Abstract
A series of 3-pyrrol-3-yl-3H-isobenzofuran-1-ones was synthesized and assessed for the ability to inhibit cytosolic phospholipase A2α (cPLA2α). Several of these compounds were found to be active in both a cell based assay and an isolated enzyme assay. The most potent inhibitor was the thiazolidine-2,4-dione substituted derivative 35. With IC50-values of 0.7 μM and 7.3 μM in the cellular and isolated enzyme assay, respectively, it possesses similar inhibitory potency as the known cPLA2α inhibitor arachidonyltrifluoromethyl ketone (AACOCF3). Structure–activity relationship studies revealed that the evaluated isobenzofuran-1-ones seem to exert their cellular activities not only by a direct interaction with the enzyme but also by other as yet unknown mechanisms.
Introduction
Cytosolic phospholipase A2α (cPLA2α) is an esterase that selectively cleaves the sn-2 position of arachidonoyl-glycerophospholipids of biomembranes to generate free arachidonic acid and lysophospholipids [Citation1]. Arachidonic acid in turn is metabolized to a variety of inflammatory mediators including prostaglandins and leukotrienes. Lysophospholipids with an alkyl ether moiety at the sn-1 position can be acetylated to platelet activating factor (PAF), another mediator of inflammation [Citation2]. Thus, inhibition of cPLA2α is considered as an attractive target for the design of new anti-inflammatory drugs [Citation3–5].
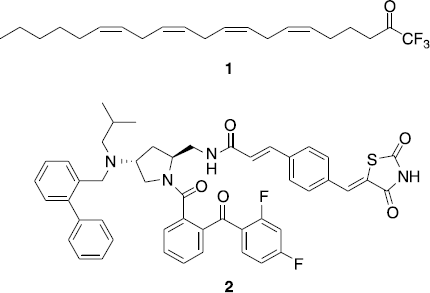
First-generation cPLA2α inhibitors were analogues of arachidonic acid with the COOH group replaced by COCF3 (AACOCF3, 1) or CH2PO(OCH3)F (MAFP) [Citation3]. Inhibitors of cPLA2α with very high in vitro potency reported later are thiazolidinediones from Shionogi, such as compound 2 () [Citation6], and propan-2-ones from AstraZeneca [Citation7].
Figure 1. Structures of known cPLA2α inhibitors.
We here describe the synthesis and biological evaluation of a series of 3-pyrrol-3-yl-3H-isobenzofuran-1-ones. Some of these compounds show inhibition of cPLA2α in a cell free as well as in a cellular test system with IC50 values in the low micromolar range.
Material and methods
Chemical synthesis
Column chromatography was performed on silica gel 60, 230–400 mesh ( = flash chromatography) or 70–230 mesh from Merck, Darmstadt, Germany. Preparative HPLC was performed on a RP18 column (Kromasil 100, 5 μm, 10 mm (I.D.) × 250 mm protected with an analogously filled guard column 10 mm (I.D.) × 50 mm, CS-chromatographie service, Langerwehe, Germany). Melting points were determined on a Büchi B-540 apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were recorded on a Varian Mercury Plus 400 spectrometer (400 MHz). Mass spectra were obtained on Finnigan GCQ and LCQ apparatuses applying electron beam ionization (EI) and electrospray ionisation (ESI), respectively. The purity of the target compounds was determined using two diverse HPLC systems with UV detection at 254 nm. The first one applied a reversed phase C18 column (Nucleosil 100 RP18, 10 μm, 4.0 mm (I.D.) × 250 mm, Macherey & Nagel, Düren, Germany) eluting the compounds isocratically with CH3CN/H2O containing 0.1% H3PO4 at a flow rate of 1 mL/min. In the second system separation was performed using a cyano phase (LiChrospher 100 CN, 10 μm, 4.0 mm (I.D.) × 250 mm, Merck, Darmstadt, Germany) under reversed phase conditions, eluting with CH3CN/H2O containing 0.1% H3PO4 at a flow rate of 0.8 mL/min. With exception of 17 and 30, all target compounds showed purities greater than 95% in both systems. The purities evaluated for 17 and 30 were 91 and 93%, respectively, in each case. The reference inhibitor arachidonyltrifluoromethyl ketone (AACOCF3) was purchased from Biomol, Hamburg, Germany.
2,5-Dimethyl-1-phenylpyrrole-3-carbaldehyde oxime (4)
2,5-Dimethyl-1-phenylpyrrole-3-carbaldehyde (3) (2.5 g, 12.5 mmol) was dissolved in methanol (50 mL) by heating. After addition of an aqueous solution (20 mL) of hydroxylamine-HCl (0.87 g, 12.5 mmol) and sodium carbonate (1.33 g, 12.5 mmol) the mixture was refluxed for 3 h. The precipitate formed was filtered off by suction and washed with cold methanol to yield 4 [Citation8] (2.36 g, 88%); mp 196°C. 1H-NMR (DMSO-d6): δ 1.90 (s, 3H), 2.06 (s, 3H), 6.08 (s, 1H), 7.24–7.50 (m, 5H), 8.03 (s, 1H), 10.37 (s, 1H). 13C-NMR (DMSO-d6): δ 10.86, 12.64, 103.89, 113.06, 127.85, 127.94, 128.69, 128.78, 129.20, 137.27, 143.10.
(2,5-Dimethyl-1-phenylpyrrol-3-yl)methylamine (5)
A solution of TiCl4 (3.3 mL, 20 mmol) in 1,2-dimethoxyethane (20 mL) was treated under a nitrogen atmosphere at 0°C with NaBH4 (2.27 g, 60 mmol). A suspension of 4 (3.0 g, 14 mmol) in 1,2-dimethoxyethane (10 mL) was added slowly and the resulting mixture was allowed to warm up to room temperature. After being stirred for 14 h at this temperature, water (40 mL) and 25% ammonium hydroxide (40 mL) were added with ice cooling. The precipitate formed was filtered off with suction. The filtrate was extracted with CH2Cl2 and the precipitate was washed with CH2Cl2. The CH2Cl2 phases were combined and the solvent distilled off. The residue was dissolved in dilute HCl and washed with diethyl ether. The aqueous phase was alkalized with dilute aqueous KOH-solution and extracted with diethyl ether. The organic layer was dried (Na2SO4) and concentrated to give 5 as an oil (1.90 g, 68%). 1H-NMR (CDCl3): δ 1.68 (s, broad, 2H), 1.99 (s, 3H), 2.02 (s, 3H), 3.69 (s, 2H), 5.94 (s, 1H), 7.18–7.47 (m, 5H). 13C-NMR (CDCl3): δ 10.92, 13.13, 38.52, 105.88, 121.16, 124.74, 127.67, 128.13, 128.36, 128.44, 129.04, 129.10, 138.96. MS (EI): m/z (%) = 200 (12) [M+], 184 (100).
