Abstract
Pyrazole carboxylic acid amides of 5-amino-1,3,4-thiadiazole-2-sulfonamide were synthesized from 4-benzoyl-1,5-diphenyl-1H-pyrazole-3-carbonyl chloride and 4-benzoyl-1-(3-nitrophenyl)-5-phenyl-1H-pyrazole-3-carbonyl chloride. Carbonic anhydrase isoenzymes (hCA-I and hCA-II) were purified from human erythrocyte cells by the affinity chromatography method. The inhibitory effects of 5-amino-1,3,4-thiadiazole-2-sulfonamide 1, acetazolamide 2 and new synthesized amides on these isozymes have been studied in vitro. The I50 concentrations (the concentration of inhibitor producing a 50% inhibition of CA activity) against hydratase activity ranged from 1.2 to 2.2 nM for hCA-I and from 0.4 to 2 nM for hCA-II. The I50 values against esterase activity ranged from 1.4 to 8 nM for hCA-I and from 1.3 to 6 nM for hCA-II. The Ki values were observed between 8.2·10− 5 to 6.2·10− 4 M for hCA-I and between 2.9·10− 4 to 8.2·10− 4 M for hCA-II. The comparison of new synthesized amides to 5-amino-1,3,4-thiadiazole-2-sulfonamide 1, acetazolamide 2 indicated that the new synthesized compounds (18–23) inhibit CA activity more potently than the parent compounds.
Introduction
Carbonic anhydrase (CA, EC 4.2.1.1) catalyzes the reversible hydration of carbon dioxide, a very simple but critically important physiological reaction [Citation1–5]. This enzyme has 16 different isoenzymes presently known in human. Some of these isoenzymes (CA-I, CA-II and CA-IV) are expressed in the human eyes [Citation6–9].
Glaucoma is a group of diseases characterized by gradual loss of visual field due to intraocular pressure (IOP) and thus this disease is the second leading cause of blindness worldwide [Citation10]. Carbonic anhydrase inhibitors have been used for the treatment of glaucoma for years [Citation11,12].
Sulfonamides are the best known inhibitors of carbonic anhydrase enzyme in the treatment of glaucoma in clinical medicine. These inhibitors are very effective in this treatment of the disease by reducing the elevated intraocular pressure (IOP) [Citation1–13].
5-amino-1,3,4-thiadiazole-2-sulfonamide, 1, has several biological activites and antibacterial properties [Citation9]. Derivatives of this ring have been used in treatment of glaucoma for years, one of the drugs is acetazolamide 2 having a number of side effects. However, many of these drugs (e.g. acetazolamide) have a number of side effect on account of systemic use. Therefore, such systemic inhibitors have not been used in the treatment of glaucoma for decades. Recently dorzolamide 3 and brinzolamide 4 have been available for clinical use as topically acting inhibitors [Citation14–16]. These drugs have been shown to reduce intraocular pressure exclusively by lowering the aqueous humour flow [Citation2,17–19]. The common ocular side effects of dorzolamide and brinzolamide are stinging, burning, blurred vision, itching and tearing [Citation2,16,17,20].
Due to side effects of these drugs, it is required to develop new compound and new drugs. In this respect, in this study we studied the synthesis of new CA inhibitors with 5-substitued-1,3,4-thiadiazole and determined their effects on hCA-I and hCA-II purified from human erythrocytes. We investigated the role of the substituted pyrazole carboxylic acid derivatives which have antipyretic, analgesic and anti-inflammatory effects, moiety attached to a 1,3,4-thiadiazole ring on CA inhibition [Citation21,22].
The I50 and Ki values were determined to compare the inhibitory effects of 5-amino-1,3,4-thiadiazole-2-sulfonamide 1, acetazolamide 2 and newly synthesized compounds on human erythrocyte carbonic anhydrase I and II [Citation23].
Materials and methods
Materials
Sepharose 4B for affinity column and electrophoresis reagents were obtained from Sigma Chem. Co. All other used chemicals were analytical grade and purchased from either Sigma or Merck.
