Abstract
Oleanolic acid has been isolated from chloroform extract of Olea ferruginea Royle after removal of organic bases and free acids. The literature survey revealed it to be biologically very important. In this review the biological significance of oleanolic acid and its derivatives has been discussed. The aim of this review is to update current knowledge on oleanolic acid and its natural and semisynthetic analogs, focussing on its cytotoxic, antitumer, antioxidant, anti-inflamatory, anti-HIV, acetyl cholinesterase, alpha-glucosidase, antimicrobial, hepatoprotective, anti-inflammatory, antipruritic, spasmolytic activity, anti-angiogenic, antiallergic, antiviral and immunomodulatory activities. We present in this review, for the first time, a compilation of the most relevant scientific papers and technical reports of the chemical, pre-clinical and clinical research on the properties of oleanolic acid and its derivatives.
Introduction
Oleanolic acid (1) along with other triterpenoidal compounds like, ursolic acid (2, Scheme–), oleanolic acid glycosides and few sugers were isolated from Olea ferruginea [Citation1] and also from R. stricta growing in Kashmir [Citation2]. A group of scientists from Japan isolated a hypoglycaemic component, elatoside E, and elatoside F along with oleanolic acid and its glycosides from the root cortex of Aralia elata Seem. (Araliaceae) growing in Japan [Citation3]. Chemical investigation of Eugenia jumbolana flowers by scientists also revealed the presence of oleanolic acid [Citation4]. Isolation of oleanolic acid from various plants by different workers has been known. The plants were Beta vulgaris L. (Suger Beet,Chenopodiaceae) [Citation5], Kochiae fructus [Citation6], Kochia scoparia Schrad, Momordica cochinchinensis Spreng.) [Citation7], Tiarella polyphylla [Citation8], Clerodendranthus spicatus [Citation9], Perilla frutescens [Citation10], Glechoma hederaceae [Citation11], Olea europaea [Citation12,13], Couepia polyandra [Citation14], Liquidambar formosana [Citation15], Pistacia terebinthus galls[Citation16], Rosmarinus officinalis L. [Citation17], Crataegus pinnatifida Bunge [Citation18], Luffa cylindricall [Citation19], Rosa woodsii, (leaves), Prosopis glandulosa (leaves and twigs), Phoradendron juniperinum (whole plant), Syzygium claviflorum (leaves), Hyptis capitata (whole plant), Ternstromia gymnanthera (aerial part) [Citation20], Plantago major [Citation21] and Ludwigia octovalvis [Citation22]. A number of oleanolic acid derivatives have been prepared. Oleanolic acid derivatives (4-14) showed anti-HIV activity. 3-O-fatty acid ester derivatives (22-27) showed antifeedant activity. Semi synthetic oleanolic acid derivatives (2-7) showed gastro protective and ulcer-healing activity. Oleanolic acid glycosides (41-45) showed gastrointestinal transit accelerating activity. Three oleanane type triterpene (15-17) showed significant cytotoxicity against two human tumor cell lines. These derivatives have attracted considerable interest due to there biological activity. Individual presentations on various aspects of oleanolic acid and its derivatives have been appeared as articles in journals [Citation8–16] but only few publications in a compiled form have appeared on oleanolic acid and its derivatives.
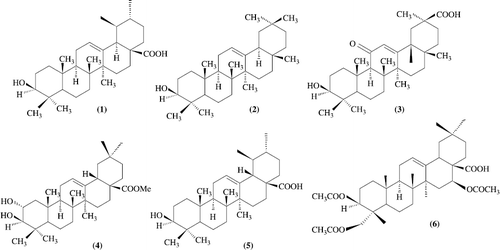
Biological and pharmacological significance of oleanolic acid:
Oleanolic acid is a triterpenoid which is quite common in nature in the form either of free acid or in triterpenoid saponin glycosides found in food, medicinal herbs and various other plants in free form or bound to glycosides and is distributed wildly in plants all over the world. In recent years, it was found that it had marked biological and pharmacological significance.
Oleanane triterpenoids with inhibitory activity against NFAT transcription factor from Liquidambar formosana
In a search for inhibitory components from natural products against NFAT transcription factor, this study investigated the ethyl acetate extract of the fruits of Liquidambar formosana. Four oleanane triterpenoids were isolated and identified to be liquidambaric acid, oleanolic acid, 3 alpha-acetoxy-25-hydroxy-olean-12-en-28-oic acid and lantanolic acid. Of these compounds, 3 alpha-acetoxy-25-hydroxy-olean-12-en-28-oic acid (IC50: 4.63 microM) and lantanolic acid (IC50: 12.62 microM) exhibited strong inhibitory activity against the NFAT transcription factor [Citation15].
Oleanolic acid as a trypanocidal constituent in Rosemary
The MeOH extract of the leaves of Rosemary (Rosmarinus officinalis L.) completely inhibited the motility of cultured epimastigotes of Trypanosoma cruzi at the concentration of 2 mg/mL after 2 hours of incubation. Activity-guided fractionation of the MeOH extract has resulted in the isolation of three triterpene acids, betulinic, oleanolic and ursolic acids. Ursolic acid stopped the movement of all T. cruzi epimastigotes at the minimum concentration (MC (100) of 40 micro g/mL (88 micro M) after 48 hours of incubation. Oleanolic acid was less active (MC (100): 250 micro g/mL, 550 micro M) and betulinic acid was practically inactive [Citation17].
Two triterpenoid compounds, ursolic acid and uvaol, were isolated from Crataegus pinnatifida Bunge leaves. Ursolic acid inhibits chitin synthase II from S. cerevisiae with an IC50 value of 0.84 microgram/mL and the inhibition appears to be selective for chitin synthase II, whereas uvaol has no inhibitory activity up to 280 micrograms/mL. Oleanolic acid, alpha-hederin hydrate and betulic acid inhibited the chitin synthase II activity under the same conditions with an IC50 of 5.6, 64.3 and 98.7 micrograms/mL, respectively [Citation18].
Oleanolic acid, isolated from Luffa cylindrica, inhibits the in-vitro immunohaemolysis of antibody-coated sheep erythrocytes by guinea-pig serum. In further experiments this reduced immunohaemolysis was found to be due to inhibition of the C3-convertase of the classical complement pathway. The threshold concentration for inhibition of C3-convertase was 100 micrograms mL− 1. However, higher concentrations of oleanolic acid showed constant inhibitory effects on immunohaemolysis. Oleanolic acid also exhibited weak inhibitory effects on individual components of the complement system [Citation19].
A hexane extract of Plantago major was investigated by bioactivity-directed fractionation, using an in vitro cyclooxygenase-2 catalyzed prostaglandin biosynthesis inhibition assay, and resulted in the isolation of ursolic acid. This triterpenoid showed a significant cyclooxygenase -2 inhibitory effect, directly on the enzyme activity, with an IC50 value of 130 microM and a cyclooxygenase-2/ cyclooxygenase-1 selectivity ratio of 0.6. The structural isomer oleanolic acid was found to be less active than ursolic acid, with an IC50 value of 295 microM, but showed a similar selectivity ratio (0.8). Furthermore, no significant inhibition on cyclooxygenase -2 or cyclooxygenase-1 was observed by the triterpenoid, 18 beta-glycyrrhetinic acid (3). The direct inhibitory effect of oleanolic acid and ursolic acid on cyclooxygenase-2 catalyzed prostaglandin biosynthesis increased with preincubation, indicating a time-dependent inhibition, while the effect on cyclooxygenase-1 was found to be independent of preincubation time [Citation20].
The antifertility activity of oleanolic acid, isolated from the flowers of Eugenia jambolana, was evaluated in male albino rats. The administration of the compound for 60 days decreased the fertilizing capacity of the animals without any significant changes in body weight or reproductive organ weights. The compound produced arrest of spermatogenesis but did not cause any abnormality to spermatogenic cells, Leydig interstitial cells and Sertoli cells. Oleanolic acid may prove to be a promising antifertility agent devoid of undesirable side effects [Citation4].
Hepatoprotective effects of momordin Ic and oleanolic acid obtained from Kochiae Fructus, the fruit of a traditional Oriental medicinal plant, were evaluated against carbon tetrachloride (CCl4)-induced liver damage in rats. Male Sprague-Dawley rats were divided into four groups: control, CCl4-treated, CCl4 plus momordin Ic-treated (MMDIc-CCl4), and CCl4 plus oleanolic acid-treated (OAA-CCl4). Momordin Ic (30 mg/kg of body weight) and oleanolic acid (30 mg/kg of body weight) were orally administered once a day for 14 days. A mixture of 0.2 mL/100 g of body weight of CCl4 in olive oil (1:1, vol/vol) was injected 30 minutes after the final administration of momordin Ic and oleanolic acid. The momordin Ic and oleanolic acid pretreatments resulted in significantly lower serum transaminase, lactic dehydrogenase, and gamma-glutamyltransferase levels in the CCl4-treated rats. The CCl4-treated rats had significantly lower activities of glutathione, glutathione reductase, glutathione S-transferase, superoxide dismutase, catalase, and glutathione peroxidase. However, pretreatment with momordin Ic and oleanolic acid reduced the effect of CCl4 and helped maintain levels of the enzymes. Pretreatment with momordin Ic and oleanolic acid resulted in significantly lower production of aminopyrine N-demethylase and aniline hydroxylase in the CCl4-treated rats. Pretreatment with momordin Ic resulted in lower catalase and aminopyrine N-demethylase activity induction by CCl4, towards normalization. Momordin Ic and oleanolic acid obtained from Kochiae Fructus appear to contribute to alleviating the adverse effects of CCl4 treatment by enhancing the hepatic antioxidant defense system [Citation6].