N-(2,5-Dimethyl-1-phenylpyrrol-3-ylmethyl)-2,2,2-trifluoroacetamide (6)
A solution of 5 (750 mg, 3.7 mmol) in dry THF (10 mL) was treated under a nitrogen atmosphere at 0°C with ethyl trifluoroacetate (0.45 mL, 3.7 mmol). After being stirred at this temperature for 10 min, the solvent was distilled off and the residue was purified by chromatography on silica gel (hexane/ethyl acetate, 4:1) to give 6 as an oil (0.96 g, 87%). 1H-NMR (CDCl3): δ 1.98 (s, 3H), 2.01 (s, 3H), 4.37 (d, 2H, J = 5 Hz), 5.91 (s, 1H), 6.37 (s, broad, 1H), 7.19–7.47 (m, 5H). 13C-NMR (CDCl3): δ 10.78, 12.91, 36.78, 106.29, 112.90, 115.88 (q), 127.01, 128.02, 128.09, 128.84, 129.18, 138.28, 156.55 (q). MS (EI): m/z (%) = 522 (95) [M+], 463 (100).
3-Chloro-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (7)
A mixture of 2-(4-fluorobenzoyl)benzoic acid (5 g, 19 mmol) and thionyl chloride (5 mL) was heated with a catalytical amount of DMF under reflux for 1 h. The remaining thionyl chloride was distilled off under reduced pressure to yield 7 [Citation16] (5.3 g, 98%) as solid. The crude product was used without further purification. It was stable under a nitrogen atmosphere at − 18°C for several weeks.
2,2,2-Trifluoro-N-{4-[1-(4-fluorophenyl)-3-oxo-1H-isobenzofuran-1-yl]-2,5-dimethyl-1-phenylpyrrol-3-ylmethyl}acetamide (8)
A solution of SnCl4 (0.43 mL, 3.7 mmol) in dry 1,2-dichloroethane (25 mL) was treated under a nitrogen atmosphere with 7 (813 mg, 3.1 mmol). After dissolution of 7, compound 6 (915 mg, 3.1 mmol) was added and the mixture was heated under reflux for 2 h. The reaction mixture was cooled, treated with ice-water and extracted with diethyl ether. The organic layer was dried (Na2SO4) and the solvent was evaporated. The residue was purified by silica gel chromatography (hexane/ethyl acetate, 4:1) to give 8 (0.80 g, 50%) as solid; mp 173–174°C. 1H-NMR (CDCl3): δ 1.27 (s, 3H), 1.86 (s, 3H), 3.74 (dd, 1H, J = 5 Hz and 15 Hz), 3.99 (dd, 1H, J = 5 Hz and 15 Hz), 6.97 (t, 2H, J = 8 Hz), 7.02–7.09 (m, 3H), 7.33–7.42 (m, 5H), 7.45 (t, 1H, J = 8 Hz), 7.56 (t, 1H, J = 8 Hz), 7.85 (d, 1H, J = 8 Hz). 13C-NMR (CDCl3): δ 11.18, 13.22, 35.31, 90.46, 113.13, 115.89, 116.05 (q), 116.10, 116.67, 124.80, 125.05, 126.23, 127.54, 127.78, 127.87, 128.64, 128.66, 128.71, 129.46, 129.52, 129.61, 129.70, 134.19, 137.86, 138.23, 138.26, 152.52, 156.36 (q), 161.44, 163.91, 169.35. MS (EI): m/z (%) = 522 (5) [M+], 301 (100).
3-(4-Aminomethyl-2,5-dimethyl-1-phenylpyrrol-3-yl)-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (9)
To a solution of KOH (2.5 g) in water (7 mL) and methanol (10 mL) was added 8 (400 mg, 0.76 mmol). The mixture was stirred at room temperature for 30 min, diluted with water and extracted with diethyl ether and CH2Cl2. The organic phases were combined and the solvent distilled off. The residue was dissolved in dilute HCl and washed with diethyl ether. The aqueous phase was alkalized with dilute aqueous KOH-solution and extracted with diethyl ether. The organic layer was dried (Na2SO4) and concentrated to give 9 (257 mg, 78%) as solid; mp 193–195°C. 1H-NMR (DMSO-d6): δ 1.03 (s, 3H), 1.86 (s, 3H), 3.20 (d, 1H, J = 13 Hz), 3.28 (d, 1H, J = 13 Hz), 6.34 (d, 1H, 8 Hz), 6.95–7.16 (m, 8H), 7.34–7.58 (m, 4H). 13C-NMR (DMSO-d6): δ 10.48, 12.07, 37.10, 78.52, 113.50, 113.70, 121.48, 123.52, 123.83, 125.04, 125.74, 126.13, 126.24, 127.07, 127.99, 128.71, 130.52, 138.60, 143.72, 144.72, 144.74, 146.25, 159.21, 161.61, 173.04.
4-(2,4-Dioxothiazolidin-5-ylidenemethyl)-N-{4-[1-(4-fluorophenyl)-3-oxo-1H-isobenzofuran-1-yl]-2,5-dimethyl-1-phenylpyrrol-3-ylmethyl}benzamide (10)
To a solution of 9 (250 mg, 0.59 mmol) in dry DMF (10 mL) was added 4-(2,4-dioxothiazolidin-5-ylidenemethyl)benzoic acid [Citation9] (147 mg, 0.59 mmol). After addition of EDC-HCL (123 mg, 0.64 mmol) and 1-hydroxybenzotriazole (79 mg, 0.59 mmol), the mixture was stirred at room temperature for 1 h. Ethyl acetate was added and the organic solution was extracted twice with water and three times with brine. The organic layer was dried (Na2SO4) and the solvent was evaporated. The residue was purified by silica gel chromatography (CHCl3/methanol, 49:1) to give 10 (282 mg); 33 mg of the obtained solid were further purified by RP-HPLC applying acetonitrile/H2O (73:27, v/v) as mobile phase. The eluates were concentrated under reduced pressure until most of the acetonitrile was distilled off. The remaining solvent was removed by freeze drying yielding pure 10 (26 mg). 1H-NMR (CDCl3): δ 1.35 (s, 3H), 2.01 (s, 3H), 3.80 (dd, 1H, J = 4 Hz and 15 Hz), 4.21 (dd, 1H, J = 6 Hz and 15 Hz), 6.89 (t, 1H, J = 8 Hz), 7.01–7.05 (m, 2H), 7.12–7.17 (m, 2H), 7.42–7.67 (m, 10H), 7.88 (s, 1H), 7.94 (d, 1H, J = 8 Hz), 8.01 (d, 2H, J = 8 Hz). 13C-NMR (CDCl3): δ 10.97, 13.05, 35.13, 90.67, 114.65, 115.56, 115.77, 116.31, 124.71, 124.82, 125.89, 127.21, 127.56, 127.64, 127.72, 128.34, 128.47, 128.93, 129.16, 129.21, 129.42, 130.00, 131.56, 133.97, 135.55, 135.69, 137.85, 137.93, 137.96, 152.54, 161.15, 163.61, 164.94, 167.05, 167.42, 169.44. MS (EI): m/z (%) = 657 (1) [M+], 163 (100); MS (ESI-): m/z (%) = 656 (100) [M+ − 1].