Synthesis of organic compounds
The parent compound 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 required for synthesis of inhibitors was produced by acid hydrolysis of 5-acetylamino-1,3,4-thiadiazole-2-sulfonamide 2.On the other hand pyrazole carboxylic acides were obtained from the hydrazine of 4,5-substitue-2,3-furandions (14–17) which reactes with hydrazines such as benzaldehyte hydrazone as without solvent (Scheme–) [Citation21,22].
Scheme 1 Synthesis of title compounds.
In order to synthesize the target inhibitors carboxly group of pyrazol carboxylic acides were reacted with the amino group of 5-amino-1,3,4-thiadiazole-2-sulfonanide 1 and thus it were obtained amide derivatives of pyrazole carboxylic acids (18–23) (Scheme–) [Citation14,15]. To prepare these compounds, firstly carboxyl group was activated by thionyl chloride Later amino thiadiazole with activated pirazole carboxylic acid were refluxed to obtain amides of pirazole carboxylic acids (18–23) in dry THF (Scheme–) [Citation1,21,22]. The structures of these synthesized compounds have been identified on the basis of IR, 400 MHz 1H and 100 MHz 13C-NMR data, and satisfactory mass spectral analysis.
General procedure for the preparation of 5-amino-1,3,4-thiadiazole-2-sulfonamide and amides of pirazole carboxylic acids
The parent compound 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 required for synthesis of inhibitors was produced by acidic hydrolysis of 5-acetylamino-1,3,4-thiadiazole-2-sulfonamide 2.On the other hand, pyrazole carboxylic acids were obtained from the hydrazine of 4,5-substitue-2,3-furandions (14–17) which react with hydrazines such as benzaldehyte hydrazone as without solvent (Scheme–).
For synthesis of target compounds, pyrazole carboxylic acids 12 (12–17) (432 mg,1 mmol) was dissolved in 50 mL of dry and freshly distilled THF. To this solution, 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 (360 mg, 2 mmol) was added. After this mixture was stirred and refluxed for 4 h, cooled and the precipitated solid was filtrated. After removal of the solvent, residue (580 mg) was crystallized from ethanol-water mixture: colorless solids 18 (18–23) (471 mg, % 82, m.p: 226°C).
4-Benzoyl-1-(3-nitrophenyl)-5-phenyl-N-(5-sulfomoyl-1,3,4-thiadiazole-2-yl)-1H-pyrazole-3-carboxamide (18)
As white crystals, m.p:226°C. IR (KBr), cm-1; 3270 (NH), 3064 (Ar-CH), 1693 (benzoyl C = O), 1651(amide C = O), 1588, 1520, 1494, 1423 (Ar C = C), 1350, 1167 (SO2) MS (CI) m/z 576.1(M + 1). 1H NMR (400 MHz, DMSO-d6): δ 13.20 (m, 1H, CONH), 8.28 (m, 2H, -SO2NH2), 8.49–7.29 (m, 14H, ArH),. 13C NMR (100 MHz, DMSO-d6) δ 191.71 (benzoyl C = O), 166.63 (amide CONH), 162.72, 161.51, 149.65, 148.87, 145.98, 144.84, 140.82, 138.95, 135.50, 133.19, 132.29, 131.69, 130.95, 130.48, 130.41, 128.69, 125.26, 124.72, 122.29.
4-Benzoyl-1-(4-nitrophenyl)-5-phenyl-N-(5-sulfamoyl-1,3,4-thiadiazole-2-yl)-1H-pyrazole-3-carboxamide (19)
As white crystals, mp 275°C. IR(cm-1): 3261 (NH), 3062 (Ar-CH), 1685 (benzoyl C = O), 1647 (amide C = O), 1593, 1523, 1495,1430 (Ar C = C), 1352, 1167 (SO2), MS(CI) m/z 576.1(M + 1). 1H NMR (400 MHz, DMSO-d6): δ 13.22 (m, 1H, CONH), 8.03 (m, 2H, SO2NH2), 8.33–7.25 (m, 14H, ArH), 13C NMR (100 MHz, DMSO-d6): δ 190.53 (benzoyl C = O), 165.51(amide CONH), 161.78, 160.43, 147.50, 144.89, 144.17, 143.85, 137.80, 134.40, 130.63, 130.53, 129.88, 129.48, 129.32, 127.70, 126.97, 125.27, 123.93.
