Abstract
The synthesis of dideoxy-6-azathymidine 4′-thionucleoside 1-(2,3-dideoxy-4-thio-β-D-erythro-pentofuranosyl)-(6-azathymidine) (2), and the L-nucleoside, 1-(4-thio-β-L-erythro-pentofuranosyl)-(6-azathymidine) (3) and their evaluation against a wide panel of antiviral assays are described. The L-thionucleoside (3) was devoid of antiviral activity. The dideoxy-thionucleoside (2) was moderately active against vaccinia virus (VV) and the herpes simplex virus strains HSV-1 (strain KOS) and HSV-2 (strain G) (MIC 12 μM) and retained inhibitory activity vs a thymidine kinase-deficient strain HSV-1/TK–, suggesting that (2) is not dependent on viral TK-catalysed phosphorylation for antiviral activity and/or may use an alternative metabolic activation pathway.
Introduction
We have recently shown that 1-(2-deoxy-4-thio-β-D-erythro-pentofuranosyl)-(6-azathymidine) (1) (, R = CH3) displayed pronounced activity against herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2), varicella-zoster virus (VZV) and vaccinia virus [Citation1]. Thionucleoside 1 displayed activity comparable with acyclovir (ACV) (EC50∼1 μM) but was considerably less active than brivudin ((E)-5-(2-bromovinyl)-2′-deoxyuridine, BVDU) (EC50: 0.003 μM). However, importantly 1 showed a good retention of antiviral activity against VZV/TK– strains, whereas BVDU lost activity by at least 4 orders of magnitude [Citation1]. These observations suggest that the 4′-thionucleoside 1 may not entirely depend on viral TK-catalysed phosphorylation for antiviral activity and/or use an alternative metabolic activation pathway, and/or display a unique mechanism of antiviral action by the unmetabolised nucleoside analogue. 1-(2-Deoxy-4-thio-β-D-erythro-pentofuranosyl)-(6-azauracil) (, R = H), which has previously been described by us [Citation2], was devoid of antiviral activity. Likewise base modification at the 5-position generated the 5-ethyl and 5-propyl-6-azauridine 4′-thio-2′-deoxynucleosides (, R = Et/Pr) that also lacked any antiviral activity [Citation1].
Figure 1 Structures of 1, 2 & 3.
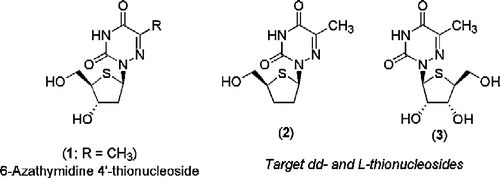
This paper describes the synthesis and antiviral evaluation of two novel 4′-thiosugar modified derivatives of 6-azathymidine, namely the dideoxy, 1-(2,3-dideoxy-4-thio-β-D-erythro-pentofuranosyl)-(6-azathymidine) (2), and the L-nucleoside analogue, 1-(4-thio-β-L-erythro-pentofuranosyl)-(6-azathymidine) (3) ().
Materials and methods
1H and 13C NMR spectra were recorded with a Brucker Avance DPX500 spectrometer operating at 500 and 125 MHz, with Me4Si as internal standard. Mass spectra were determined by the EPSRC mass spectrometry centre (Swansea, UK). Microanalyses were determined by Medac Ltd (Surrey, UK). Flash column chromatography was performed with silica gel 60 (230–400mesh) (Merck) and TLC was carried out on precoated silica plates (kiesel gel 60 F254, BDH). Melting points were determined on an electrothermal instrument and are uncorrected. Compounds were visualised by illumination under UV light (254 nm) or by the use of vanillin stain followed by charring on a hotplate. All solvents were dried prior to use as described by the handbook Purification of Laboratory Chemicals [Citation3] and stored over 4 Å molecular sieves, under nitrogen.