Chikusetsusaponin IV and V, whose genin is oleanolic acid, exhibited weak hemolytic activities. Removal of glucose residue at position 29 of chikusetsusaponin V by partial hydrolysis increased the activity more than 30-fold. Methylation of the carboxyl group at position 28 increased the activity furthermore by about 10-fold, showing HD50 value of 3.77 microM. On the other hand, removal of the sugar chain at position 3 of chickusetsusaponin V by partial hydrolysis completely lost the activity. These facts suggest that the sugar chain at position 3 of oleanolic acid is essential but that at position 29 is pernicious for the activity. The cytolytic agents, whose target has been regarded as membrane cholesterol, were inactivated not only by cholesterol but also by sapogenins such as oleanolic acid, gitogenin and hederagenin. Among saponins tested, akebia saponin B and C were inactivated by cholesterol, but not by the genins, probably because their affinities for the genins are too low to form complexes [Citation23].
Oleanolic acid and ursolic acid were examined for anti-angiogenic activities by using the chick embryo chorioallantoic membrane assay. The presence of ursolic acid or oleanolic acid inhibited angiogenesis in a dose-dependent manner; the doses required for half-maximal inhibition (ID50) were 5 micrograms and 40 micrograms per chorioallantoic membrane, respectively. Ursolic acid was a more potent angiogenic inhibitor than oleanolic acid. It was also tested for inhibitory effect on the proliferation of bovine aortic endothelial cell. They effectively inhibited the proliferation of bovine aortic endothelial cell in a concentration-dependent manner. The IC50 values of anti-proliferative effects were determined to be 5 microM for ursolic acid and 20 microM for oleanolic acid. Based on these results, it was speculated that the inhibitory effects on bovine aortic endothelial cell proliferation of ursolic acid and oleanolic acid might be important for anti-angiogenesis [Citation24].
In vitro anti-HIV activity of oleanolic acid and its derivatives on infected human mononuclear cells
Oleanolic acid was identified as an anti-HIV principle. It inhibited HIV-1 replication in acutely infected H9 cells with an EC50 value of 1.7 microg/mL, and inhibited H9 cell growth with an IC50 value of 21.8 microg/mL [therapeutic index (T. I.) 12.8]. Pomolic acid, isolated from R. woodsii and H. capitata, was also identified as an anti-HIV agent (EC50 1.4 microg/mL, T. I. 16.6). Although ursolic acid did show anti-HIV activity (EC50 2.0 microg/mL), it was slightly toxic (IC50 6.5 microg/mL, T. I. 3.3). A new triterpene, 1beta-hydroxy-2-oxopomolic acid was also isolated from the CHCl3-soluble fraction of R. woodsii, though it showed no anti-HIV activity. Based on these results, the examination of the anti-HIV activity of oleanolic acid- or pomolic acid-related triterpenes isolated from several plants were carriedout. In addition, the derivatives of betulinic acid, isolated from the leaves of S. claviflorum as an anti-HIV principle, exhibited extremely potent anti-HIV activity. Accordingly, derivatives of oleanolic acid (4-14) showed their anti-HIV activity (Scheme − ) [Citation21].
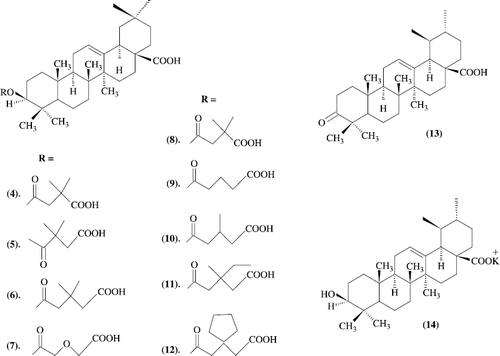
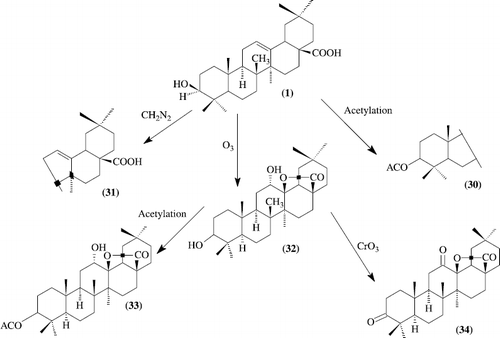
Anti-AIDS agents. anti-HIV activity of oleanolic acid and structurally related triterpenoids
As seen in previous betulinic acid derivatives, oleanolic acid 3-O-3′, 3′-dimethylsuccinate (4) demonstrated a most potent anti-HIV activity, with an EC50 value of 0.0005 Ìg/mL, and showed a greatly improved T. I. value (22 400), while the lack of any discernible anti-HIV activity in its isomer (5) was similar to the corresponding betulinic acid derivative. Compounds 6, 11, and 12 displayed relatively potent anti-HIV activity, with EC50 values of 1.5, 1.1, and 1.2 Ìg/mL, respectively, but were not as potent as 4. Moreover, the small T. I. values of 11 and 12 (4.1 and 3.9, respectively) suggested that they are slightly toxic. Oxidation of oleanolic acid (1) with pyridinium chlorochromate (PCC) yielded a 3-oxo- derivative (13), which was more toxic against uninfected H9 cells than its parent compound, but inhibited HIV-1 replication, with an improved EC50 value of 0.11 Ìg/mL. Treatment of 1 with a molar equivalent of potassium hydroxide furnished the potassium salt of oleanolic acid (14). Compound 14 exhibited potent anti-HIV activity, with an EC50 value of 0.5 Ìg/mL and a T.I value of 68.6. Such enhanced anti-HIV activity with the potassium salt (Scheme ).
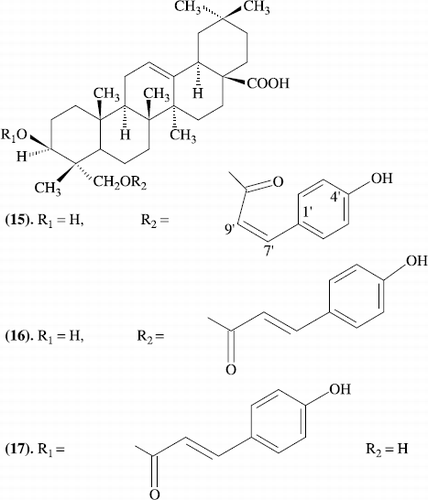
Three new oleanane-type triterpenes from Ludwigia octovalvis with cytotoxic activity against two human cancer cell lines
Three new oleanane-type triterpenes, (23Z)-coumaroylhederagenin (15), (23E)-coumaroylhederagenin (16) and (3Z)-coumaroylhederagenin (17) (Scheme–) together with two known triterpene acids, oleanolic acid and ursolic acid, have been isolated from the whole plant of Ludwigia octovalvis, and their structures have been elucidated by spectroscopic methods. All three new triterpenes showed significant cytotoxicity against two human tumor cell lines, namely, oral epidermoid carcinoma KB and colorectal carcinoma HT29, and gave IC50 values in the range 1.2-3.6 microM [Citation22].
Oleanolic acid derivatives with different lengths of 3-O-acidic acyl chains were synthesized and evaluated for their inhibitory activity against HIV-1 protease. The lengths of the acidic chains were optimized to 6 and 8 carbons. Changing a 3-ester bond to an amide bond or dimerization of the triterpenes retained their inhibitory activity against HIV-1 protease. Introduction of an additional acidic chain to C-28 of oleanolic acid increased the inhibitory activity appreciably, though a derivative with only one acidic chain linked at C-28 also showed potent activity against HIV-1 protease. The inhibitory mechanism was proved directly by size exclusion chromatography to be inhibition of dimerization of the enzyme polypeptides. The ester bonds of the triterpene derivatives were found to be stable to lipase under mild alkaline conditions [Citation26].
3-O-acyl-betulinic and oleanolic acids, especially the 3-O-(3′, 3′-dimethyl)-succinyl derivatives demonstrated potent anti-HIV activity [EC50 < 0.00035 and 0.00086 microM; therapeutic index (TI) > 20 000 and 22 326, respectively]. Several 3-O-acyl-ursolic acids were prepared and evaluated for anti-HIV activity. Ursolic acid was equipotent (EC50 4.4 microM) with oleanolic acid (EC50, 3.7 microM), although it was slightly toxic (IC50 14.3 microM, TI 3.3). 3-O-Diglycoryl-ursolic acid, demonstrated relatively potent anti-HIV activity with an EC50 of 0.31 microM and a TI of 155.5. In contrast, 3-O-(3′, 3′-dimethylsuccinyl)-ursolic acid, which is analogous to the extremely potent anti-HIV betulinic acid and oleanolic acid derivatives, displayed only weak anti-HIV activity (EC50, 2.1 microM, TI 23.6) [Citation41].
The effect of oleanolic acid on the growth of human immunodeficiency virus-1 (HIV-1) in cultures of human peripheral mononuclear cells and of monocyte/macrophages (M/M) was studied. Its inhibitory activity was also evaluated on peripheral mononuclear cells obtained from HIV-1 infected patients. Results obtained show that oleanolic acid inhibits the HIV-1 replication in all the cellular systems used (EC50 values: 22.7 microM, 24.6 microM and 57.4 microM for in vitro infected peripheral mononuclear cells, naturally infected peripheral mononuclear cells and M/M, respectively). As regards the mechanism of action, oleanolic acid inhibits in vitro the HIV-1 protease activity [Citation42].