4-Benzoyl-1,5-diphenyl-N-(5-sulfamoyl-1,3,4-thiadiazole–2-yl)-1H-pyrazole-3-carboxamide (20)
As white crystals, mp 162°C. IR(cm− 1): 3300 (NH), 3066 (Ar-CH), 1685 (benzoyl C = O), 1657 (amid C = O), 1582, 1528, 1497, 1428 (Ar C = C) 1370, 1174 (SO2). 1H NMR (200 MHz, CDCl3): δ 13.20 (m, 1H, CONH), 6.39 (m, 2H, SO2NH2), 7.07–7.77 (m, 15 H, ArH), 13C NMR (100 MHz, CDCl3): δ 193.18 (benzoyl C = O), 166.50 (amide CONH), 163.30, 160.32, 147.01, 143.83, 140.39, 139.30, 135.60, 131.84, 131.63, 131.62, 131.16, 130.96, 130.60, 130.40, 129.40, 127.24, 124.70.
Ethyl-1,5–diphenyl-3-(5-sulfamoyl-1,3,4-thiadiazole-2–ylcarbamoyl)-1H-pyrazole-4-carboxylate (21)
As white crystals, mp 190°C. IR(cm− 1): 3375 (NH), 3067(Ar-CH), 1735 (ester C = O), 1665 (amide C = O), 1547, 1486, 1457, 1429 (Ar C = C), 1364, 1171 (SO2), MS(CI) m/z 499.1(M + 1). 1H NMR (400 MHz, CDCl3): δ 13.13 (m, 1H, CONH), 8.07 (s, 2H, SO2NH2), 7.37–7.17 (m, 10 H, ArH), 4.10 (q, 2H, –CH2–) 1.08 (t, 3H, –CH3).
Ethyl-1-(3–nitrophenyl)–5–phenyl-3-(5-sulfamoyl-1,3,4-thiadiazol-2-ylcarbamoyl)-1H-pyrazole-4-carboxylate (22)
As white crystals, mp 193°C. IR(cm− 1): 3227 (NH), 3094 (Ar-CH), 1738 (ester C = O), 1659 (amide C = O), 1523, 1478,1431 (Ar C = C), 1345, 1165 (SO2), MS(CI) m/z 544.0 (M + 1). 1H NMR (400 MHz, DMSO-d6): δ 12.95 (m, 1H, CONH), 8.32 (s, 2H, SO2NH2), 8.11–7.29 (m, 9H, ArH), 4.11 (q, 2H, –CH2–), 0.99 (t, 3H, CH3), 13C NMR (100 MHz, DMSO-d6): 170.24 (ester C = O), 167.09 (amide C = O), 166.15, 164.51, 153.06, 151.33, 149.98, 144.08, 135.56, 135.20, 135.06, 133.48, 133.47, 132.82, 132.21, 125.17, 119.79, 66.12, 18.49.
Ethyl-1-(4-nitrophenyl)-5-phenyl-3-(5-sulfamoyl-1,3,4-thiadiazol-2-ylcarbamoyl)-1H-pyrazole-4-carboxylate (23)
As white crystals, mp 214 oC. IR(cm− 1): 3234 (NH), 1695 (ester C = O), 1593 (amide C = O), 1525, 1495,1440 (Ar C = C), 1367, 1174 (SO2), MS(CI) m/z 544.0 (M + 1). 1H NMR (400 MHz, DMSO-d6): δ?13.94 (m, 1H, CONH), 8.43 (s, 2H, SO2NH2), 8.27–7.40 (m, 9H, ArH), 4.13 (q, 2H, –CH2–) 1.01 (t, 3H, –CH3), 13C NMR (100 MHz, DMSO-d6): δ 65.55 (ester C = O), 161.76 (amide C = O), 161.40, 160.99, 147.26, 146.56, 146.11, 143.37, 130.83, 130.50, 128.94, 127.51, 126.85, 125.03, 114.88, 61.23, 13.94.