Synthesis
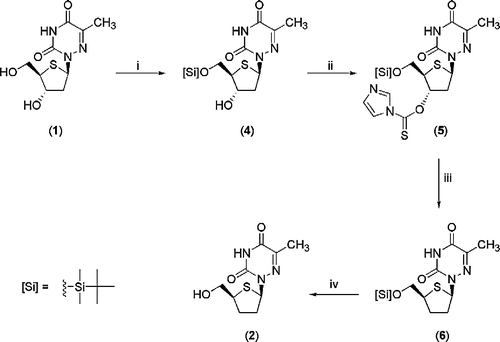
5′-O-tert-Butyldimethylsilyl-2′-deoxy-4′-thio-β-D-erythro-pentofuranosyl-1′-(6-azathymine) (4)
To a solution of 1-(2-deoxy-4-thio-β-D-erythro-pentofuranosyl)-(6-azathymidine) (1) [Citation1] (0.30 g, 1.16 mmol) and imidazole (0.20 g, 2.89 mmol) in dry N,N-dimethylformamide (DMF) (10 mL) was added a solution of tert-butyldimethylsilyl chloride (TBDMSCl) (0.22 g, 1.45 mmol) in dry DMF (2 mL) and the reaction stirred at room temperature under nitrogen for 3.5 h. The reaction mixture was quenched with MeOH (6 mL), diluted with EtOAc (12 mL) and washed with H2O (6 × 12 mL). Then the organic layer was dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product as a pale yellow solid. Purification by column chromatography (EtOAc-petroleum ether 1:1 v/v) yielded 0.23 g (53%) of compound 4 as a white solid. Rf 0.38 (EtOAc-petroleum ether 3:2 v/v); m.p. 36–38 ˚C 1H-NMR (CDCl3): δ 8.85 (bs, 1, NH), 6.27 (dd, 1, J = 7.6, 3.3 Hz, H-1′), 4.75 (m, 1, OH), 3.84 (dd, 1, J = 10.0, 5.4 Hz, H-5′), 3.73 (t, 1, J = 9.8 Hz, H-5′), 3.35 (quintet, 1, J = 5.2 Hz, H-3′), 2.65 (m, 1, H-4′), 2.57 (ddd, 1, J = 13.6, 5.1, 3.4 Hz, H-2′), 2.37 (dd, 1, J = 13.6, 7.9 Hz, H-2′), 2.26 (s, 3, CH3), 0.88 (s, 9, 3 × CH3-Si), 0.07 (s, 3, CH3-Si), 0.06 (s, 3, CH3-Si). 13C-NMR (CDCl3): δ 156.08 (C = O, C-2), 148.25 (C, C-5), 144.85 (C = O, C-4), 78.43 (CH, C-3′), 67.10 (CH2, C-5′), 62.28 (CH, C-1′), 55.60 (CH, C-4′), 41.31 (CH2, C-2′), 26.28 (CH3, 3 × CH3-Si), 18.67 (C, SiCCH3), 17.08 (CH3), − 4.92 (CH3, 2 × CH3-Si). IRVmax/cm − 1 (NaCl, film): 1690.1 (C = O). HRMS (ES+) calcd for C15H28N3O4SSi (M + H)+374.1564, found, 374.1565.
3′-O-Thiocarbonylimidazole-5′-O-tert-butyldimethylsilyl-2′-deoxy-4′-thio-β-D-erythro-pentofuranosyl-1′-(6-azathymine) (5)
Thiocarbonyldiimidazole (0.43 g, 2.40 mmol) was added to a solution of 4 (0.45 g, 1.20 mmol) in dry CH2Cl2 (35 mL) and the reaction stirred at 40°C for 8 h and at room temperature for 12 h. The reaction mixture was concentrated under reduced pressure to give the crude product as a yellow syrup. Purification by column chromatography (EtOAc-petroleum ether 1:1 v/v) yielded 0.80 g (69%) of compound 5 as a white solid. Rf 0.24 (EtOAc-petroleum ether 1:1 v/v); m.p. 160 ˚C (sharp); 1H-NMR (CDCl3): δ 