Oleanolic acid glycosides, saponins, oleanolic acid synthetic and semi-synthetic derivatives
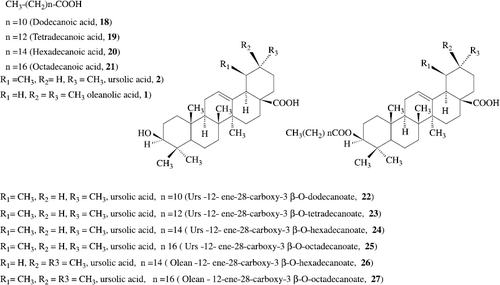
Antifeedant Activity of Some Pentacyclic Triterpene Acids and their Fatty Acid Ester Analogues: The 3-O-fatty acid ester derivatives (22-27) C (12)-C (18) of two pentacyclic triterpenic acids, ursolic acid and oleanolic acid, were synthesized under mild esterification conditions in excellent yields (80-85%) and screened for their antifeedant activity, together with the parent acids, against the agricultural pest tobacco caterpillar larvae (Spodoptera litura F) in a no-choice laboratory study. Antifeedants are potential slimming agents. The Urs-12-ene-28-carboxy-3beta-octadecanoate (26) and olean-12-ene-28-carboxy-3beta-hexadecanoate (25) (Scheme– ) were found to exhibit exceptionally potent antifeedant activities at 50 microg/cm (2) concentration, even after 48 hours [Citation27].
The antipruritic effects of various oleanolic acid glycosides from natural medicines such as Kochiae Fructus (the fruit of Kochia scoparia Schrad.) and Momordicae Radix (the roots of Momordica cochinchinensis Spreng.) was examined by using a compound 48/80-induced pruritic model in mice. Oleanolic acid 3-O-monodesmosides showed an antipruritic effect, while oleanolic acid 3, 28-O-bisdesmosides and their common sapogenol oleanolic acid lacked the activity. This evidence indicated that the 3-O-glycoside moiety and the 28-carboxyl group in oleanolic acid glycosides were essential for exhibiting the antipruritic effect. Furthermore, it was found that the 3-O-glucuronides showed more potent activity than the corresponding 3-O-glucosides [Citation7].
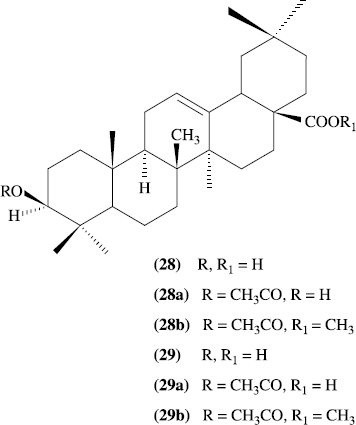
A new triterpenoid acid named eucalyptanoic acid has been isolated from the fresh uncrushed leaves of Eucalyptus camaldulensis var. obtusa along with two known constituents, beta-sitosterol and betulinic acid. The structure of eucalyptanoic acid has been established as 3beta-hydroxyolean-9 (11), 12-dien-28-oic acid. eucalyptanoic acid and its acetyl (28a) and acetylmethyl (28b) derivatives were tested for spasmolytic activity. Acetylmethyl (28b) derivatives was found to be the most active spasmolytic, mediated through blockade of calcium influx at 1 mg/mL. In this study 1b was also prepared starting from oleanolic acid. Acetylation of 29 gave 29a, which on methylation afforded 29b (Scheme–). Reaction of 29b with N-bromosuccinimide furnished 28b. Hence 29 may be regarded as the biogenetic precursor of 1. Compounds 29 and 29a were found inactive at 1 mg/mL, while 29b was moderately active in showing spasmolytic activity [Citation29].
Anticomplement activities of oleanolic acid monodesmosides and bisdesmosides isolated from Tiarella polyphylla
Seven known oleanolic acid glycosides were isolated from the MeOH extract of Tiarella polyphylla. The structures were identified to be 3-O-(beta-D-glucopyranosyl) oleanolic acid, 3-O-[beta-D-glucopyranosyl-(1 → 3)-beta-D-glucopyranosyl] oleanolic acid, 3-O-[beta-D-glucopyranosyl-(1 → 2)-beta-D-glucopyranosyl] oleanolic acid, 3-O-[beta-D-glucopyranosyl-(1 → 3)-beta-D-glucopyranosyl] oleanolic acid 28-O-beta-D-glucopyranosyl ester, 3-O-[beta-D-glucopyranosyl-(1 → 2)-beta-D-glucopyranosyl] oleanolic acid 28-O-beta-D-glucopyranosyl ester, 3-O-[a-L-rhamnopyranosyl-(1 → 3)-beta-D-glucuronopyranosyl] oleanolic acid and 3-O-[alpha-L-rhamnopyranosyl-(1 → 3)-beta-D-glucuronopyranosyl] oleanolic acid 28-O-beta-D-glucopyranosyl ester on the basis of physicochemical and spectral data. These triterpene glycosides were tested for the anticomplement activity and hemolytic activity. Bisdesmosidic saponins, 3-O-[beta-D-glucopyranosyl-(1 → 3)-beta-D-glucopyranosyl] oleanolic acid 28-O-beta-D-glucopyranosyl ester, 3-O-[beta-D-glucopyranosyl-(1 → 2)-beta-D-glucopyranosyl] oleanolic acid 28-O-beta-D-glucopyranosyl ester, and 3-O-[alpha-L-rhamnopyranosyl-(1 → 3)-beta-D-glucuronopyranosyl] oleanolic acid 28-O-beta-D-glucopyranosyl ester, showed anticomplement activity; in contrast, monodesmosidic saponins, 3-O-(beta-D-glucopyranosyl) oleanolic acid, 3-O-[beta-D-glucopyranosyl-(1 → 3)-beta-D-glucopyranosyl] oleanolic acid, 3-O-[beta-D-glucopyranosyl-(1 → 2)-beta-D-glucopyranosyl] oleanolic acid and 3-O-[a-L-rhamnopyranosyl-(1 → 3)-beta-D-glucuronopyranosyl] oleanolic acid, showed direct hemolytic activity. Methyl esterified monodesmosidic saponins showed anticomplement activity at a low concentration and hemolytic activity at a high concentration [Citation8]. Hemolytic activities are not good for a potential drug.
A new acylated oleanane triterpenoid from Couepia polyandra that inhibits the lyase activity of DNA polymerase
Bioassay-directed fractionation of a n-hexane extract of Couepia polyandra using an assay to detect inhibitors of the lyase activity of DNA polymerase resulted in the isolation of the new triterpene 3, 16, 23-triacetoxyolean-12-en-28-oic acid (6) (Scheme–) and four known compounds,oleanolic acid, betulinic acid, stigmasterol, and, sitosterol. The structure of the new compound was established on the basis of extensive 1D and 2D NMR spectroscopic interpretation. All five compounds inhibited DNA polymerase lyase activity [Citation14].
Antifungal activity was tested in 49 pentacyclic triterpenoids and their glycosides, of plant and semisynthetic origin. Several of these compounds inhibited the multiplication of the yeast Saccharomyces carlsbergensis. The highest antifungal activity was found in the triterpene glycosides oleanolic acid and hederagenin, which have a free carboxyl group at C-28. Triterpenes of the meristotropic acid, macedonic acid, and lupan types had no fungistatic activity at concentrations up to 100 microgram/mL [Citation63].
Inhibition of a-glucosidase by oleanolic acid and its synthetic derivatives
Oleanolic acid and five synthetic derivatives (30-34) were tested spectrophotometrically for inhibition of urease, beta-lactamase, acetyl cholinesterase and alpha-glucosidase (Scheme–). All products showed a positive response only against alpha-glucosidase but not against the other enzymes; IC50 calculations (30, IC50 = 55.097 ± 2.635), (31, IC50 = 19.012 ± 0.835), (32, IC50 = 7.97 ± 0.214), (33, IC50 = 89.71 ± 2.105) and (34, IC50 = 21.63 ± 2.3352) showed that the dihydroxy-olide derivative (32) was the most potent among all tested samples [Citation30].
Cardiotonic and antidysrhythmic effects of oleanolic and ursolic acids, methyl maslinate and uvaol: The cardiotonic and antidysrhythmic effects of four triterpenoid derivatives, namely oleanolic acid, ursolic acid and uvaol, isolated from the leaves of African wild olive (Olea europaea, subsp. africana) as well as methyl maslinate (4) isolated from the leaves of Olea europaea (Cape cultivar) were examined. The derivatives showed low toxicity on brine shrimp test. They displayed significant, dose-response vasodepressor effect and sinus bradicardia, most prominent for oleanolic acid and methyl maslinate. The derivatives acted as beta-adrenergic antagonists, blocking the effect of adrenaline and isoprenaline. The established positive inotropic and dromotropic effects were most distinctive for oleanolic acid and methyl maslinate. The antidysrhythmic effects were evaluated on CaCl2 and adrenaline-induced chemical arrhythmias, and on ischemia-reperfusion arrhythmia. oleanolic acid and ursolic acid displayed antidysrhythmic effects on both types of chemical arrhythmia; oleanolic acid and uvaol (5) (Scheme–) in dose 40 mg/kg conferred significant antidysrhythmic activity on ischemia and reperfusion arrhythmias. The effect was comparable to that of propranolol and suggestive of beta-adrenergic antagonistic activity [Citation62].
Anti - gastric ulcer activity of hemisuccinates of derivative of oleanolic acid: The synthesis of hemisuccinates of some derivatives of oleanolic acid is reported, their inhibitory response to experimentally induced gastric ulcers in rats is examined. The hemisuccinates (sodium salt of hemisuccinates) are more effective inhibitors than carbenoxolon-sodium [Citation25].