3-(2,5-Dimethyl-1-phenylpyrrol-3-yl)-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (11)
Dry 1,2-dichloroethane (10 mL) was subsequently treated under a nitrogen atmosphere with SnCl4 (0.18 mL, 1.55 mmol) and 7 (373 mg, 1.42 mmol). The mixture was stirred at room temperature for 10 min. Then 2,5-dimethyl-1-phenylpyrrole (250 mg, 1.35 mmol) was added and the resulting mixture heated at 50°C for 2 h. After cooling, water was added and the mixture was extracted twice with diethyl ether. The combined organic phases were washed with water, dried (Na2SO4) and concentrated. The residue was purified by silica gel chromatography (hexane/ethyl acetate, 4:1) to give 11 as solid (213 mg, 36%); mp 144–146°C. 1H-NMR (CDCl3): δ 1.60 (s, 3H), 1.85 (s, 3H), 5.49 (s, 1H), 6.92 (t, 2H, J = 9 Hz), 7.08–7.10 (m, 3H), 7.30–7.46 (m, 6H), 7.58 (t, 1H, J = 8 Hz), 7.83 (d, 1H, J = 8 Hz). 13C-NMR (CDCl3): δ 12.40, 12.98, 89.82, 107.20, 115.41, 115.62, 117.97, 123.86, 125.51, 125.98, 127.24, 127.95, 128.03, 128.37, 128.53, 128.98, 129.15, 129.42, 134.15, 138.32, 138.35, 138.42, 153.93, 161.33, 163.78, 170.72. MS (EI): m/z (%) = 397 (100) [M+].
3-(2,5-Dimethyl-1-phenylpyrrol-3-yl)-3-methyl-3H-isobenzofuran-1-one (12)
A mixture of 2,5-dimethyl-1-phenylpyrrole (1.0 g, 5.84 mmol) and 2-acetylbenzoic acid (0.96 g, 5.84 mmol) was heated at 120–140°C for 2 h. After being cooled, the reaction mixture was purified by silica gel chromatography (hexane/ethyl acetate, 9:1) to give 12 as solid (370 mg, 20%); mp 153°C. 1H-NMR (CDCl3): 1.69 (s, 3H), 1.96 (s, 3H), 2.01 (s, 3H), 5.95 (s, 1H), 7.20–7.25 (s, 2H), 7.40–7.44 (m, 3H), 7.48–7.52 (m, 2H), 7.63–7.67 (m, 1H), 7.88–7.90 (m, 1H). 13C-NMR (CDCl3): δ 12.10, 13.03, 28.41, 86.95, 105.63, 117.64, 122.49, 125.69, 126.09, 126.99, 127.65, 128.32, 128.63, 128.89, 129.37, 134.17, 138.43, 155.22, 170.60. MS (EI): m/z (%) = 317 (51) [M+], 302 (100).
3-(2,5-Dimethyl-1-phenylpyrrol-3-yl)-3H-isobenzofuran-1-one (13)
A solution of 2,5-dimethyl-1-phenylpyrrole (300 mg, 1.75 mmol) and 2-formylbenzoic acid (263 mg, 1.75 mmol) in toluene (10 mL) was refluxed under a nitrogen atmosphere for 2 h. The solvent was distilled off and the residue was purified by silica gel chromatography (hexane/ethyl acetate, 9:1) followed by preparative RP-HPLC (acetonitrile/water, 715:285). Recrystallization from ethanol/H2O gave 13 as solid (45 mg, 8%); mp 120–121°C. 1H-NMR (CDCl3): 1.92 (s, 3H), 2.09 (s, 3H), 5.48 (s, 1H), 6.50 (s, 1H), 7.21–7.23 (m, 2H), 7.43–7.51 (m, 4H), 7.55 (t, 1H, J = 7 Hz), 7.66–7.70 (m, 1H), 7.95 (d, 1H, J = 8 Hz). 13C-NMR (CDCl3): δ 11.16, 12.96, 78.44, 105.24, 114.01, 123.39, 125.54, 127.05, 128.39, 128.48, 129.12, 129.48, 129.58, 134.02, 138.48, 150.27, 171.05. MS (EI): m/z (%) = 303 (100) [M+], 244 (63).
3-(4-Fluorophenyl)-3-(1,2,5-trimethylpyrrol-3-yl)-3H-isobenzofuran-1-one (14)
Under a nitrogen atmosphere AlCl3 (573 mg, 4.3 mmol) and 7 (985 mg, 3.7 mmol) were added to 1,2-dichloroethane (20 mL) at 0°C. Then the mixture was treated dropwise with 1,2,5-trimethylpyrrole (0.5 mL, 3.7 mmol) ensuring that the temperature did not exceed 20°C. After being stirred at room temperature for 1 h, the reaction mixture was treated with ice water and extracted with diethyl ether. The organic phase was dried (K2CO3) and the solvent evaporated. The residue was purified by silica gel chromatography (hexane/ethyl acetate, 4:1) to give 14 as solid (750 mg, 60%); mp 158–160°C. 1H-NMR (CDCl3): δ 1.88 (s, 3H), 2.13 (s, 3H), 3.35 (s, 3H), 5.45 (s, 1H), 6.95–7.00 (m, 2H), 7.38–7.51 (m, 4H), 7.61–7.65 (m, 1H), 7.88–7.90 (m, 1H). MS (EI): m/z (%) = 335 (66) [M+], 276 (100).
3-(2,5-Dimethylpyrrol-3-yl)-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (15)
2,5-Dimethylpyrrole (1.1 mL, 10 mmol) was reacted with 7 (2.8 g, 10 mmol) and SnCl4 (1.5 mL, 12 mmol) in 1,2-dichloroethane (30 mL) in a similar manner as described for the synthesis of 11. The product was recrystallized from ethanol to yield 15 (1.9 g, 57%); mp 185–186°C. 1H-NMR (CDCl3): δ 1.87 (s, 3H), 2.15 (s, 3H), 5.43 (s, 1H), 6.98 (t, 2H, J = 9 Hz), 7.39–7.49 (m, 4H), 7.63 (t, 1H, J = 8 Hz), 7.68 (s, br, 1H), 7.88 (d, 1H, J = 8 Hz). MS (EI): m/z (%) = 321 (66) [M+], 262 (100).