9.17 (bs, 1, NH), 8.36 (s, 1, H-5″), 7.61 (s, 1, H-2″), 7.04 (s, 1, H-3″), 6.47 (t, 1, J = 6.6 Hz, H-1′), 6.40 (q, 1, J = 4.0 Hz, H-3′), 3.79 (d, 2, J = 8.9 Hz, H-5′), 3.72 (q, 1, J = 8.0 Hz, H-4′), 3.00 (dd, 1, J = 15, 5.3 Hz, H-2′), 2.61 (ddd, 1, J = 14.3, 7.3, 4.7 Hz, H-2′), 2.29 (s, 3, CH3), 0.85 (s, 9, 3 × CH3-Si), 0.03 (s, 6, 2 × CH3-Si). 13C-NMR (CDCl3): δ 183.24 (C = S, OCSN), 156.40 (C = O, C-2), 148.78 (C, C-5), 148.78 (C = O, C-4), 137.43 (CH, C-3″), 131.25 (CH, C-2″), 118.44 (CH, C-5″), 87.05 (CH, C-3′), 65.57 (CH2, C-5′), 62.87 (CH, C-1′), 54.62 (CH, C-4′), 37.56 (CH2, C-2′), 26.22 (CH3, 3 × CH3-Si), 18.67 (C, SiCCH3), 17.15 (CH3), − 4.90 (CH3, CH3-Si), − 4.98 (CH3, CH3-Si). IRVmax/cm − 1 (NaCl, film): 3423.0 (N-H stretch), 1701.7 (C = O), 1388.0-1232.0 (C-O stretch). Anal. Calcd. for C19H29N5O4S2Si (483.6747): C, 47.18%, H, 6.04%, N, 14.48%. Found: C, 47.05%, H, 6.12%, N, 14.35%.
5′-O-tert-Butyldimethylsilyl-2′,3′-dideoxy-4′-thio-β-D-erythro-pentofuranosyl-1′-(6-azathymine) (6)
Tributyltin hydride (0.13 g, 0.45 mmol) and 1,1′-azobis(cyclohexene-carbonitrile) (0.07 g, 0.30 mmol) was added to a solution of 5 (0.15 g, 0.31 mmol) in dry toluene (14 mL) and stirred at 100°C for 30 min. The reaction mixture was concentrated under reduced pressure to give the crude product as a clear syrup. Purification by column chromatography (EtOAc-petroleum ether 3:7 v/v) yielded 0.07 g (64%) of compound 6 as a clear syrup. Rf 0.77 (EtOAc-petroleum ether 1:1 v/v); 1H-NMR (CDCl3): δ 8.56 (bs, 1, NH), 5.57 (t, 1, J = 4.0 Hz, H-1′), 3.93 (q, 2, J = 7.3 Hz, H-3′), 3.75 (dd, 1, J = 10.0, 7.9 Hz, H-5′), 3.67 (dd, 1, J = 10.0, 6.4 Hz, H-5′), 3.52 (quintet, 1, J = 7.0 Hz, H-4′), 2.43 (m, 1, H-2′), 2.38 (m, 1, H-2′), 1.51 (s, 3, CH3), 0.84 (s, 9, 3 × CH3-Si), 0.02 (s, 3, CH3-Si), 0.00 (s, 3, CH3-Si). 13C-NMR (CDCl3): δ 156.34 (C, C-5), 148.78 (C = O, C-2), 143.94 (C = O, C-4), 66.86 (CH2, C-5′), 65.44 (CH, C-1′), 52.20 (CH, C-4″), 34.55 (CH2, C-2′), 31.94 (CH2, C-3′), 25.95 (CH3, CH3-Si), 25.88 (CH3, CH3-Si), 25.85 (CH3, CH3-Si), 18.40 (C, SiCCH3), 16.20 (CH3), − 5.20 (CH3, CH3-Si), − 5.29 (CH3, CH3-Si). IRVmax/cm − 1 (NaCl, film): 3450.7 (N–H stretch), 1651.6 (C = O), 1462.3–1100.1 (C–O stretch). HRMS (ES+) calcd for C15H28N3O3SSi (M + H)+358.1615, found, 358.1614.