Gastroprotective and ulcer-healing activity of oleanolic acid derivatives: in vitro-in vivo relationships
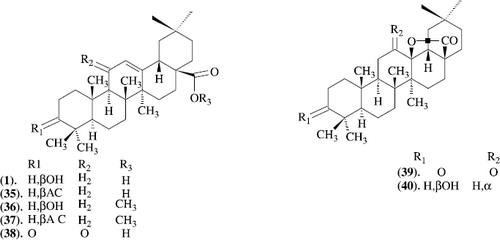
The oleanolic acid 1 and its semisynthetic derivatives 2-7 (Scheme–) were assessed for gastroprotective and ulcer-healing effect using human epithelial gastric cells and human lung fibroblasts (MRC-5). The ability of the compounds to protect the human epithelial gastric cells against the damage induced by sodium taurocholate, to stimulate the cellular reduced glutathione and prostaglandin E content, to enhance epithelial gastric cells and MRC-5 cell proliferation and to scavenge superoxide anion in vitro was studied. The cytotoxicity of the compounds was assessed towards MRC-5 and epithelial gastric cells cells. In addition, the gastroprotective activity of the compounds was assessed in vivo using the HCl/EtOH-induced ulcer model in mice. All the assayed compounds displayed a significant reduction of human epithelial gastric cells damage after incubation with sodium taurocholate. None of the studied compounds was active as a superoxide anion scavenger nor stimulated the GSH content in epithelial gastric cells cell cultures. Compounds 1, 35, 37 and 39 were able to increase the prostaglandin content in epithelial gastric cells cell cultures. Concerning the proliferation assays, a significant stimulating effect was observed for compounds 36 and 40 on epithelial gastric cells cells and for 1 and 7 on MRC-5 fibroblasts. Regarding cytotoxicity, derivatives 35, 37, 39 and 40 were less toxic than the parent compound oleanolic acid. The results strongly support the predictive capacity of the in vitro assessment of gastroprotective activity allowing the reduction of experimental animals [Citation31].
A review is made of the literature describing the structural changes to glycyrrhetic, oleanolic and ursolic acids and their influence on anti-ulcer activity. For the glycyrrhetic acid derivatives some analogues were prepared in which the ketonic group in position 11 was removed and the carboxylic function at position 30 was either intact, reduced to alcohol or transformed into ketone. This first series of compounds suggests the possibility of obtaining compounds devoid of the conjugated ketonic group, maintaining anti-ulcer activity but with reduced or lacking mineralocorticoid activity. Based on these findings, a series of carbenoxolone analogues in the beta-amyrin series of glycyrrhetic and oleanolic acid was prepared. In particular, the delta 9,11 unsaturated compounds and the 11-methylene derivative 18 present advantages in terms of acute toxicity and mineralocorticoid activity as compared to the reference compound. The derivative delta 9,11 unsaturated compounds in the volunteer showed an increase of gastric PGE2 levels with minor pseudoaldosteronic effect. Among the ursolic acid derivatives, the dihemisuccinate sodium salt demonstrated a good separation between anti-ulcer and mineralocorticoid activities. Nevertheless, kidney and liver toxicity was observed in the monkey thus jeopardizing its further development. Better results were obtained with the uvaol dihemiphthalate sodium salt and the diene analogue. In particular, diene analogue showed a potent anti-ulcer activity, 3- to 25-fold higher than carbenoxolone [Citation33].
The gastroprotective effect of the triterpene oleanolic acid was assessed on gastric ulceration in rats. The effect of a single oral dose of oleanolic acid was evaluated at 50, 100 and 200 mg kg ( − 1) in the following models: pylorus ligature (Shay) and aspirin- and ethanol-induced gastric ulcers. A single oral administration of oleanolic acid at doses of 50, 100 and 200 mg kg-′ inhibited the appearance of gastric lesions induced by ethanol, aspirin and pylorus ligature. In the pylorus ligature and aspirin models, the effect of oleanolic acid at the selected concentrations was comparable with that of ranitidine at 50 mg kg ( − 1). In the ethanol-induced gastric lesion model, oleanolic acid showed a dose-dependent activity, and at 100 and 200 mg kg ( − 1) was as active as omeprazole at 20 mg kg ( − 1). The effect of oleanolic acid, its acetylated and methoxylated derivatives, oleanonic acid and its methyl ester were assessed on HCI/ethanol-induced ulcers in mice at 200 mg kg ( − 1). oleanolic acid and its methoxylated and acetylated derivatives proved to be active in this animal model. The semisynthetic acetylated and methoxylated derivatives had the greatest gastroprotective activity, but their effect was not significantly greater than oleanolic acid. In an acute toxicity test on mice, intraperitoneal administration of oleanolic acid showed no toxicity at doses up to 600 mg kg ( − 1) [Citation40].
Effects of oleanolic acid glycosides on gastrointestinal transit and ileus in mice
The effects of various oleanolic acid glycosides obtained from medicinal herbs on gastrointestinal transit and ileus were investigated in fasted mice. Ileus was induced by the peritoneal-irritation or by the laparotomy with manipulation. One hour after the oral administration, three oleanolic acid 3-O-monodesmosides (oleanolic acid 3-O-glucuronide (42, 50 mg/kg), momordin Ic (43, 25 and 50 mg/kg), and momordin I (45, 25 mg/kg) significantly accelerated gastrointestinal transit, but two oleanolic acid 3-O-monodesmosides (28-deglucosyl-chikusetsu saponins IV (47) and V (49), oleanolic acid 3,28-O-bisdesmosides (momordin IIc (5), chikusetsu saponins IV (46) and V (48), and their common aglycon (oleanolic acid (1) (50 mg/kg) showed no significant effect. On the other hand, oleanolic acid 28-O-monodesmoside (compound O (2, 50 mg/kg)) significantly inhibited gastrointestinal transit. 43 (5-25 mg/kg) and 45 (12.5 and 25 mg/kg) also significantly prevented the inhibition of gastrointestinal transit induced by the peritoneal injection of acetic acid. 2 and 9 (50 mg/kg) significantly potentiated the inhibition of gastrointestinal transit, whereas 1, 42, 44, 46, 47, and 49 (50 mg/kg) showed no significant effect. 42, 43, 45, and 49 (50 mg/kg) significantly prevented the inhibition of gastrointestinal transit induced by laparotomy with manipulation, while 1, 41, 44, 46, 47, and 48 (50 mg/kg) showed no significant effect. These results indicate that the 3-O-glycoside moiety seems to be essential to show the gastrointestinal transit accelerating activity, and the 28-O-glucoside moiety reduce the activity. The accelerations of gastrointestinal transit by 42, 43 and 45 were completely abolished by the pretreatment with streptozotocin (100 mg/kg, i.v.), but not by the pretreatment with capsaicin (75 mg/kg in total, s.c.). These results suggest that sympathetic nervous system, but not capsaicin-sensitive sensory nerves, be involved in the enhancements of gastrointestinal transit by 42, 43 and 45 (Scheme–). It is worthy to study their therapeutical effect in the prevention of the inhibition of gastrointestinal transit, including ileus, in clinic [Citation48].
Structure related hypoglycemic activity of oleanolic acid oligoglycoside: The effects of various oleanolic acid oligoglycosides obtained from traditional herbs on gastric emptying in non-nutrient meal- or nutrient meal-loaded mice were examined. Test samples were given orally to fasted mice 0.5 hours before loading of test meals. Oleanolic acid 3-O-monodesmosides [oleanolic acid 3-O-glucuronide (51, 12.5-50 mg/kg), momordin Ic (52, 25 and 50 mg/kg), momordin I (54, 12.5-50 mg/kg), and 28-O-deglucosyl-chikusetsusaponins IV (56, 12.5-50 mg/kg) and V (58, 50 mg/kg)] were found to show inhibitory effects on gastric emptying in 1.5% CMC-Na test meal-loaded mice. 52, 54, and 56 also inhibited gastric emptying in mice given 40% glucose test meal, milk test meal, and 60% ethanol test meal. 51 inhibited gastric emptying in mice given milk test meal or 60% ethanol test meal, but lacked significant inhibition in 40% glucose test meal-loaded mice. 58 (50 mg/kg) also slightly inhibited gastric emptying in milk test meal-loaded mice, but lacked the significant inhibition in mice given 40% glucose or 60% ethanol test meal. Whereas oleanolic acid 1, 28-O-bisdesmosides [momordin IIc (53), chikusetsusaponins IV (55) and V (57)], oleanolic acid 28-O-monodesmoside [compound O (50)], and their common aglycon [oleanolic acid (1)] showed no such effects at dose of 50 mg/kg. 28-O-Deglucosyl-chikusetsusaponin V (58) (Scheme–) showed a little inhibition in these experiments. These results indicate that both the 3-O-monodesmoside structure and 28-carboxyl group were confirmed to be essential for such activity and the 28-ester glucoside moiety and 2′-O-beta-D-glucopyranoside moiety reduce the activity [Citation49].
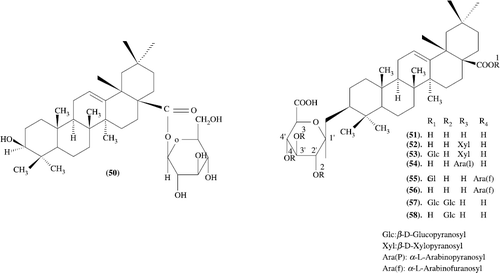
Synthesis and biological evaluation of oleanolic acid derivatives as novel inhibitors of glycogen phosphorylase: Oleanolic acid and its synthetic derivatives have been identified as novel inhibitors of glycogen phosphorylase. Within this series of compounds, 63 () (IC50 = 3.3 microM) is the most potent glycogen phosphorylase inhibitor. Preliminary structure-activity relationships of the oleanolic acid derivatives are discussed [Citation50].