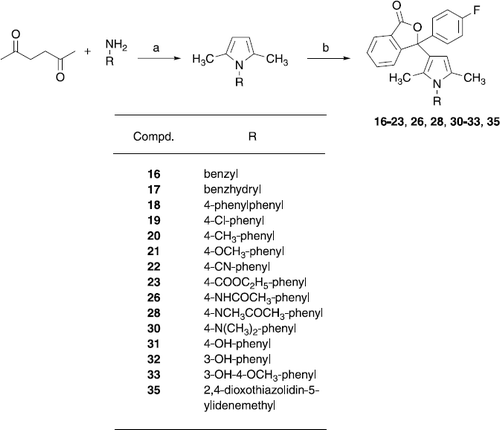
Compounds 16–23, 26, 28, 30–33 and 35 were synthesized by reacting the appropriate substituted 2,5-dimethylpyrroles with 3-chloro-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (7) in a similar manner as described for the synthesis of 11. The 1-aryl-2,5-dimethylpyrroles needed for the synthesis of these compounds were obtained by refluxing benzylamine, benzhydrylamine and substituted anilines, respectively, with hexane-2,5-dione and a catalytic amount p-toluenesulfonic acid in toluene at a water separator for 2 h followed by purification with silica gel chromatography.
The carboxylic acid derivative 24 was obtained by saponification of its ethyl ester 23 with aqueous KOH in ethanol. The amide 25 was prepared by refluxing the carbonitrile 22 with solid KOH in tert-butanol. The aniline derivatives 27 and 29 were synthesized by hydrolyzing their acetates 26 and 28, respectively, with half concentrated HCl in methanol or ethanol. The dihydroxy-derivative 34 was obtained by cleaving the methylether of the corresponding 3-hydroxy-4-methoxy compound 33 with 2-bromobenzo[1,3,2]dioxaborol in dry CH2Cl2.
3-(1-Benzyl-2,5-dimethylpyrrol-3-yl)-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (16)
Mp 141°C. 1H-NMR (CDCl3): δ 1.83 (s, 3H), 2.07 (s, 3H), 4.99 (s, 2H), 5.56 (s, 1H), 6.86 (d, 2H, J = 7 Hz), 6.96–7.01 (m, 2H), 7.23 (t, 1H, J = 8 Hz), 7.28–7.32 (m, 2H), 7.39–7.44 (m, 2H), 7.48–7.53 (m, 2H), 7.63–7.67 (m, 1H), 7.90–7.92 (m, 1H). 13C-NMR (CDCl3): δ 11.88, 12.64, 47.19, 89.85, 107.12, 115.36, 115.57, 117.78, 123.80, 125.38, 125.67, 125.90, 126.43, 127.40, 127.74, 127.83, 128.20, 128.98, 129.06, 134.05, 137.73, 138.41, 138.44, 153.79, 161.13, 163.58, 170.70. MS (EI): m/z (%) = 411 (100) [M+].
3-(1-Benzhydryl-2,5-dimethylpyrrol-3-yl)-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (17)
Mp 84°C. 1H-NMR (CDCl3): δ 1.68 (s, 3H), 1.79 (s, 3H), 5.53 (s, 1H), 6.72 (s, 1H), 6.93–6.99 (m, 2H), 7.06–7.09 (m, 4H), 7.28–7-37 (m, 8H), 7.44–7.46 (m, 1H), 7.48–7.52 (m, 1H), 7.61–7.65 (m, 1H), 7.90–7.92 (m, 1H). 13C-NMR (CDCl3): δ 13.28, 14.49, 62.52, 89.79, 108.34, 115.38, 115.59, 118.02, 123.91, 125.46, 125.97, 127.51, 127.72, 127.81, 128.45, 128.59, 128.69, 128.76, 129.08, 129.60, 134.05, 139.04, 139.22, 153.96, 163.71, 170.98. MS (EI): m/z (%) = 487 (8) [M+], 167 (100).
3-(1-Biphenyl-4-yl-2,5-dimethylpyrrol-3-yl)-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (18)
Mp 91°C. 1H-NMR (CDCl3): δ 1.73 (s, 3H), 1.99 (s, 3H), 5.60 (s, 1H), 6.99–7.05 (m, 2H), 7.25 (d, 2H, J = 9 Hz), 7.38 (t, 1H, J = 7 Hz), 7.44–7.56 (m, 6H), 7.61–7.69 (m, 5H), 7.92–7.94 (m, 1H). 13C-NMR (CDCl3): δ 12.49, 13.08, 89.83, 107.34, 115.43, 115.64, 118.12, 123.88, 125.53, 126.00, 127.36, 127.97, 127.99, 128.05, 128.39, 128.82, 129.05, 129.15, 129.18, 134.18, 137.52, 138.31, 138.34, 140.21, 141.28, 153.93, 161.35, 163.81, 170.73. MS (EI): m/z (%) = 473 (68) [M+], 414 (100).
3-[1-(4-Chlorophenyl)-2,5-dimethylpyrrol-3-yl]-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (19)
Mp 190°C. 1H-NMR (CDCl3): δ 1.67 (s, 3H), 1.92 (s, 3H), 5.58 (s, 1H), 6.98–7.03 (m, 2H), 7.12 (d, 2H, J = 9 Hz), 7.42–7.47 (m, 4H), 7.50–7.54 (m, 1H), 7.65–7.69 (m, 1H), 7.91–7.93 (m, 1H). 13C-NMR (CDCl3): δ 12.37, 12.95, 89.64, 107.55, 115.45, 115.67, 118.45, 123.82, 125.46, 126.02, 127.22, 127.91, 127.99, 128.86, 129.22, 129.72, 129.82, 134.21, 134.37, 136.92, 138.21, 153.81, 161.35, 163.81, 170.64. MS (EI): m/z (%) = 433 (26), 431 (73) [M+], 372 (100).
