bind to a hydrophobic pocket that is proximal to, but distinct from the transcriptase active site and is located at the palm-thumb subdomain interface. Binding of these inhibitors causes restriction on the movement of the thumb, conformational changes of the residues at the transcriptase active site, and displacement of the primer grip [Citation20]. In the hepatitis C virus RNA polymerase non-nucleoside inhibitors bind to a hydrophobic pocket on the surface of the thumb subdomain and have an allosteric effect that interferes with the conformational change of the thumb [Citation21]. Thus, it is conceivable that aphidicolin can inhibit pol α in a similar allosteric manner. Since the molecular model, we have presented here, includes the active site only and is not a complete representation of the whole pol α molecule, this question still needs to be addressed.
In summary, the lack of effective therapeutics against actinic keratosis and cutaneous squamous cell carcinoma has encouraged us to investigate - on the basis of molecular modelling methods - the inhibition properties of nucleotide inhibitors and aphidicolin on human DNA pol α. We were able to build a three-dimensional homology model of the pol α active site. This model offered molecular insights into the structural requirements for enzyme inhibition and allowed us in a forthcoming study to postulate new pol α inhibitors.
Acknowledgements
This work was financially supported by RIEMSER Arzneimittel AG, Greifswald - Insel Riems, Germany.
References
- Chakrabaty A, Geisse JK. Medical therapies for non-melanoma skin cancer. Clin Dermatol 2004; 22: 183–188
- Alba M. Replicative DNA polymerises. Genome Biol 2001; 2, reviews3002.1–3002.4
- Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem 1999; 274: 17395–17398
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994; 22: 4673–4680
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389–3402
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999; 292: 195–202
- SYBYL 7.0., Tripos Inc., 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA.
- Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell 2001; 105: 657–667
- Laskowski R, MacArthur M, Moss D, Thornton J. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst 1993; 26: 283–291
- Introduction to Comparative Protein Modeling. Molecular modeling: Basic principles and applicationsSecond Edition, HD Höltje, W Sippl, D Rognan, G Folkers. Wiley-VCH Verlag, Weinheim 2003; 87–142
- Rupp B, Raub S, Marian C, Höltje HD. Molecular design of 2 sterol-14 a-demethylase homology models and their interactions with the azole antifungals ketoconazole and bifonazole. J Comput Aided Mol Des 2005; 19: 149–163
- Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J Mol Model 2001; 7: 306–317
- Cihlar T, Chen M. Incorporation of selected nucleoside phosphonates and anti-human immunodeficiency virus nucleotide analogues into DNA by human DNA polymerases α, β, and γ. Antivir Chem Chemother 1997; 8: 187–195
- Khan NN, Wright GE, Dudycz LW, Brown NC. Butylphenyl dGTP: A selective and potent inhibitor of mammalian DNA polymerase alpha. Nucleic Acids Res 1984; 12: 3695–3706
- Arabshahi L, Brown N, Khan N, Wright G. Inhibition of DNA polymerase alpha by aphidicolin derivatives. Nucleic Acids Res 1988; 16: 5107–5113
- Selmi B, Boretto J, Sarfati SR, Guerreiro C, Canard B. Mechanism-based suppression of dideoxynucleotide resistance by K65R human immunodeficiency virus reverse transcriptase using an alpha-boranophosphate nucleoside analogue. J Biol Chem 2001; 276: 48466–48472
- Yang G, Franklin M, Li J, Lin TC, Konigsberg W. A conserved Tyr residue is required for sugar selectivity in a pol alpha DNA polymerase. Biochemistry 2002; 41: 10256–10261
- Hall JD, Wang YS, Pierpont J, Berlin MS, Rundlett SE, Woodward S. Aphidicolin resistance in herpes simplex virus type I reveals features of the DNA polymerase dNTP binding site. Nucleic Acids Res 1989; 17: 9231–9244
- Copeland WC, Wang TS. Mutational analysis of the human DNA polymerase alpha. The most conserved region in alpha-like DNA polymerases is involved in metal-specific catalysis. J Biol Chem 1993; 268: 11028–11040
- Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol 1995; 2: 303–308
- Love RA, Parge HE, Yu X, Hickey MJ, Diehl W, Gao J, Wriggers H, Ekker A, Wang L, Thomson JA, Dragovich PS, Fuhrman SA. Crystallographic identification of a noncompetitive inhibitor binding site on the hepatitis C virus NS5B RNA polymerase enzyme. J Virol 2003; 77: 7575–7581