Purification of carbonic anhydrase I and II from human erythrocytes
Erythrocytes were obtained from human blood. The blood samples were centrifuged at 1500 rpm for 20 min and plasma and buff coat were removed. Cell membranes were removed by centrifugation at 4°C, 20,000 rpm for 30 min. The homogenate was applied to affinity column having a structure of Sepharose-4B-l-tyrosine-p-aminobenzen sulfonamide. Carbonic anhydrase isoenzymes were purified from this column [Citation23,24].
SDS polyacrylamide gel electrophoresis
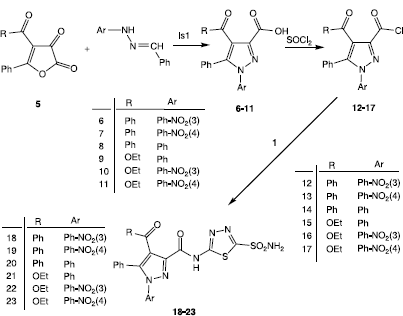
SDS polyacrylamide gel electrophoresis was performed after the purification of the enzyme. It was carried out with 10% separating gel and 4% stacking gel concentrations for running and the stacking gel, respectively, containing 0.1% SDS according to Laemmli [Citation25]. Equal amounts of each sample were applied to the electrophoresis gel. Gels were stained for 45 min in 0.1% Coommassie Brillant Blue R-250 dissolved 50% methanol and 10% acetic acid and then detained with several changes of the same solvent above without the dye (see ).
Figure 1. SDS-PAGE analysis of CA isoenzymes. Standard enzymes: Lane-1 (hCA-I),3(BCA),5(hCA-II). Enzymes used here: Lane-2(hCA-I),4(BCA),6(hCA-II).
Measurement of CA activity
CO2-hydratase activity of the enzyme was determined at 0°C in a veronal buffer (pH = 8.15) with pH-state method and saturated carbon dioxide solution as substrate in a final volume of 4.2 mL. The time (in seconds) taken for the solution to change from 8.15 to 6.3 in pH was measured. The enzyme unit (EU) is the enzyme amount resulting in 50% decrease of the non enzymatic reaction time. Activity as an enzyme unit was calculated by using the Equation (t0 − tc/tc) where to and tc are times for pH change of the non enzymatic and enzymatic reactions, respectively Citation26–30].
Esterase activities of carbonic anhydrase enzymes were assayed by the hydrolysis of p-nitro phenyl acetate. 1.2 mL of 3 mM p-nitro phenyl acetate was used as substrate. The substrate was added to a 1.5 mL volume of five different concentrations of inhibitors and water. Reaction was started by adding of 1.4 mL of 0.05 M Tris–SO4 (pH 7.4) and 0.1 mL enzyme solution for a final volume of 3 mL. The absorbance was determined at 348 nm after 3 min [Citation30].
Determination of I50 and Ki values of compounds
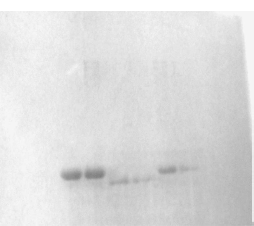
The % carbonic anhydrase activity values were assayed by following the hydration of CO2. I50 values for 5-amino-1,3,4-thiadiazole-2-sulfonamide 1, acetazolamide (2) and the synthesized compounds were determined on hCA-I and hCA-II. In order to determine I50 values, a saturated solution of CO2 was used as substrate. To a total volume of 1.7 mL of various aliquots (10–250 μL) of a suitable concentration of inhibitors and water were added to 1 mL of Tris–HCl and 0.1 mL enzyme solution. The reaction was started, by adding 2.5 mL of CO2 solution. The activity was determined by using the Equation (t0 − tc/tc) where t0 and tc are times for pH change of enzymatic reactions for each inhibitor concentration, respectively [Citation26–30]. Regression analysis graphs were drawn on X axis of inhibitor concentrations and Y axis of % enzyme activity by using Microsoft Excell Program. The data were then fitted with nonlinear regression using second degree polynomial equation. The I50 value was obtained by solving the equation derived from second degree polynomial as well as by simply extrapolating the X value (compound concentration) corresponding to the Y axis indicating the 50% inhibition [Citation1,26–30] (see ).