Oleanolic acid glycosides from several medicinal foodstuffs were found to show potent inhibitory activity on the increase of serum glucose levels in oral glucose-loaded rats. By examination of the structure-activity relationships, the 3-O-glucuronide moiety and the 28-carboxyl group in oleanolic acid glycosides were required to exert the hypoglycemic activity. Oleanolic acid glycosides were found to have neither insulin-like nor insulin-releasing activity, but they inhibited gastric emptying and glucose-uptake in the small intestine. Investigation of the mode of action revealed that the inhibition of gastric emptying was mediated by capsaicin-sensitive sensory nerves and the central nervous system. Furthermore, oleanolic acid glycosides were suggested to suppress the gastric emptying by stimulating the release and/or production of dopamine to act through dopamine 2 receptors, which in turn causes the release of prostaglandins [Citation36].
Mode of action of oleanolic acid and its derivatives
Inhibitory effect on serum glucose levels
The action mechanism of oleanolic acid 3-O-monodesmoside, momordin Ic and oleanolic acid 3-O-glucuronide for the inhibitory effect on the increase in serum glucose levels in oral glucose-loaded rats was examined. Although momordin Ic and oleanolic acid 3-O-glucuronide dose-dependently inhibited the increase in serum glucose levels in oral glucose-loaded rats, these compounds showed no significant effects on serum glucose levels in normal rats, intraperitoneal glucose-loaded rats, and alloxane-induced diabetic mice. Furthermore, momordin Ic and oleanolic acid 3-O-glucuronide were found to suppress gastric emptying in rats, and also to inhibit the glucose uptake in rat small intestine concentration dependently in vitro. These results indicate that momordin Ic and oleanolic acid 3-O-glucuronide given orally have neither insulin-like activity nor insulin releasing-activity. momordin Ic and oleanolic acid 3-O-glucuronide apparently inhibited glucose absorption by suppressing the transfer of glucose from the stomach to the small intestine and by inhibiting the glucose transport system at the small intestinal brush border of the small intestine [Citation37,60].
Protective effects of oleanolic acid in leukemic cells
Protective effects of ursolic acid and oleanolic acid against H2O2-induced DNA damage in leukemic L1210, K562 and HL-60 cells using single-cell gel electrophoresis was carriedout. Scientists compared their protective effects (antioxidant activities) with respect to the different position of the methyl group in their chemical structures. After 24 hours pre-treatment of cells both compounds investigated inhibited significantly the incidence of DNA single strand breaks induced by H2O2. The concentration range of ursolic acid and oleanolic acid was in all experiments 2.5–10 micromol/l. The antioxidant activity of oleanolic acid determined by single-cell gel electrophoresis was significantly higher compared to ursolic acid in L1210 (+) P < 0.05) and K562 cells (+++) P < 0.001). Significant difference of the antioxidant activities of the two compounds was evidently connected with the different position of the methyl group. The protective effect of oleanolic acid was in HL-60 cells slightly lower compared to the activity of ursolic acid, but the difference between the protective effects of ursolic acid and oleanolic acid was not significant. In conclusion we can say that both natural pentacyclic triterpenoic acids investigated, ursolic acid and oleanolic acid, manifested potent antioxidant effects. The different position of one methyl group in their chemical structures caused moderately different biological activities of these compounds on three leukemic cell lines [Citation55].
The protective effects of oleanolic acid on carbon tetrachloride-induced hepatotoxicities and the possible mechanisms involved in this protection were also investigated in mice. Pretreatment with oleanolic acid prior to the administration of carbon tetrachloride significantly prevented the increase in serum alanine aminotransferase and lactate dehydrogenase activity and liver lipid peroxidation in a dose-dependent manner. Hepatic glutathione levels and glutathione-S-transferase activities were not affected by treatment with oleanolic acid alone but pretreatment with oleanolic acid protects carbon tetrachloride-induced depletion of hepatic glutathione levels. The effects of oleanolic acid on the cytochrome P450 2E1, the major isozyme involved in carbon tetrachloride bioactivation were investigated. Treatment of mice with oleanolic acid resulted in a significant decrease of P450 2E1-dependent p-nitrophenol and aniline hydroxylation in a dose-dependent manner. Consistent with these observations, the P450 2E1 expressions were also decreased, as determined by immunoblot analysis. These results show that the protective effects of oleanolic acid against the carbon tetrachloride-induced hepatotoxicity may, at least in part, be due to its ability to block bioactivation of carbon tetrachloride mainly by the inhibition of expression and activities of P450 2E1 [Citation38].
The protective effects of oleanolic acid-type saponins and their derivatives on in vitro immunological liver injury of primary cultured rat hepatocytes were studied. A known antihepatotoxic saponin (chikusetsusaponin IVa) showed hepatoprotective activity in this model. Although a rhamnosyl derivative of chikusetsusaponin IVa, similarly showed hepatoprotective activity, its prosapogenin did not show any hepatoprotective activity. On the contrary, prosapogenin exhibited cytotoxicity toward liver cells. In the absence of antiserum, monodesmosyl saponins showed hepatotoxicity, while the bisdesmosyl saponins except for chikusetsusaponin IVa, did not show such hepatotoxicity. In order to clarify the effects of the sugar residues at C-3 and C-28 responsible for hepatoprotective and hepatotoxic actions, oleanolic acid 3-O-glucuronide and oleanolic acid 28-O-glucoside were prepared and tested. 28-O-glucoside showed neither hepatoprotective action nor hepatotoxicity. In contrast, oleanolic acid 3-O-glucuronide was effective at 90 microM on hepatoprotection, although it showed strong hepatotoxicity. Oleanolic acid (2c) itself showed both hepatoprotective action and weak hepatotoxicity. Therefore, the hepatoprotective activity of these types of saponins could represent a balance between hepatoprotective action and hepatotoxicity [Citation28].
Phytocomponents of triterpenoids, oleanolic acid and ursolic acid, regulated differently the processing of epidermal keratinocytes via PPAR-α pathway
There is little study on the mechanism of triterpenoids involved in the differentiation of keratinocytes as well as their effects on epidermal permeability barrier. A study was therefore conducted to determine whether oleanolic acid and ursolic acid could stimulate the differentiation of epidermal keratinocytes through peroxisome proliferator-activated receptor-α activation. This work was then extended to investigate the rate of formation of cornified envelope as a marker in the terminal differentiation of keratinocytes and the amount of transglutaminase in human keratinocytes treated with oleanolic acid and ursolic acid. It was shown that oleanolic acid induced the differentiation of keratinocytes, whereas ursolic acid had little effect. In addition, reporter gene assay using peroxisome proliferator-activated receptor response element activity demonstrated that oleanolic acid might be related to the increase of PPAR-α activity in CV-1 cells. Moreover, it enhanced the recovery of epidermal permeability barrier function as well as increased ceramides in epidermis after topical application. It was therefore propose that the effect of oleanolic acid on the stimulation of differentiation in epidermal keratinocytes seems to be highly related to activation of peroxisome proliferator-activated receptor (PPAR–α) [Citation39].
Structure activity relationship of oleanolic acid
Oleanolic acid has been shown to protect against a number of hepatotoxicants, and is used in China to treat hepatitis. The effect of oleanolic acid on acetaminophen -induced acute liver injury in mice and the mechanism of protection was examined. Oleanolic acid pretreatment (25-100 mg/kg s.c. for 3 days) remarkably decreased acetaminophen (500 mg/kg i.p.)-induced liver damage in mice, as indicated by decreased serum activities of alanine aminotransferase and sorbitol dehydrogenase, as well as by histopathological observation. Additionally, oleanolic acid pretreatment mitigated acetaminophen (300-450 mg/kg i.v.)-induced depletion in liver glutathione content. The protective effect was not evident until 24 hours after a single s.c. injection of oleanolic acid (300 mg/kg) and lasted for 72 hours. To examine the mechanism of this protection, the biliary and urinary excretion of acetaminophen and acetaminophen metabolites were measured for 2 hours after acetaminophen administration (150 mg/kg i.v.) in bile duct-cannulated mice. Oleanolic acid pretreatment resulted in an increased urinary excretion of AA-glucuronide and a decreased biliary excretion of AA-GSH. Microsomes from oleanolic acid pretreated mice, incubated in vitro with acetaminophen, produced less benzoquinoneimine intermediate than controls, as determined by the formation of AA-GSH. Hepatic subcellular distribution of [3H] acetaminophen to the nuclear fraction was also decreased by oleanolic acid. Oleanolic acid pretreatment of mice had no influence on liver UDP-glucuronic acid concentration, but increased hepatic glucuronosyltransferase activity toward AA. In summary, Oleanolic acid pretreatment dramatically protects against acetaminophen -induced hepatotoxicity in mice [Citation34].
By monitoring the inhibitory effect on ethanol absorption in rats, new active saponins named elatosides A and B were isolated from the bark of Aralia elata Seem. together with elatosides C and D. The inhibitory effects of several oleanolic acid oligoglycosides on ethanol absorption have been examined and some structure-activity relationships have been found [Citation35].
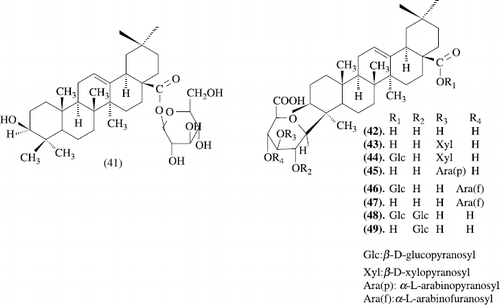
The hypoglycemic component, elatoside E, was isolated from the root cortex of Aralia elata SEEM. (Araliaceae) together with elatoside F and eight known oleanolic acid glycosides, elatosides A and C, oleanolic acid 3-O-[alpha-L-arabinofuranosyl (1 → 4)]-beta-D-glucopyranosiduronic acid, oleanolic acid 3-O-beta-D-glucopyranosiduronic acid, stipuleanosides R1 and R2, and chikusetsusaponins IV and IVa. The structures of elatosides E and F were determined on the basis of chemical and physicochemical evidence as oleanolic acid 3-O-[beta-D-xylopyranosyl (1 → 2)][beta-D-glucopyranosyl (1 → 3)]-alpha-L-arabinopyranoside and its 28-O-beta-D-glucopyranosyl ester, respectively. The hypoglycemic activity of oleanolic acid and nine oleanolic acid oligoglycosides from the root cortex of Aralia elata was determined by monitoring inhibition effect on the elevation of plasma glucose level by oral sucrose tolerance test in rats, and some structure-activity relationships of oleanolic acid glycoside were obtained [43].