3-[2,5-Dimethyl-1-(4-methylphenyl)pyrrol-3-yl]-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (20)
Mp 191°C. 1H-NMR (CDCl3): δ 1.67 (s, 3H), 1.93 (s, 3H), 2.40 (s, 3H), 5.57 (s, 1H), 6.98–7.03 (m, 2H), 7.05 (d, 2H, J = 8 Hz), 7.24 (d, 2H, J = 8 Hz), 7.45–7.50 (m, 2H), 7.52–7.54 (m, 1H), 7.65–7.69 (m, 1H), 7.91–7.93 (m, 1H). 13C-NMR (CDCl3): δ 12.38, 12.97, 21.38, 89.89, 107.03, 115.39, 115.61, 117.78, 123.88, 125.52, 125.95, 127.30, 127.96, 128.04, 128.24, 129.05, 129.14, 130.04, 134.15, 135.78, 138.28, 138.36, 138.39, 153.98, 161.32, 163.78, 170.75. MS (EI): m/z (%) = 411 (59) [M+], 352 (100).
3-[2,5-Dimethyl-1-(4-methoxyphenyl)pyrrol-3-yl]-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (21)
Mp 188°C. 1H-NMR (CDCl3): δ 1.67 (s, 3H), 1.92 (s, 3H), 3.85 (s, 3H), 5.56 (s, 1H), 6.94–7.03 (m, 4H), 7.09 (d, 2H, J = 8 Hz), 7.45–7.54 (m, 4H), 7.64–7.69 (m, 1H), 7.91–7.92 (m, 1H). 13C-NMR (CDCl3): δ 12.51, 13.10, 55.74, 89.85, 106.81, 114.44, 115.29, 115.50, 117.54, 123.76, 125.35, 125.81, 127.37, 127.81, 127.89, 129.01, 129.09, 129.35, 130.97, 134.04, 138.19, 153.75, 159.17, 161.08, 163.53, 170.52. MS (EI): m/z (%) = 427 (13) [M+], 368 (100).
3-[1-(4-Cyanophenyl)-2,5-dimethylpyrrol-3-yl]-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (22)
Mp 83°C. 1H-NMR (CDCl3): δ 1.70 (s, 3H), 1.94 (s, 3H), 5.63 (s, 1H), 6.98–7.03 (m, 2H), 7.31 (d, 2H, J = 9 Hz), 7.42–7.47 (m, 2H), 7.50–7.54 (m, 2H), 7.65–7.69 (m, 1H), 7.77 (d, 2H, J = 9 Hz), 7.90–7.92 (m, 1H). 13C-NMR (CDCl3): δ 12.68, 13.24, 89.38, 108.40, 112.33, 115.49, 115.70, 118.18, 119.34, 123.72, 125.27, 125.99, 126.98, 127.78, 127.86, 128.48, 129.26, 129.34, 133.39, 134.24, 137.85, 137.88, 142.29, 153.46, 161.21, 163.66, 170.35. MS (EI): m/z (%) = 422 (68) [M+], 363 (100).
Ethyl 4-{3-[1-(4-fluorphenyl)-3-oxo-1,3-dihydroisobenzofuran-1-yl]-2,5-dimethylpyrrol-1-yl}benzoate (23)
Mp 61°C. 1H-NMR (CDCl3): δ 1.41 (t, 3H, J = 7 Hz), 1.70 (s, 3H), 1.94 (s, 3H), 4.40 (q, 2H, J = 7 Hz), 5.60 (s, 1H), 6.98–7.04 (m, 2H), 7.26 (d, 2H, J = 8 Hz), 7.44–7.48 (m, 2H), 7.48–7.54 (m, 2H), 7.66–7.70 (m, 1H), 7.92 (m, 1H), 8.17 (d, 2H, J = 8 Hz). 13C-NMR (CDCl3): δ 12.61, 13.19, 14.73, 61.61, 89.57, 107.83, 115.39, 115.61, 118.66, 123.73, 125.31, 125.91, 126.99, 127.79, 127.87, 128.37, 128.60, 129.13, 130.34, 130.70, 134.12, 137.99, 138.02, 142.19, 153.59, 161.15, 163.60, 165.79, 170.41. MS (EI): m/z (%) = 469 (27) [M+], 410 (100).
4-{3-[1-(4-Fluorphenyl)-3-oxo-1,3-dihydroisobenzofuran-1-yl]-2,5-dimethylpyrrol-1-yl}benzoic acid (24)
A mixture of 23 (560 mg, 1.19 mmol), ethanol (10 mL) and 10% aqueous KOH (6 mL) was heated under reflux for 5 min, cooled, acidified with dilute H3PO4 and extracted with diethyl ether. The organic phase was dried (Na2SO4) and the solvent evaporated. The residue was purified by silica gel chromatography eluting with diethyl ether to give 24 as solid (464 mg, 88%); mp 144°C. 1H-NMR (CDCl3): δ 1.71 (s, 3H), 1.96 (s, 3H), 5.62 (s, 1H), 6.98–7.04 (m, 2H), 7.29 (d, 2H, J = 9 Hz), 7.44–7.48 (m, 2H), 7.51–7.54 (m, 2H), 7.66–7.70 (m, 1H), 7.91–7.93 (m, 1H), 8.20 (d, 2H, J = 9 Hz). 13C-NMR (CDCl3): δ 12.68, 13.25, 89.62, 108.03, 115.45, 115.66, 118.86, 123.74, 125.31, 125.98, 127.03, 127.81, 127.89, 128.58, 128.61, 129.15, 129.19, 131.41, 134.19, 137.94, 137.97, 143.12, 153.59, 161.19, 163.64, 170.53, 170.93. MS (EI): m/z (%) = 441 (9) [M+], 382 (100).
4-{3-[1-(4-Fluorophenyl)-3-oxo-1,3-dihydroisobenzofuran-1-yl]-2,5-dimethylpyrrol-1-yl}benzamide (25)
A mixture of 22 (350 mg, 0.83 mmol), powdered KOH (85%) (210 mg, 3.3 mmol) and tert-butanol (15 mL) was heated under reflux for 10 min. The reaction mixture was cooled, diluted with water, acidified with dilute HCl and extracted with diethyl ether. The organic phase was washed twice with water, dried (Na2SO4) and concentrated to give 25 as solid (338 mg, 92%); mp 121°C. 1H-NMR (CDCl3): δ 1.68 (s, 3H), 1.93 (s, 3H), 5.59 (s, 1H), 6.28 (s, broad, 2H), 6.98–7.03 (m, 2H), 7.25 (d, 2H, J = 8 Hz), 7.42–7.47 (m, 2H), 7.50–7.54 (m, 2H), 7.65–7.69 (m, 1H), 7.89–7.93 (m, 3H). 13C-NMR (CDCl3): δ 12.64, 13.21, 89.67, 107.85, 115.44, 115.65, 118.66, 123.75, 125.27, 125.94, 127.06, 127.79, 127.87, 128.63, 128.66, 129.19, 133.15, 134.20, 137.93, 137.96, 141.54, 153.61, 161.18, 163.63, 168.84, 170.55. MS (EI): m/z (%) = 440 (100) [M+].