Figure 2. I50 graph which were obtained from in vitro studies for compound 18 on hydratase activity of hCA-II.
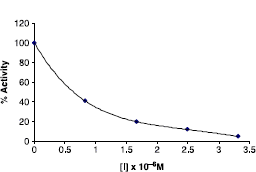
Esterase activities of carbonic anhydrase enzymes were assayed by the hydrolysis of p-nitro phenyl acetate. I50 and Ki values of the inhibitors (1,2,18–23) were determined on human erythrocyte hCA I and hCA II. In order to determine I50 values, 1.2 mL of 3 mM p-nitro phenyl acetate was used as substrate. The volume of substrate was added to 1.5 mL with five different concentrations of inhibitors (50, 100, 150, 200, and 300 μL) and water. Reaction was started by addition of 1.4 mL of 0.05 M Tris–SO4 (pH 7.4) and 0.1 mL enzyme solution for total volume of 3 mL. The absorbance was determined at 348 nm after 3 min. The experiment was repeated three times for each inhibitor. In order to determine I50 values, regression analysis graphs were drawn by using % inhibition values by a statistical packing program on a computer. The I50 concentrations of the compounds (1,2,18–23) were determined from graphs (see ). This way was followed to determine Ki values. In the media with or without inhibitor, the substrate concentrations were 0.6, 0,8, 1,0 and 1.2 mM. For this purpose, the substrate was used between 0.6 and 1.2 mL. The solutions of the compounds (1,2,18–23) were added as inhibitors to the reaction medium as 0.1, 0.2 and 0.3 mL resulting in three different fixed concentrations of inhibitor. Ki values of these compounds (1,2,18–23) were obtained by calculating from the Lineweaver–Burk graphs [Citation30] (see ).
Figure 3. Kİ graph which were obtained from in vitro studies for compound 18 on hCA-I.
Results and discussion
The treatment of glaucoma with inhibitors of the metalloenzyme carbonic anhydrase (CA, EC 4.2.1.1) is used in reducing elevated intraocular pressure (IOP) characteristic of this disease. In addition to the classical inhibitors used via the systemic route of administration (acetazolamide 2, methazolamide 24, ethoxzolamide 25, and dichlorophenamide 26) two topically acting inhibitors presently available for clinical are effective dorzolamide 3 and brinzolamide 4 [Citation1,2]. But, these inhibitors provoke a wide range of deleterious side effects due to inhibition of the enzyme present in other tissues (kidneys, lungs, red cells, stomach, etc.) than in the eye. The peripheral inhibition of carbonic anhydrase with systemic inhibitors produce a wide array of side effects which most of the patients are unable to tolerate and hence they could not continue the therapy [Citation2,16,17,20,31]. Important drugs in this field have then been discovered by the Merck group, with the discovery compounds of the first clinically used, topically effective anti glaucoma sulfonamide, dorzolamide 3 and brinzolamide 4 known as the ‘ring approach [Citation17–20]. The two compounds also had side effects such as stinging, burning, blurred vision, itching and tearing [Citation16–20]. Therefore, new compounds and drugs have been investigated rigorously for decades.