2′,3′-Dideoxy-4′-thio-β-D-erythro-pentofuranosyl-1′-(6-azathymine) (2)
To a solution of 6 (0.13 g, 0.36 mmol) in MeOH (10 mL), was added Dowex 50 W (H+) (0.50 g) [Dowex 50 W (H+) was washed with 10% aq. HCl (10 mL), MeOH (10 mL), CH2Cl2 (10 mL) and another 10% aq. HCl (10 mL) prior to use] and the reaction mixture was stirred at room temperature for 24 h. The resulting light yellow solution was filtered to remove Dowex 50W (H+) and washed with MeOH. The filtrate was concentrated in vacuo to give the crude product as a light yellow syrup. Purification by column chromatography (MeOH-CH2Cl2 1:19 v/v) yielded 0.06 g (67%) of compound 2 as a clear syrup. Rf 0.28 (EtOAc-petroleum ether 1:1 v/v); 1H-NMR (DMSO-d6): δ 12.01 (bs, 1, NH), 6.15 (dd, 1, J = 7.0, 3.5 Hz, H-1′), 4.90 (bs, 1, OH), 3.81 (m, 1, H-3′), 3.60 (m, 1, H-3′), 3.48 (dd, 1, J = 13.6, 6.9 Hz, H-5′), 3.44 (m, 1, H-5′), 3.38 (m, 1, H-4′), 2.37 (m, 1, H-2′), 2.24 (m, 1, H-3′), 2.08 (s, 3, CH3). 13C-NMR (DMSO-d6): δ 148.47 (C, C-5), 143.23 (2 × C = O, C-2 & C-4), 65.35 (CH2, C-5′), 64.38 (CH, C-1′), 52.31 (CH, C-4′), 33.91 (CH2, C-2′), 31.82 (CH2, C-3′), 16.41 (CH3). LRMS (CI+) m/z: 117.0 (M-heterocyclic base)+, 128.0 (M-2,3-dideoxy-thiosugar)+, 244.1 (M + H)+. HRMS (ES+) calcd for C9H14N3O3S (M + H)+244.0750, found, 244.0751.
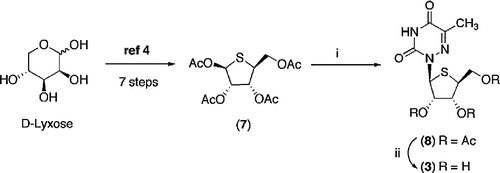
2′,3′,5′-Tri-O-acetyl-4′-thio-β-L-6-azathymidine (8)
To a suspension of 6-azathymine (0.17 g, 1.35 mmol) in dry CH3CN (5 mL) was added BSA (0.67 mL, 2.70 mmol) and the reaction mixture was stirred at room temperature under nitrogen for 30 min. A solution of 1,2,3,5-tetra-O-acetyl-4-thio-β-L-ribofuranose 7 [Citation4] (0.52 g, 1.55 mmol) in dry CH3CN (5 mL) was then added. The reaction mixture was cooled in an ice bath before TMSOTf (0.57 mL, 2.56 mmol) was added dropwise. The reaction mixture was stirred at 50°C under nitrogen for 8 h, then at room temperature for 12 h. The reaction mixture was diluted with CH2Cl2 (50 mL) and washed with saturated aqueous sodium bicarbonate (2 × 30 mL). The organic layer was dried (MgSO4) and concentrated under reduced pressure to give a yellow syrup. Purification by column chromatography (EtOAc-petroleum ether 2:3 v/v) yielded 0.35 g (65%) of compound 8 as a thick syrup. Rf 0.62 (EtOAc-petroleum ether 2:1 v/v); 1H-NMR (CDCl3): δ 8.86 (bs, 1, NH), 6.25 (d, 1, J = 4.4 Hz, H-1′), 5.83 (t, 1, J = 4.2 Hz, H-2′), 5.73 (dd, 1, J = 5.6, 4.1 Hz, H-3′), 4.40 (dd, 1, J = 11.6, 6.8 Hz, H-5′), 4.31 (dd, 1, J = 11.6, 6.6 Hz, H-5′), 3.77 (q, 1, J = 6.3 Hz, H-4′), 2.14 (s, 3, CH3Ac), 2.12 (s, 3, CH3Ac), 2.11 (s, 3, CH3Ac), 2.07 (s, 3, CH3). 13C-NMR (CDCl3): δ 170.46 (C = O, Ac), 169.81 (C = O, Ac), 169.64 (C = O, Ac), 155.46 (C = O, C-2), 147.94 (C = O, C-5), 145.54 (C, C-4), 75.56 (CH, C-2′), 74.07 (CH, C-3′), 64.70 (CH2, C-5′), 64.01 (CH, C-1′), 47.10 (CH, C-4′), 20.64 (CH3, CH3Ac), 20.42 (CH3, CH3Ac), 16.67 (CH3, CH3Ac), 14.20 (CH3). IRVmax/cm − 1 (NaCl, film): 1642.7 (C = O), 1226.8 (C–O stretch). HRMS (ES+) calcd for C15H20O8N3S (M + H)+419.1231, found, 419.1229.