N-(4-{3-[1-(4-Fluorophenyl)-3-oxo-1,3-dihydroisobenzofuran-1-yl]-2,5-dimethylpyrrol-1-yl}phenyl)acetamide (26)
Mp 240°C. 1H-NMR (CDCl3): δ 1.63 (s, 3H), 1.90 (s, 3H), 2.23 (s, 3H), 5.55 (s, 1H), 6.97–7.03 (m, 2H), 7.04–7.13 (m, 2H), 7.42–7.47 (m, 2H), 7.50–7.54 (m, 4H), 7.66–7.70 (m, 1H), 7.76 (s, broad, 1H), 7.90–7.92 (m, 1H). 13C-NMR (CDCl3): δ 12.56, 13.16, 24.98, 90.15, 107.10, 115.42, 115.64, 117.67, 120.37, 123.85, 125.22, 125.87, 127.35, 127.78, 127.87, 128.82, 128.96, 129.16, 133.77, 134.27, 137.99, 138.02, 138.21, 153.82, 161.16, 163.62, 168.82, 170.96. MS (EI): m/z (%) = 454 (5) [M+], 227 (100).
3-[1-(4-Aminophenyl)-2,5-dimethylpyrrol-3-yl]-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (27)
A solution of 26 (250 mg, 0.55 mmol) in methanol (5 mL) was adjusted to pH 1 with dilute HCl. After heating under reflux for 6 h, the reaction mixture was cooled, neutralized with 25% aqueous KOH solution and extracted twice with diethyl ether. The combined organic phases were washed with water, dried (Na2SO4) and concentrated. The residue was purified by silica gel chromatography (hexane/ethyl acetate, 2:3) to give 27 as solid (64 mg, 28%); mp 213°C. 1H-NMR (CDCl3): δ 1.66 (s, 3H), 1.92 (s, 3H), 3.82 (s, 2H), 5.53 (s, 1H), 6.70 (d, 2H, J = 9 Hz), 6.92 (d, 2H, J = 9 Hz), 6.97–7.02 (m, 2H), 7.44–7.52 (m, 4H), 7.64–7.68 (m, 1H), 7.90–7.91 (m, 1H). 13C-NMR (CDCl3): δ 12.33, 12.92, 89.80, 106.37, 115.09, 115.30, 117.04, 123.59, 125.19, 125.61, 127.25, 127.64, 127.72, 128.70, 128.81, 128.97, 129.04, 133.83, 138.06, 138.09, 146.15, 153.63, 160.90, 163.34, 170.42. MS (EI): m/z (%) = 412 (14) [M+], 353 (100).
N-(4-{3-[1-(4-Fluorophenyl)-3-oxo-1,3-dihydroisobenzofuran-1-yl]-2,5-dimethylpyrrol-1-yl}phenyl)-N-methylacetamide (28)
Mp 73°C. 1H-NMR (CDCl3): δ 1.70 (s, 3H), 1.95 (s, 3H), 2.00 (s, 3H), 3.34 (s, 3H), 5.59 (s, 1H), 6.98–7.03 (m, 2H), 7.22–7.32 (m, 4H), 7.49–7.54 (m, 2H), 7.65–7.69 (m, 1H), 7.90–7.92 (m, 1H). MS (EI): m/z (%) = 468 (100) [M+].
3-[2,5-Dimethyl-1-(4-methylaminophenyl)pyrrol-3-yl]-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (29)
A solution of 28 (180 mg, 0.38 mmol) in a small amount of ethanol was treated with half concentrated HCl (5 mL) and heated under reflux for 4 h. The reaction mixture was poured into ice-water, alkalized with aqueous KOH and extracted three times with diethyl ether. The combined organic phases were dried (Na2SO4) and concentrated. The residue was purified by silica gel chromatography (hexane/ethyl acetate, 7:3) to give 29 as solid (83 mg, 51%); mp 210°C. 1H-NMR (CDCl3): δ 1.66 (s, 3H), 1.93 (s, 3H), 2.87 (s, 3H), 3.90 (s, broad, 1H), 5.53 (s, 1H), 6.62 (d, 2H, J = 9 Hz), 6.95–7.02 (m, 4H), 7.44–7.52 (m, 4H), 7.63–7.67 (m, 1H), 7.89–7.91 (m, 1H). 13C-NMR (CDCl3): δ 12.60, 13.20, 31.09, 102.93, 106.53, 112.45, 115.33, 115.55, 117.19, 123.85, 125.49, 125.88, 127.60, 127.76, 127.92, 128.00, 129.04, 129.15, 129.44, 134.05, 149.01, 153.93, 163.60, 167.50, 169.47. MS (EI): m/z (%) = 426 (100) [M+].
3-[1-(4-Dimethylaminophenyl)-2,5-dimethylpyrrol-3-yl]-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (30)
Mp 74°C. 1H-NMR (CDCl3): δ 1.70 (s, 3H), 1.95 (s, 3H), 3.00 (s, 6H), 5.58 (s, 1H), 6.73 (d, 2H, J = 9 Hz), 6.98–7.05 (m, 4H), 7.48–7.56 (m, 4H), 7.65–7.69 (m, 1H), 7.91–7.92 (m, 1H). 13C-NMR (CDCl3): δ 12.48, 13.08, 40.68, 89.97, 106.43, 112.20, 115.19, 115.41, 117.05, 123.75, 125.32, 125.69, 126.81, 127.52, 127.79, 127.88, 128.76, 128.92, 129.27, 133.96, 149.88, 153.80, 161.00, 163.44, 170.52. MS (EI): m/z (%) = 440 (27) [M+], 213 (100).
3-(4-Fluorophenyl)-3-[1-(4-hydroxyphenyl)-2,5-dimethylpyrrol-3-yl]-3H-isobenzofuran-1-one (31)
Mp 238°C. 1H-NMR (CDCl3): δ 1.61 (s, 3H), 1.91 (s, 3H), 5.54 (s, 1H), 6.92–7.02 (m, 7H), 7.42–7.46 (m, 2H), 7.51–7.55 (m, 2H), 7.67–7.71 (m, 1H), 7.92–7.94 (m, 1H). 13C-NMR (CDCl3): δ 12.49, 13.11, 90.82, 106.71, 115.43, 115.64, 116.22, 117.08, 123.84, 125.09, 125.99, 127.48, 127.78, 127.86, 129.21, 129.26, 130.43, 134.43, 137.82, 137.85, 153.96, 156.26, 161.18, 163.63, 171.75. MS (EI): m/z (%) = 413 (100) [M+].