Effects of ursolic acid and oleanolic acid on human colon carcinoma cell line HCT15
There is little literature currently available regarding oleanolic acid effects on colon carcinoma cells. This study was designed to investigate their inhibitory effects on human colon carcinoma cell line HCT15. HCT15 cells were cultured with different drugs. The treated cells were stained with hematoxylin-eosin and their morphologic changes observed under a light microscope. The cytotoxicity of these drugs was evaluated by tetrazolium dye assay. Cell cycle analysis was performed by flow cytometry. Data were expressed as means +/ − SEM and analysis of variance and student' t-test for individual comparisons. Twenty-four to 72 hours after ursolic acid or oleanolic acid 60 micromol/L treatment, the numbers of dead cells and cell fragments were increased and most cells were dead at the 72nd hour. The cytotoxicity of ursolic acid was stronger than that of oleanolic acid. Seventy-eight hours after 30 micromol/L of ursolic acid or oleanolic acid treatment, a number of cells were degenerated, but cell fragments were rarely seen. The IC50 values for ursolic acid and oleanolic acid were 30 and 60 micromol/L, respectively. Proliferation assay showed that proliferation of ursolic acid and oleanolic acid -treated cells was slightly increased at 24hours and significantly decreased at 48 hours and 60 h, whereas untreated control cells maintained an exponential growth curve. Cell cycle analysis by FCM showed HCT15 cells treated with ursolic acid 30 and oleanolic acid 60 for 36 hours and 72 hours gradually accumulated in G(0)/G (1) phase (both drugs P < 0.05 for 72 h), with a concomitant decrease of cell populations in S phase (both drugs P < 0.01 for 72 h) and no detectable apoptotic fraction. Ursolic acid and oleanolic acid have significant anti-tumor activity. The effect of ursolic acid is stronger than that of oleanolic acid. The possible mechanism of action is that both drugs have an inhibitory effect on tumor cell proliferation through cell-cycle arrest [Citation44].
Anti-tumor activity of oleanolic acid
Oleanolic acid and ursolic acid were examined for their ability to inhibit the tumor growth and modify hematopoiesis after irradiation in three experimental systems: (a) in vivo anti-tumor activity of implanted tumor by ascitic cells was found to be augmented by addition of oleanolic acid and ursolic acid at a high concentration and inhibited in a dose-dependent manner; (b) in the sublethal whole-body irradiated mice treated with the drugs in the 30 min preirradiation period, enhanced effects of oleanolic acid and ursolic acid on peripheral leukocytes were observed by a different significance, and (c) when these chemicals were administered i.p. to mice 30 min before 4 Gy irradiation, both oleanolic acid and ursolic acid enhanced the postirradiation responses of splenic blastogenesis by PHA. ursolic acid was a more potent tumorigenic inhibitor than oleanolic acid. Combining with the gamma-irradiation, however, there was no significant synergetic effect on their anti-tumor activity. The beneficial effects of oleanolic acid and ursolic acid on hematopoiesis and immunocompetence under this study, suggested they might partially play a role in anti-cancer and, furthermore, with the ability to decrease undesirable radiation damage to the hematopoietic tissue after radiotherapy [Citation45,46].
Anti-tumor activity of a 3-oxo derivative of oleanolic acid
It was found that oleanolic acid (3beta-hydroxy-olea-12-en-28-oic acid, oleanolic acid) had marked anti-tumor effects and exhibited cytotoxic activity towards many cancer cell lines in culture. In this article, the anti-tumor and differentiation-inducing effects of a derivative of oleanolic acid modified at C-3, 3-oxo oleanolic acid (3-oxo-olea-12-en-28-oic acid, 3-oxo-OA, 3-7-1) was reported. In vitro, 3-7-1 were found to inhibit significantly the growth of cancer cells derived from different tissues. And 3-7-1 had inhibitory effect on melanoma in vivo. This selection may relate to the differentiation induced by 3-7-1. The inhibition of 3-7-1 on B16-BL6 suggests that 3-7-1 may be a useful anti-cancer agent for melanoma [Citation52,53].
To examine whether oleanolic acid modulates hepatic toxicant-activating and detoxifying systems as a means of protection, a study was designed. Mice were treated with oleanolic acid (100 and 200 mumol/kg s.c.) for 3 days, and liver microsomes and cytosols were prepared 24 hours after the last dose. oleanolic acid produced a dose-dependent reduction in liver microsomal cytochrome P450 (P450) levels (25–37%) and cytochrome b5 (15–21%) content, but had no effect on NADPH-cytochrome c reductase activity. Oleanolic acid treatment also decreased several P450 enzyme activities, such as coumarin 7-hydroxylation (45%), 7-pentoxyresorufin O-dealkylation (35%), 7-ethoxyresorufin O-dealkylation (25%) and chlorzoxazone 6-hydroxylation (20%). Treatment of mice with oleanolic acid decreased caffeine N3-demethylation (40%), but had no effect on caffeine 8-hydroxylation. Oleanolic acid treatment decreased testosterone 6 alpha- and 15 alpha-hydroxylation (40–50%) and androstenedione formation (35%), but slightly increased testosterone 1 alpha/beta-, 2 beta- and 6 beta-hydroxylation. Consistent with enzyme activities, oleanolic acid decreased the amounts of mouse liver CYP1A and CYP2A enzymes, but had no appreciable effect on CYP3A enzymes, as determined by immunoblotting with antibodies against rat P450 enzymes. oleanolic acid treatment slightly increased liver glutathione content and the activity of glutathione S-transferases toward 1-chloro-2, 4-dinitrobenzene, but had no effect on glutathione peroxidase and glutathione reductase. The activities of superoxide dismutase and DT-diaphorase were unaffected by oleanolic acid treatment. At the high dose of oleanolic acid, catalase activity was decreased by 20% [Citation47].
Oleanolic acid displayed anti-inflammatory activity in carrageenan and dextran-induced oedema in rats. It elicited marked anti-arthritic action in adjuvant-induced polyarthritis in rats and mice and in formaldehyde-induced arthritis in rats. Oleanolic acid checked the inflammation-induced increased serum transaminase levels. It reduced exudate volume and inhibited leucocyte infiltration in carrageenan-induced pleurisy in rats. It is devoid of any analgesic, antipyretic or ulcerogenic action. Oleanolic acid did not affect the parturition time in pregnant rats or castor oil-induced diarrhoea in rats. Oral LD50 was found to be greater than 2 g kg− 1 in mice and rats [Citation61].
In order to determine whether further oxidation of carbon 3 of oleanolic acid affects anti-inflammatory activity in mice, different tests were carried out on oleanolic acid and its 3-oxo-analogue oleanonic acid, which was obtained from Pistacia terebinthus galls. The last one showed activity on the ear oedema induced by 12-deoxyphorbol-13-phenylacetate, the dermatitis induced by multiple applications of 12-O-tetradecanoyl-13-acetate and the paw oedemas induced by bradykinin and phospholipase A2. The production of leukotriene B4 from rat peritoneal leukocytes was reduced by oleanonic acid with an IC50 of 17 microM. Negligible differences were observed in the response of both triterpenes to12-deoxyphorbol-13-phenylacetate, bradykinin, and phospholipase A2, while oleanonic acid was more active on the dermatitis by 12-O-tetradecanoyl-13-acetate and on the in vitro leukotriene formation. The presence of a ketone at C-3 implies an increase in the inhibitory effects on models related to 5-lipoxygenase activity and on associated in vivo inflammatory processes [Citation16].
Oleanolic acid inhibits the activity of the multidrug resistance protein ABCC1 (MRP1) but not of the ABCB1 (P-glycoprotein). Possible use in cancer chemotherapy
The effects of oleanolic acid on ABCB1 and ABCC1 activities were studied in a cell line constitutively expressing both proteins. It was observed that oleanolic acid did not alter ABCB1 activity, but inhibited the activity of ABCC1 protein. This inhibition was reversible and only occurred in the presence of oleanolic acid. In addition, oleanolic acid did not alter the expression of ABCC1 mRNA. These results suggest that oleanolic acid could be a good choice in the treatment of MDR tumours, either as a chemotherapic itself in tumours bearing ABCB1, or as an adjuvant in the chemotherapy of ABCC1 expressing tumours [Citation32].
Antimutagenicity of ursolic acid and oleanolic acid against doxorubicin-induced clastogenesis in Balb/c mice
The antimutagenic potential of ursolic acid and oleanolic acid using the micronucleus test in peripheral blood and bone marrow of Balb/c mice was evaluated. The animals were divided into 10 treatment groups: mice treated with ursolic acid (80 mg/kg b.w.); oleanolic acid (80 mg/kg b.w.); a mixture of ursolic acid and oleanolic acid (80 mg/kg b.w.); the antineoplastic agent doxorubicin (90 mg/kg b.w.); DMSO and doxorubicin; ursolic acid and doxorubicin; oleanolic acid and doxorubicin; ursolic acid, oleanolic acid and doxorubicin, and negative and solvent controls. ursolic acid, oleanolic acid and a mixture of ursolic acid and oleanolic acid were administered to the animals by gavage, followed by the intraperitoneal injection of doxorubicin. The results showed a significant reduction in micronucleus frequency in the groups concomitantly treated with the triterpenoid compounds and doxorubicin compared to that treated with doxorubicin alone. The results demonstrate the antimutagenic activity of ursolic acid and oleanolic acid [Citation51].