4′-Thio-β-L-6-azathymidine (3)
Aqueous methylamine (7 mL) was added to compound 8 (0.30 g, 0.75 mmol) and the resulting clear solution was stirred at 50°C for 30 min. On completion, the clear solution changed colour to a light brown solution and was concentrated in vacuo to give a thick light brown syrup. Purification by column chromatography (MeOH-CH2Cl2 1:9 v/v) yielded 0.19 g (90%) of compound 3 as a light yellow syrup. Rf 0.46 (MeOH-CH2Cl2 1:9 v/v); 1H-NMR (DMSO-d6): δ 12.13 (bs, 1, NH), 5.87 (d, 1, J = 5.5 Hz, H-1′), 5.32 (d, 1, J = 5.0 Hz, OH, H-2′), 5.13 (d, 1, J = 4.8 Hz, OH, H-3′), 4.97 (m, 1, OH, H-5′), 4.36 (q, 1, J = 3.9 Hz, H-2′), 4.18 (q, 1, J = 3.8 Hz, H-3′), 3.69 (ddd, 1, J = 13.2, 6.6, 3.9 Hz, H-5′), 3.45 (ddd, 1, J = 17.9, 10.6, 6.5 Hz, H-5′), 3.24 (ddd, 1, J = 14.4, 7.0, 4.4 Hz, H-4′), 2.12 (s, 3, CH3). 13C-NMR (DMSO-d6): δ 156.53 (C = O, C-2), 148.89 (C, C-5), 143.69 (C = O, C-4), 75.63 (CH, C-2′), 73.54 (CH, C-3′), 64.65 (CH, C-1′), 64.40 (CH2, C-5′), 53.16 (CH, C-4′), 16.45 (CH3). LRMS (ES+) m/z: 298.05 (M + Na)+; LRMS (ES-) m/z: 274.1 (M–H)+, 275.1 (M)+. HRMS (ES+) calcd for C9H17N4O5S (M + NH4)+293.0914, found, 293.0912.
Thymidine kinase assay [Citation1]
The radiolabeled substrate [methyl-3H]dThd (70 Ci/mmol) was obtained from Amersham Pharmacia Biotech. The thymidine kinase activity using purified cytosolic TK-1, recombinant mitochondrial TK-2, recombinant herpes simplex virus type 1 TK, and recombinant varicella-zoster virus TK was assayed in a 50 μL reaction mixture containing 50 mM Tris HCl, pH 8.0, 2.5 mM MgCl2, 10 mM DTT, 0.5 mM CHAPS, 3 mg/mL bovine serum albumin, 2.5 mM ATP, 1 μM [methyl-3H]dThd, and varying concentrations of compound 2 or 3 and enzyme. The samples were incubated at 37°C for 30 min. Aliquots of 45 μL of the reaction mixtures were spotted on Whatman DE-81 filter paper disks. The filters were washed three times for 5 min in 1 mM HCOONH4 and once for 5 min in ethanol. The radioactivity was determined by scintillation counting.
Cell cultures
The antiviral assays were based on an inhibition of virus-induced cytopathicity in either E6SM, HeLa, Vero, or HEL cell cultures, following previously established procedures Citation5-8. Briefly, confluent cell cultures in 96-well microtiter plates were inoculated with 100 CCID50 of virus, 1 CCID50 being the virus dose required to infect 50% of the cell cultures. After a 1 h virus adsorption period, residual virus was removed, and the cell cultures were incubated in the presence of varying concentrations (400, 200, 100,. μg/mL) of the test compounds. Viral cytopathicity was recorded as soon as it reached completion in the control virus-infected cell cultures.