3-(4-Fluorophenyl)-3-[1-(3-hydroxyphenyl)-2,5-dimethylpyrrol-3-yl]-3H-isobenzofuran-1-one (32)
Mp 217°C. 1H-NMR (CDCl3): δ 1.66 (s, 3H), 1.93 (s, 3H), 5.53 (s, 1H), 6.52 (s, broad, 1H), 6.68–6.75 (m, 2H), 6.92–7.01 (m, 3H), 7.25–7.29 (m, 1H), 7.41–7.44 (m, 2H), 7.50–7.53 (m, 2H), 7.66–7.70 (m, 1H), 7.90–7.93 (m, 1H). 13C-NMR (CDCl3): δ 12.54, 13.15, 107.05, 115.41, 115.62, 115.74, 117.44, 120.41, 123.85, 125.17, 125.95, 127.21, 127.85, 127.93, 128.99, 129.21, 130.12, 134.36, 137.87, 137.90, 139.32, 153.91, 156.85, 161.18, 163.63. MS (EI): m/z (%) = 413 (36) [M+], 354 (100).
3-(4-Fluorophenyl)-3-[1-(3-hydroxy-4-methoxyphenyl)-2,5-dimethylpyrrol-3-yl]-3H-isobenzofuran-1-one (33)
Mp 88°C. 1H-NMR (CDCl3): δ 1.67 (s, 3H), 1.93 (s, 3H), 3.94 (s, 3H), 5.53 (s, 1H), 5.76 (s, broad, 1H), 6.66 (dd, 1H, J = 3 Hz and 8 Hz), 6.75 (d, 1H, J = 3 Hz), 6.88 (d, 1H, J = 8 Hz), 6.97–7.03 (m, 2H), 7.44–7.53 (m, 4H), 7.64–7.68 (m, 1H), 7.91–7.92 (m, 1H). 13C-NMR (CDCl3): δ 12.52, 13.10, 56.37, 89.91, 106.80, 110.58, 114.85, 115.32, 115.54, 117.52, 120.10, 123.80, 125.40, 125.86, 127.35, 127.86, 127.94, 129.04, 131.67, 134.05, 138.24, 145.91, 146.41, 153.80, 161.13, 163.58, 170.60. MS (EI): m/z (%) = 443 (8) [M+], 261 (100).
3-[1-(3,4-Dihydroxyphenyl)-2,5-dimethylpyrrol-3-yl]-3-(4-fluorophenyl)-3H-isobenzofuran-1-one (34)
To a solution of 2-bromobenzo[1,3,2]dioxaborol (560 mg, 2.82 mmol) in dry CH2Cl2 (10 mL) was added dropwise under a nitrogen atmosphere at − 70°C a solution of 33 (250 mg, 0.56 mmol) in dry CH2Cl2 (5 mL). The reaction mixture was allowed to warm up to room temperature and held there for 24 h. After addition of water, the organic phase was separated and the aqueous phase was extracted exhaustively with diethyl ether. The combined organic phases were dried (Na2SO4) and concentrated. The residue was purified by silica gel chromatography (hexane/ethyl acetate/formic acid, 6:4:0.05). The product fractions were washed with dilute NaHCO3 solution and water. After drying over Na2SO4, the solvent was evaporated to yield 34 as solid (123 mg, 51%); mp 116°C. 1H-NMR (DMSO-d6): δ 1.53 (s, 3H), 1.93 (s, 3H), 5.43 (s, 1H), 6.44–6.47 (m, 1H), 6.51 (s, 1H), 6.78 (d, 1H, J = 8 Hz), 7.15–7.20 (m, 2H), 7.42–7.45 (m, 2H), 7.58–7.62 (m, 1H), 7.68–7.70 (m, 1H), 7.76–7.80 (m, 1H), 7.85–7.87 (m, 1H). 9.27 (s, broad, 2H). 13C-NMR (DMSO-d6): δ 12.10, 12.72, 89.08, 106.33, 115.24, 115.35, 115.59, 115.68, 116.93, 118.83, 119.28, 124.04, 124.15, 125.24, 126.75, 127.42, 127.71, 127.79, 128.70, 129.51, 134.79, 138.20, 145.32, 145.50, 153.13, 160.32, 162.61, 169.33. MS (EI): m/z (%) = 429 (95) [M+], 370 (100).
5-(4-{3-[1-(4-Fluorophenyl)-3-oxo-1,3-dihydroisobenzofuran-1-yl]-2,5-dimethyl-pyrrol-1-yl}benzylidene)thiazolidine-2,4-dione (35)
1H-NMR (DMSO-d6): δ 1.61 (s, 3H), 1.92 (s, 3H), 5.57 (s, 1H), 7.16–7.21 (m, 2H), 7.42–7.48 (m, 4H), 7.60–7.64 (m, 1H), 7.70–7.73 (m, 3H), 7.78–7.82 (m, 1H), 7.85 (s, 1H), 7.89–7.90 (m, 1H), 12.68 (s, 1H). 13C-NMR (DMSO-d6): δ 12.05, 12.62, 88.58, 107.27, 115.20, 115.41, 117.91, 123.84, 123.91, 124.34, 125.09, 126.57, 127.03, 127.55, 127.63, 128.77, 129.37, 130.37, 130.74, 132.57, 134.65, 137.74, 137.77, 138.46, 152.76, 160.16, 162.59, 167.05, 167.42, 169.01. MS (EI): m/z (%) = 524 (63) [M+], 465 (100).
Biological assays
Assay with the isolated enzyme
The ability of test compounds to inhibit cPLA2α isolated from human platelets was performed as previously described [Citation10]. Briefly, sonicated covesicles consisting of 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (0.2 mM) and 1,2-dioleoyl-sn-glycerol (0.1 mM) were used as substrate. Enzyme reaction was terminated by addition of a mixture of acetonitrile, methanol and 0.1 M aqueous EDTA-Na2 solution, which contained 4-undecyloxybenzoic acid as internal standard and nordihydroguaiaretic acid as oxygen scavenger. cPLA2α activity was determined by measuring the arachidonic acid released by the enzyme with reversed phase HPLC and UV-detection at 200 nm after cleaning up the samples by solid phase extraction.
Cell assay
The ability of compounds to inhibit cPLA2α activity in intact cells was determined by measuring the calcium ionophore A23187-induced arachidonic acid release from human platelets with HPLC/UV-detection according to a method previously described [Citation12]. Deviating from this procedure, for HPLC-separation of arachidonic acid a RP18 multospher 100 column, 3 μm, 3.0 mm (I.D.) × 125 mm, with a RP18 multospher 100 guard column, 5 μm, 3.0 mm (I.D.) × 20 mm (CS-chromatographie service, Langerwehe, Germany) was applied. The mobile phase consisted of acetonitrile/10 mM (NH4)2HPO4 buffer adjusted to pH 7.4 with ortho-phosphoric acid (50:50, v/v). The flow rate was 0.33 mL/min and the injected sample volume was 300 μL. The detection wavelength was 200 nm applying a Waters 2487 UV-detector. After each run the column was washed with 0.6 mL methanol. 3-(4-Decyloxyphenyl)propanoic acid was applied as internal standard.