Oleanolic acid inhibitor of human DNA ligase I
Enzymatic activity mediated by recombinant human DNA ligase I (hLI), in conjunction with tannin removal procedures, has been applied to a natural-product screen involving approximately 1000 plant extracts and various pure compounds. The primary hLI activity assay involved the measurement of the amount of radiolabelled phosphate in a synthetic nucleic acid hybrid that becomes resistant to alkaline phosphatase as a result of ligation. A bioactivity-guided fractionation scheme resulted in the isolation of ursolic [IC50 = 100 micrograms/mL (216 microM)] and oleanolic [IC50 = 100 micrograms/mL (216 microM)] acids from Tricalysia niamniamensis Hiern (Rubiaceae), which demonstrated similar DNA ligase inhibition profiles to other triterpenes such as aleuritolic acid. Protolichesterinic acid [IC50 = 6 micrograms/mL (20 microM)], swertifrancheside [IC50 = 8 micrograms/mL (11) microM)] and fulvoplumierin [IC50 = 87 micrograms/mL (357 microM)] represent three additional natural-product structural classes that inhibit hLI. Fagaronine chloride [IC50 = 10 micrograms/mL (27 micronM] and certain flavonoids are also among the pure natural products that were found to disrupt the activity of the enzyme, consistent with their nucleic acid intercalative properties. Further analyses revealed that some of the hLI-inhibitory compounds interfered with the initial adenylation step of the ligation reaction, indicating a direct interaction with the enzyme protein. However, in all cases, this enzyme-inhibitor interaction did not disrupt the DNA relaxation activity mediated by hLI. These results indicate that, although the same enzyme active site may be involved in both enzyme adenylation and DNA relaxation, inhibitors may exert allosteric effects by inducing conformational changes that disrupt only one of these activities. Studies with inhibitors are important for the assignment of specific cellular functions to these enzymes, as well as for their development into clinically useful antitumour agents [Citation54].
Oleanolic acid, a pentacyclic triterpene attenuates capsaicin-induced nociception in mice: Possible mechanisms
The anti-inflammatory pentacyclic triterpene, oleanolic acid was examined on acute nociception induced by intraplantar injection of capsaicin in mice. Oleanolic acid administered orally to mice at 10, 30 and 100 mgkg ( − 1), significantly attenuated the paw-licking response to capsaicin (1.6 microg/paw) by 53%, 68.5% and 36.6%, respectively. Ruthenium red (3 mgkg ( − 1), s.c.), a non-competitive vanilloid receptor (V1, TRPV1)-antagonist also suppressed the capsaicin nociception by 38.6%. The maximal antinociception produced by 30 mgkg ( − 1) oleanolic acid was significantly blocked in animals pre-treated with naloxone (2 mgkg ( − 1), i.p.), the opioid antagonist; l-arginine (600 mgkg ( − 1), i.p.), the substrate for nitric oxide synthase; or glibenclamide (2 mgkg ( − 1), i.p.), the K (ATP)-channel blocker, but was unaffected by yohimbine (2 mgkg ( − 1), i.p.), an alpha (2)-adrenoceptor antagonist. In open-field and rota-rod tests that detect motor deficits, mice received 30 mgkg ( − 1) oleanolic acid did not manifest any effect per se, indicating that the observed antinociception is not a consequence of motor abnormality. These data suggest that oleanolic acid inhibits capsaicin-evoked acute nociception due to mechanisms possibly involving endogenous opioids, nitric oxide, and K (ATP)-channel opening [Citation56].
This study evaluated the antinociceptive potential of oleanolic acid, in the mouse model of colonic nociception induced by mustard oil. The possible participation of opioid, alpha-2-adrenergic, and transient receptor potential vanilloid 1 (TRPV1)-receptors in its mechanism was further examined. Mice were pretreated orally with oleanolic acid (3, 10, 30 mg/kg) or vehicle, and the pain-related behavioral responses to intracolonic injection of mustard oil was analysed. Oleanolic acid significantly suppressed the mustard oil-induced nociceptive behaviors at test doses of 10 and 30 mg/kg, in a dose-related manner. The antinociceptive effect of oleanolic acid (30 mg/kg) was significantly blocked by pretreatment with the opioid antagonist, naloxone (2 mg/kg, i.p.), while the alpha2-adrenoceptor antagonist, yohimbine (2 mg/kg, s.c.), had no effect. Pretreatment with ruthenium red (3 mg/kg, s.c.), a non-competitive TRPV1 antagonist alone caused significant inhibition of mustard oil-induced nociception but its co-administration with oleanolic acid produced neither antagonism nor potentiation of oleanolic acid antinociception. In the open-field test that detects sedative or motor abnormality, mice received 30 mg/kg oleanolic acid did not show any per se influence, but significantly inhibited the mustard oil-induced decrease in ambulation frequency. These data demonstrate the visceral antinociceptive potential of oleanolic acid that involves an opioid mechanism and possibly a modulatory influence on vanilloid-receptors [Citation58].
Cardiovascular, antihyperlipidemic and antioxidant effects of oleanolic acid in experimental hypertension
Cardiovascular (systolic and diastolic blood pressure, heart rate), antihyperlipidemic (tryglycerides, total cholesterol and lipoprotein fractions), antioxidant (glutathione peroxidase—GPx, and superoxide dismutase—SOD), diuretic/saluretic and hypoglycemic activity of 98% pure oleanolic and ursolic acid were studied in Dahl salt-sensitive, insulin resistant rat model of genetic hypertension. Both oleanolic acid and ursolic acid displayed low toxicity, with LC50 0.10 and 0.95 mg/mL, respectively. Although both triterpenoids did not have direct hypotensive effect, after 6-week application in a daily dose 60 mg/kg b.w., i.p., they prevented the development of severe hypertension. The F effect was attributed to their potent diuretic-natriuretic-saluretic activity; direct cardiac effect (heart rate decrease by 34% and 32%, respectively); antihyperlipidemic (more than two times decrease of LDL and triglycerides); antioxidant (GPx increase by 12% and 10%, respectively; SOD increase by 12% and 22%, respectively), and hypoglycemic (blood glucose decrease by 20% and 50%, respectively) effects on the Dahl salt-sensitive rats [Citation57].
The antihypertensive, diuretic, antiatherosclerotic, antioxidant and hypoglycemic effects of authentic oleanolic and ursolic acid and the three isolates (GO, AO and CT) were studied on Dahl salt-sensitive, insulin-resistant rat genetic model of hypertension. All three isolates, in a dose 60 mg/kg b.w. for 6 weeks treatment, prevented the development of severe hypertension and atherosclerosis and improved the insulin resistance of the experimental animals [Citation59].
Bioassay-directed fractionation of a n-hexane extract of Couepia polyandra using an assay to detect inhibitors of the lyase activity of DNA polymerase beta resulted in the isolation of the new triterpene 3 beta,16 beta, 23-triacetoxyolean-12-en-28-oic acid and four known compounds, oleanolic acid, betulinic acid, stigmasterol, and beta-sitosterol. All five compounds inhibited DNA polymerase beta lyase activity [Citation13].
Conclusion
The literature survey reveals that oleanolic acid is a highly potent compound which shows variety of biological activities. It is known for their antimicrobial, hepatoprotective, anti-inflammatory, antiallergic, antiviral and cytotoxic activities. It can be an active ingredient in treating urease, beta-lactamase, acetyl cholinesterase, alpha-glucosidase, antimicrobial, hepatoprotective, anti-inflammatory, antipruritic effects, spasmolytic activity, anti-angiogenic activities, antiallergic, antiviral and cytotoxic activities. In recent years, it was found that oleanolic acid had marked anti-tumor effects. It was shown that it protects mice against the hepatotoxicity of carbon tetrachloride, acetaminophen, bromobenzene, thioacetamide, furosemide, phalloidin, colchicine, cadmium, D-galactosamine and endotoxin. It lower serum transaminase, lactic dehydrogenase, and gamma-glutamyltransferase levels in the CCl4-treated rats. It enhances the bioavailability of the active ingredient of the pharmaceutical compound.
It has been found to be an active ingredient from Olea ferruginea Royle and Rubus species, which attributes to antimicrobial activity. It has also been found to be inhibitory to the growth of intestinal bacteria Bacteroides fragilis, Clostridium clostridiiforme, C. perfringens, C. paraputrificum, Escherichia coli, Enterobacter cloacae and Salmonella typhimurium. It shows antioxidant and pro-oxidant properties. It protects mammalian and bacterial cells from cytotoxicity induced by hydroperoxides. On the basis of the vasodepressor, cardiotonic and antidysrhythmic effects of this compound, it was concluded that oleanolic acid, isolated from wild African olive leaves can provide a cheap and accessible source of additive to conventional treatment of hypertension, complicated by stenocardia and cardiac failure. Oleanolic acid could provide an effective and cheap treatment of most common type of salt-sensitive hypertension in the African population.
Finally we can suggest that Olea ferruginea which is a widely grown, in many countries, could be used as a source of oleanolic acid. Further studies involving crude extracts of Olea ferruginea containing oleanolic acid would be highly economic advantageous.
References
- N Sultana, and Atta-ur-Rahman. (2007). Fitoterapia (Submitted)
- N Sultana, Attr-ur-Rahman, and MI Choudhary. (2007). Biotransformation of oleanolic acid. Natural Product Research (in press)
- M Yoshikawa, T Murakami, E Harada, N Urakami, J Yamahara, and H Matsuda. (1996). Chem Pharm Bul (Tokyo) 44 (10):1923–1927.