We have synthesized new amides of 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 with some pyrazole carboxylic acides an alternative approach for the design of topically acting anti glaucoma sulfonamides (18–23) as shown Scheme . Some compounds contain pyrazole carboxylic acid derivatives with nitro phenyl and phenyl. And some also have pyrazole carboxylic acid derivatives without nitro phenyl but have only phenyl groups. In the new synthesized six compounds, the inhibitor concentrations causing 50% inhibition (I50 values) were determined from regression analysis graphs (). I50 values obtained, for erythrocyte hCA-I and hCA-II purified by affinity chromatography are shown in .
Table I. I50 values which were obtained from in vitro studies for the compounds (1, 2, 18–23) on hydratase activity of hCA-I and II.
According to in vitro studies, I50 values of newly synthesized compounds (18–23) are generally lower than I50 values (respectively 0.4, 0.42, 0.98, 1.1, 0.9 and 2 nM) of 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 (5 nM) and acetazolamide 2 (13 nM) on hydratase activity of erythrocyte hCA-II (). At the same time these new compounds (18–23) also have the similar I50 values (respectively 3, 1.3, 4.7, 4.2, 5.6 and 6 nM) with 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 (2 nM) and acetazolamide 2 (4.9 nM) on esterase activity of this enzyme inhibition (). Nevertheless Ki values of these compounds (18–23) are approximately similar (respectively 3.10− 4, 2.6.10− 4, 3.10− 4, 8.2.10− 4, 5.3.10− 4 and 2.9.10− 4) with 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 (0.82.10− 4) and acetazolamide 2 (2.10− 4) (). Therefore the whole obtained inhibition values for these compounds on human erythrocyte CA-II are very appropriate to use on glaucoma treatment.
Table II. I50 values which were obtained from in vitro studies for the compounds (1, 2, 18–23) on esterase activity of hCA-I and II.
Table III. Ki values which were obtained from in vitro studies for synthesized compounds (1, 2, 18–23) on esterase of carbonic anhydrase isoenzymes.
For inhibition of human erythrocyte CA-I, obtained I50 values of new compounds (18–23) are generally the closely I50 values (respectively 1.2, 2.2, 2, 1.2, 1.4 and 1.4 nM) with 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 (5.1 nM) and acetazolamide 2 (1.1 nM) on hydratase activity (). Nevertheless the new compounds (18–23) also have the lower I50 values (respectively 1.4, 2.9, 8, 5.4, 2.8 and 5.9 nM) than 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 (9 nM) and acetazolamide 2 (9 nM) on esterase activity of this enzyme for enzyme inhibition (). At the same time Ki values of these compounds (18–23) are the same (respectively 1.4·10− 4, 8.2·10− 5, 3.9·10− 4, 6.2·10− 4, 1·10− 4 and 1·10− 4) as 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 (5.7·10− 4) and acetazolamide 2 (6.1·10− 4) (). Therefore since the values of I50 and Ki for hCA-I of these compounds are higher than values of hCA-II, obtained values of hCA-II are very important to use glaucoma treatment.
Since, these compounds synthesized here (18–23) have high solubility, they are more appropriate than 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 and acetazolamide 2 for glaucoma treatment.
The present study shows that, the newly synthesized compounds (18–23) described here have very significant inhibition effects and may be use as the drugs for glaucoma treatment in future.
Acknowledgements
This research was funded by TUBITAK (The Foundation of Scientific and Technological Research of Turkey, TBAG-2376) (Ankara/Turkey).
References
- M Bülbül, N Saraçoğlu, Öİ Küfrevioğlu, and M Çiftçi. (2002). Bioorg Med Chem 10:2561–2567.
- CT Supuran, and A Scozzafava. (2001). Curr Med Chem Imm Endoc Metab Agents 1:61–97.
- E Nuti, E Orlandini, S Nencetti, A Rossello, A Innocenti, A Scozzafava, and CT Supuran. (2007). Bioorg Med Chem 15:2298–2311.
- Ö Özensoy, F Kockar, O Arslan, S Isık, CT Supuran, and M Lyon. (2007). Clin Biochem 40:835.
- CT Supuran, and A Scozzafava. (2007). Bioorg Med Chem 15:4336–4350.