The following viruses were included in the study: herpes simplex virus type 1 (HSV-1, strain KOS), HSV-2 (strain G), a thymidine kinase (TK)-deficient HSV-1 strain (HSV-1/TK-ACVr), vaccinia virus and vesicular stomatitis virus (VSV) in E6SM cell cultures, cytomegalovirus (strain AD169 and Davis), varicella-zoster virus (strains YS and OKA) and TK-deficient VZV (strains 07/1 and YS/R) in HEL cell cultures, human immunodeficiency virus (HIV) type 1 and 2, vesicular stomatitis virus, Coxsackie virus B4 and respiratory syncytial virus (RSV) in HeLa cell cultures, and parainfluenza-3 virus, reovirus-1, Sindbis virus, Coxsackie virus B4, and Punta Toro virus in Vero cell cultures.
Results and discussion
Chemistry
Synthesis of the 2′,3′-dideoxy-4′-thionucleoside (2) was achieved using the Barton-McCombie deoxygenation procedure [Citation9] (Scheme ), a method previously employed successfully with 4′-thionucleosides [Citation10]. 1-(2-Deoxy-4-thio-β-D-erythro-pentofuranosyl)-(6-azathymidine) (1) [Citation1] was selectively silylated at the 5′-position on reaction with TBDMSCl and imidazole in dry DMF [Citation11]. The 5′-protected nucleoside (4) was then transformed into a 3′-thiocarbonyl derivative (5) by reaction with thiocarbonyldiimidazole in dry CH2Cl2 at 40°C for 8 h and then at room temperature overnight. Radical deoxygenation was achieved on treatment of 5 with tributyltin hydride and the radical initiator, 1,1′-azobis(cyclohexane carbonitrile) at 100°C for 30 min to give the 2′,3′-dideoxy nucleoside (6) in 64% yield. Removal of the 5′-protecting group using TBAF was successful as shown by t.l.c. However, removal of the excess TBAF proved problematic, with traces still observed on NMR after chromatographic purification. Treatment with Dowex 50W (H+) however gave the required product (2) cleanly in 67% yield.
Scheme 1 Reagents and Conditions: (i) TBDMSCl, imidazole, DMF, 3.5 h (ii) thiocarbonyldiimidazole, CH2Cl2, 40°C, 8 h then r.t. 12 h (iii) Bu3SnH, 1,1′-azobis(cyclohexene-carbonitrile), toluene, 100°C, 30 min (iv) Dowex 50W (H+), MeOH, 24 h.
Synthesis of the L-4′-thionucleoside (3) involved coupling of the L-thiosugar (7), prepared as previously reported from D-lyxose [Citation4], with 6-azathymine using the Vorbrüggen coupling procedure (Scheme ) [Citation12] 6-Azathymine was silylated using bis(trimethylsilyl)acetamide (BSA), the silylated pyrimidine was then reacted with the L-thiosugar in the presence of the Lewis acid catalyst trimethylsilyl trifluoromethane sulfonate (TMSOTf) at 50°C for 12 h to give the β-L-thionucleoside (8). Deacylation of the protected β-L-thionucleoside (8) with aqueous methylamine at 50°C for 30 min gave the required product (3) in 90% yield.
Scheme 2 Reagents and Conditions: (i) 6-Azathymine, BSA, CH3CN, 30 min then 7, TMSOTf, CH3CN, 50°C, 12 h (ii) CH3NH2 (35% aqueous), 50°C, 30 min.
Antiviral activity
The L-thionucleoside (3) was devoid of antiviral activity, however the dideoxy-thionucleoside (2) was moderately active against vaccinia virus (MIC 12 μM) and herpes simplex virus strains, HSV-1 (strain KOS) and HSV-2 (strain G) (MIC 12 μM) (). Interestingly, it fully retained inhibitory activity against the thymidine kinase-deficient HSV-1 TK– strain, suggesting that 2 is not dependent on viral TK-catalysed phosphorylation for antiviral activity and/or may use an alternative metabolic activation pathway. This result is consistent with the (partial) retention of inhibitory activity vs HSV-1 TK– observed for 1-(2-deoxy-4-thio-β-D-erythro-pentofuranosyl)-(6-azathymidine) (1) [Citation1].