- M Rajasekaran, JS Bapna, S Lakshmanan, Nair AG Ramachandran, A J Veliath, and M Panchanadam. (1988). J Ethnopharmacol 24 (1):115–121.
- M Yoshikawa, T Murakami, M Kadoya, H Matsuda, O Muraoka, J Yamahara, and N Murakami. (1996). Chem Pharm Bull (Tokyo) 44 (6):1212–1217.
- NY Kim, MK Lee, MJ Park, SJ Kim, HJ Park, JW Choi, SH Kim, SY Cho, and JS Lee. (2005). J Med Food 8 (2):177–183.
- H Matsuda, Y Dai, Y IDo, T Murakami, H Matsuda, M Yoshikawa, and M Kubo. (1998). Biol Pharm Bull 21 (11):1231–1233.
- SH Park, SR Oh, KY Jung, IS Lee, KS Ahn, JG Kim, JJ Lee, and HK Lee. (1999). Arch Pharm Res 22 (4):428–431.
- H Yoshimura, K Sugawara, M Saito, S Saito, S Murakami, N Miyata, A Kawashima, S Morimoto, N Gao, X Zhang, and J Yang. (2003). Planta Med 69 (7):673–675.
- N Banno, T Akihisa, H Tokuda, K Yasukawa, H Higashihara, M Ukiya, K Watanabe, Y Kimura, J Hasegawa, and H Nishino. (2004). Biosci Biotechnol Biochem 68 (1):85–90.
- H Ohigashi, H Takamura, K Koshimizu, H Tokuda, and Y Ito. (1986). Cancer Lett 30 (2):143–151.
- LI Somova, FO Shode, P Ramnanan, and A Nadar. (2003). J Ethnopharmacol 84 (2–3):299–305.
- LI Somova, FO Shode, and M Mipando. (2004). Phytomedicine 11 (2-3):121–129.
- VS Prakash Chaturvedula, Z Gao, SM Hecht, SH Jones, and DG Kingston. (2003). J Nat Prod 66 (11):1463–1465.
- NT Dat, IS Lee, XF Cai, G Shen, and YH Kim. (2004). Biol Pharm Bull. 27 (3):426–428.
- EM Giner-Larza, S Manez, MC Recio, RM Giner, JM Prieto, M Cerda-Nicolas, and JL Rios. (2001). Eur J Pharmacol 428 (1):137–143.
- F Abe, T Yamauchi, T Nagao, J Kinjo, H Okabe, H Higo, and H Akahane. (2002). Biol Pharm Bull 25 (11):1485–1487.
- TS Jeong, EI Hwang, HB Lee, ES Lee, YK Kim, BS Min, KH Bae, SH Bok, and SU Kim. (1999). Planta Med 65 (3):261–263.
- A Kapil, and S Sharma. (1994). J Pharm Pharmacol 46 (11):922–923.
- T Ringbom, L Segura, Y Noreen, P Perera, and L Bohlin. (1998). J Nat Prod 61 (10):1212–1215.
- Y Kashiwada, HK Wang, T Nagao, S Kitanaka, I Yasuda, T Fujioka, T Yamagishi, LM Cosentino, M Kozuka, H Okabe, Y Ikeshiro, CQ Hu, E Yeh, and KH Lee. (1998). J Nat Prod 61 (9):1090–1095.
- CI Chang, CC Kuo, JY Chang, and YH Kuo. (2004). J Nat Prod 67 (1):91–93.
- J Hase, K Kobashi, K Mitsui, T Namba, M Yoshizaki, and T Tomimori. (1981). J Pharmacobiodyn 4 (11):833–837.
- KH Sohn, HY Lee, HY Chung, HS Young, SY Yi, and KW Kim. (1995). Cancer Lett 94 (2):213–218.
- U Wrzeciono, I Malecki, J Budzianowski, H Kierylowicz, L Zaprutko, E Beimcik, and H Kostepska. (1985). Pharmazie 40 (8):542–544.
- Nakamura N Chao-Ma, and M Hattori. (2000). Chem Pharm Bull (Tokyo) 48 (11):1681–1688.
- UV Mallavadhani, A Mahapatra, SS Raja, and C Manjula. (2003). J Agric Food Chem 51 (7):1952–1955.
- J Kinjo, M Okawa, M Udayama, Y Sohno, T Hirakawa, Y Shii, and T Nohara. (1999). Chem Pharm Bull (Tokyo). 47 (2):290–292.
- S Begum, I Sultana, BS Siddiqui, F Shaheen, and AH Gilani. (2002). J Nat Prod 65 (12):1939–1941.
- MS Ali, M Jahangir, SS Hussan, and MI Choudhary. (2002). Phytochemistry 6 (3):295–299.
- M Sanchez, C Theoduloz, G Schmeda-Hirschmann, I Razmilic, T Yanez, and JA Rodriguez. (2006). Life Sci 79 (14):1349–1356.
- F Braga, D Ayres-Saraiva, CR Gattass, and MA Capella. (2006). Cancer Lett 2: [Epub ahead of Print]
- M Farina Pinza, and G Pifferi. (1998). Farmaco. 53 (1):22–32.
- J Liu, Y Liu, C Madhu, and CD Klaassen. (1993). J Pharmacol Exp Ther 266 (3):1607–1613.
- M Yoshikawa, E Harada, H Matsuda, T Murakami, J Yamahara, and N Murakami. (1993). Chem Pharm Bull (Tokyo) 41 (11):2069–2071.
- M Yoshikawa, and H Matsuda. (2000). Biofactors 13 (1–4):231–237.
- H Matsuda, Y Li, T Murakami, N Matsumura, J Yamahara, and M Yoshikawa. (1998). Chem Pharm Bull (Tokyo) 46 (9):1399–1403.
- G Jeong. (1999). Toxicol Lett 105 (3):215–222.
- HK Lee, GW Nam, SH Kim, and SH Lee. (2006). Exp Dermatol 15 (1):66–73.
- L Astudillo, JA Rodriguez, and G Schmeda-Hirschmann. (2002). J Pharm Pharmacol 54 (4):583–588.
- Y Kashiwada, T Nagao, A Hashimoto, Y Ikeshiro, H Okabe, LM Cosentino, and KH Lee. (2000). J Nat Prod 63 (12):1619–1622.
- F Mengoni, M Lichtner, L Battinelli, M Marzi, CM Mastroianni, V Vullo, and G Mazzanti. (2002). Planta Med 68 (2):111–114.
- M Yoshikawa, T Murakami, E Harada, N Murakami, J Yamahara, and H Matsuda. (1996). Chem Pharm Bull (Tokyo) 44 (10):1923–1927.
- J Li, WJ Guo, and QY Yang. (2002). World J Gastroenterol 8 (3):493–495.
- HY Hsu, JJ Yang, and CC Lin. (1997). Cancer Lett 111 (1–2):7–13.
- G Kweifio-Okai, and TA Macrides. (1992). Res Commun Chem Pathol Pharmacol 78 (3):367–372.
- J Liu, Y Liu, A Parkinson, and CD Klaassen. (1995). J Pharmacol Exp Ther 275 (2):768–774.
- Y Li, H Matsuda, and M Yoshikawa. (1999). Bioorg Med Chem 7 (6):1201–1205.
- H Matsuda, Y Li, T Murakami, J Yamahara, and M Yoshikawa. (1999). Bioorg Med Chem 7 (2):323–327.
- J Chen, J Liu, L Zhang, G Wu, W Hua, X Wu, and H Sun. (2006). Bioorg Med Chem Lett 16 (11):2915–2919.
- F Aparecida Resende, CA De Andrade Barcala, MC da Silva Faria, FH Kato, WR Cunha, and DC Tavares. (2006). Life Sci 79 (13):1268–1273.
- D Huang, Y Ding, Y Li, W Zhang, W Fang, and X Chen. (2006). Cancer Lett 233 (2):289–296.
- G Kweifio-Okai, and TA Macrides. (1992). Res Commun Chem Pathol Pharmacol 78 (3):367–372.
- GT Tan, S Lee, IS Lee, J Chen, P Leitner, JM Besterman, AD Kinghorn, and JM Pezzuto. (1996). Biochem J 314 (Pt 3):993–1000.
- Z Ovesna, K Kozics, and D Slamenova. (2006). Mutat Res 600 (1–2):131–137.
- JL Maia, RC Lima-Junior, CM Melo, JP David, JM David, AR Campos, FA Santos, and VS Rao. (2006). Pharmacol Res 54 (4):282–286.
- LO Somova, A Nadar, P Rammanan, and FO Shode. (2003). Phytomedicine 10 (2–3):115–121.
- JL Maia, RC Lima-Junior, JP David, JM David, FA Santos, and VS Rao. (2006). Biol Pharm Bull 29 (1):82–85.
- LI Somova, FO Shode, P Ramnanan, and A Nadar. (2003). J Ethnopharmacol 84 (2–3):299–305.
- H Matsuda, T Murakami, H Shimada, N Matsumura, M Yoshikawa, and J Yamahara. (1997). Biol Pharm Bull 20 (6):717–719.
- GB Singh, S Singh, S Bani, BD Gupta, and SK Banerjee. (1992). J Pharm Pharmacol 44 (5):456–458.
- LI Somova, FO Shode, and M Mipando. (2004). Phytomedicine 11 (2–3):121–129.
- MM Anisimov, VV Shcheglov, LI Strigina, NS Chetyrina, NI Uvarova, GI Oshitok, NG Alad'ina, LP Vecherko, AD Zorina, LG Matyukhina, and IA Saltykova. (1979). Biol Bull Acad Sci USSR 6 (4):464–468.