- A Di Fiore, A Scozzafava, JY Winum, JL Montero, C Pedone, CT Supuran, and GD Simone. (2007). Bioorg Med Chem Lett 17:1726–1731.
- A Innocenti, D Vullo, J Pastorek, A Scozzafava, S Pastorekova, I Nishimori, and CT Supuran. (2007). Bioorg Med Chem Lett 17:1532–1537.
- JY Winum, A Thiry, KE Cheikh, JM Dogné, JL Montero, D Vullo, A Scozzafava, B Masereel, and CT Supuran. (2007). Bioorg Med Chem Lett 17:2685–2691.
- A Scozzafava, MD Banciu, A Popescu, and CT Supuran. (2000). J Enzyme Inhib Med Chem 15:443–453.
- JS Schuman. (2000). Clinic Ther 22:2.
- BL Wilkinson, LF Bornaghi, TA Houston, A Innocenti, D Vullo, CT Supuran, and SA Poulsen. (2007). Bioorg Med Chem Lett 17:987–992.
- MA Santos, S Marques, D Vullo, A Innocenti, A Scozzafava, and CT Supuran. (2007). Bioorg Med Chem Lett 17:1538–1543.
- FA Ashour, NS Habib, M Taibbi, S Dine, and A Dine. (1990). Farmaco 45:134.
- K Sabri, and AV Levin. (2006). J Am Assoc Pediatric Ophthalmol and Strabismus 10:464–468.
- F Mincione, M Starnotti, E Masini, L Bacciottini, C Scrivanti, A Casini, D Vullo, A Scozzafava, and CT Supuran. (2005). Bioorg Med Chem Lett 15:3821–3827.
- MF Sugrue. (2000). Prog Ret Eye Res 19:87–112.
- CJ Ingram, and RF Brubaker. (1999). Am J Ophthalmol 128:3.
- JY Winum, A Casini, F Mincione, M Starnotti, JL Montero, A Scozzafava, and CT Supuran. (2004). Bioorg Med Chem Lett 14:225–229.
- D Aggarwal, D Pal, AK Mitra, and IP Kaur. (2007 in Press). Int J Pharmaceutics
- A Konowal, JC Morrison, SVL Brown, DL Cooke, LJ Maguire, DV Verdier, FT Fraunfelder, RF Dennis, and RJ Epstein. (1998). Am J Ophthalmol 127:4.
- A Şener, R Kasımoğulları, MK Sener, İ Bildirici, and Y Akçamur. (2002). J Heterocycl Chem 39:869–875. b) Şener, A, Kasımoğulları, R, Sener, MK, Genç, H. Chem Heterocycl Compounds, (2004); 8: 1201-1208
- Y Akçamur, G Penn, E Ziegler, H Sterk, G Kollenz, K Peters, EM Peters, and HGV Scnering. (1986). Monatsh Chem 117:231–245.
- O Arslan, B Nalbantoğlu, N Demir, H Özdemir, and Öİ Küfrevioğlu. (1996). Trop J Med Sci 26:163–166.
- MM Bradford. (1976). Anal Biochem 72:248–251.
- DK Laemmli. (1970). Nature 227:680–683.
- KM Wilbur, and NG Anderson. (1976). J Biol Chem 176:147–151.
- C Landolfi, M Marchetti, G Ciocci, and C Milanese. (1998). J Pharmacol Toxicol Methods 38:169–172.
- M Bülbül, O Hisar, Ş Beydemir, M Çiftçi, and Öİ Küfrevioğlu. (2003). J Enzyme Inhib Med Chem 18:371–375.
- M Çiftçi, M Bülbül, M Gül, K Gümüştekin, Dane, Şenol, and H Süleyman. (2005). J Enzyme Inhib Med Chem 20:103–108.
- O Hisar, Ş Beydemir, M Bülbül, and T Yanık. (2006). J Appl Anim Res 30:185–188.
- JY Winum, A Cecchi, JL Montero, A Innocenti, A Scozzafava, and CT Supuran. (2005). Bioorg Med Chem Lett 15:3302–3306.