Table I. Cytotoxicity and antiviral activity of novel 4′thionucleosides 2 and 3.
In fact, compound 2 had poor, if any affinity for cytosolic TK-1 and mitochondrial TK-2, but it showed an IC50 of 34 μM and 427 μM for HSV-1 TK- and VZV TK-catalysed dThd (1 μM) phosphorylation (). Therefore, HSV-1 TK-directed phosphorylation of 2 cannot be excluded but it is unclear at this moment to what extent the metabolism of the compound, if it occurs at all, contributes to the eventual antiviral activity.
Table II. Thymidine kinase affinity of dideoxynucleoside 2.
The compounds were also evaluated against a broad range of other viruses, including varicella zoster virus, cytomegalovirus, human immunodeficiency virus types 1 and 2, reovirus-1, Coxsackie virus B4, Sindbis virus, parainfluenza-3 virus, Punta Toro virus, vesicular stomatitis virus, and respiratory syncytial virus, but were found to be inactive at subtoxic concentrations.
Acknowledgements
MJ acknowledges the Malaysian Government for the award of a PhD studentship. We would like to acknowledge the EPSRC Mass Spectrometry Centre, Swansea, UK for mass spectroscopy data.
References
- Maslen HL, Hughes D, Hursthouse M, De Clercq E, Balzarini J, Simons C. 6-Azapyrimidine-2′-deoxy-4′-thionucleosides: Antiviral agents against TK+ and TK– HSV and VZV strains. J Med Chem 2004; 47: 5482–5491
- Inguaggiato G, Hughes D, De Clercq E, Balzarini J, Simons C. Novel 6-azapyrimidine-2′-deoxy-4′-thionucleosides: Synthesis, biological evaluation and conformational analysis. Antiviral Chem Chemother 1999; 10: 241–249
- Perrin DD, Armarengo WLF. Purification of laboratory chemicals3rd ed. Pergamon Press, New York 1988
- Pejavonić V, Stokic Z, Stojanovic B, Piperski V, Popsavin M, Popsavin V. Synthesis and biological evaluation of some novel 4′-thio-L-ribonucleosides with modified nucleobase moieties. Bioorg Med Chem Lett 2003; 13: 1849–1852
- De Clercq E, Descamps J, Verhelst G, Walker RT, Jones AS, Torrence PF, Shugar D. Comparative efficacy of antiherpes drugs against different strains of herpes simplex virus. J Infect Dis 1980; 141: 563–574
- De Clercq E, Holý A, Rosenberg I, Sakuma T, Balzarini J, Maudgal PC. A novel selective broad-spectrum anti-DNA virus agent. Nature 1986; 323: 464–467
- De Clercq E, Sakuma T, Baba M, Pauwels R, Balzarini J, Rosenberg I, Holý A. Antiviral activity of phosphonylmethoxy- alkyl derivatives of purine and pyrimidines. Antiviral Res 1987; 8: 261–272
- Schols D, De Clercq E, Balzarini J, Baba M, Witvrouw M, Hosoya M, Andrei G, Snoeck R, Neyts J, Pauwels R, Nagry M, Györgyi-Edelényi J, Machovich R, Horvath I, Löw M, Görög S. Sulphated polymers are potent and selective inhibitors of various enveloped viruses, including herpes simplex virus, cytomegalovirus, vesicular stomatitis virus, respiratory syncytial virus, and toga-arena- and retroviruses. Antiviral Chem Chemother 1990; 1: 233–240
- Barton DHR, McCombie SW. A new method for the deoxygenation of secondary alcohols. J Chem Soc Perkin Trans 1975; 16: 1574–1585
- Tiwari KN, Secrist JA, Montgomery JA. Synthesis and biological evaluation of 4′-thionucleosides of 2-chloroadenine. Nucleosides & Nucleotides 1994; 13: 1819–1828
- Ogilvie KK, Schifman AL, Penny CL. The synthesis of oligonucleotides III. The use of silyl protecting groups in nucleoside and nucleotide chemistry VIII. Can J Chem 1979; 57: 2230–2238
- Vorbrüggen H, Krollikiewicz K, Bennua B. Nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalyst. Chem Ber 1981; 114: 1234–1235