Results and discussion
Chemistry
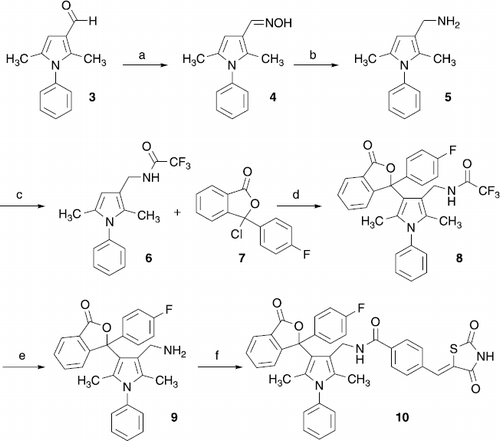
3-Pyrrol-3-yl-3H-isobenzofuran-1-one derivative 10 was synthesized by the route outlined in Scheme . 2,5-Dimethyl-1-phenylpyrrole-3-carbaldehyde (3) was reacted with hydroxylamine-HCl in the presence of Na2CO3 to give oxime 4 [Citation8]. Amine 5 was afforded by reduction of the oxime functionality of 4 by TiCl4/NaBH4. Acetylation of 5 with ethyl trifluoroacetate led to the amide 6. Treatment of this compound with 7 in the presence of SnCl4 in a Friedel-Crafts like reaction gave the intermediate 8. Compound 7 necessary for this reaction was synthesized from 2-(4-fluorobenzoyl)benzoic acid and thionyl chloride. Cleavage of the trifluoroacetyl group of 8 by KOH provided the 3-pyrrol-3-yl-3H-isobenzofuran-1-one 9, which was converted to the target compound 10 by reaction with 4-(2,4-dioxothiazolidin-5-ylidenemethyl)benzoic acid [Citation9].
Scheme 1 (a) Hydroxylamine-HCl, Na2CO3, methanol, reflux; (b) TiCl4, NaBH4, 1,2-dimethoxyethane, room temp.; (c) ethyl trifluoroacetate, THF, 0°C; (d) 7, SnCl4, 1,2-dichlororethane, reflux; (e) KOH, water, methanol, room temperature; (f) 4-(2,4-dioxothiazolidin-5-ylidenemethyl)benzoic acid, EDC, 1-hydroxybenzotriazole, DMF, room temp.
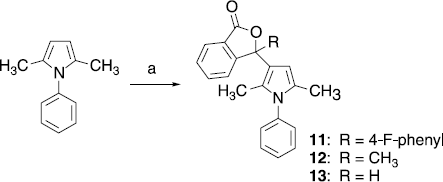
The analogous isobenzofuran-1-one 11 lacking the thiazolidine-containing side chain at the pyrrole scaffold was obtained by reaction of 2,5-dimethyl-1-phenylpyrrole with 7 applying SnCl4 as catalyst (Scheme ). Compound 12 possessing a methyl group instead of a 4-fluorophenyl substituent at position 3 of the isobenzofuran-1-one system was synthesized by heating 2,5-dimethyl-1-phenylpyrrole with 2-acetylbenzoic acid at 120–140°C. Reaction of 2,5-dimethyl-1-phenylpyrrole with 2-formylbenzoic acid led to the analogous isobenzofuran-1-one with only a pyrrole substituent at position 3.
Scheme 2 (a) 11: 7, SnCl4, 1,2-dichlororethane, 50°C; 12: 2-acetylbenzoic acid, 120–140°C; 13: 2-formylbenzoic acid, toluene, reflux.
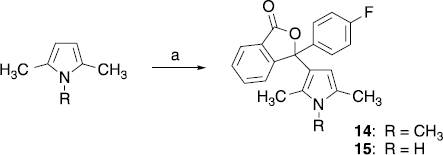
The dimethyl- and trimethylpyrrolyl-substituted 3-(4-fluorophenyl)isobenzofuran-1-ones 14 and 15 were prepared by reaction of 2,5-dimethylpyrrole and 1,2,5-trimethylpyrrole with 7, applying AlCl3 and SnCl4, respectively, as catalyst (Scheme ).
Scheme 3 (a) 14: 7, AlCl3, 1,2-dichlororethane, room temp.; 15: 7, SnCl4, 1,2-dichlororethane, 50°C.
Derivatives of 11 bearing substituents in the phenyl ring of the pyrrole nucleus (16–23,26,28,30–33,35) were prepared as shown in Scheme . Reaction of substituted aniline derivatives with hexane-2,5-dione resulted in the formation of phenyl substituted 2,5-dimethyl-1-phenylpyrroles, which were converted to the target compounds by reaction with 7 in the presence of SnCl4. The ethyl ester 23 was cleaved with aqueous KOH in ethanol to yield the carboxylic acid derivative 24. Refluxing carbonitrile 22 with solid KOH in tert-butanol led to the amide 25. The aniline derivatives 27 and 29 were synthesized by hydrolyzing their acetates 26 and 28, respectively, with half concentrated HCl in methanol or ethanol. The dihydroxy-derivative 34 was obtained by cleaving the methylether of the corresponding 3-hydroxy-4-methoxy compound 33 with 2-bromobenzo[1,3,2]dioxaborol in dry CH2Cl2.
Scheme 4 (a) p-Toluenesulfonic acid, toluene, reflux; (b) 7, SnCl4, 1,2-dichlororethane, 50°C.
Biological evaluation
All newly synthesized isobenzofuran-1-one derivatives were evaluated in an assay applying cPLA2α isolated from human platelets [Citation10]. Sonicated covesicles consisting of 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine and 1,2-dioleoyl-sn-glycerol were used as enzyme substrate. A possible problem of assays using such an aggregated form of phospholipids is that a test compound could inhibit the enzyme not by binding to its active site but merely by altering the substrate assembly and hence causing the enzyme to desorb from the lipid–water-interface. To exclude this way of action, the mole fraction of inhibitor in the interface has to be kept low [Citation11]. Thus, the highest concentration of test compounds evaluated was 33 μM, while the concentration of the vesicle forming lipids was 300 μM. The enzyme activity was determined by measuring the enzyme product arachidonic acid formed after an incubation time of 60 minutes with HPLC and UV-detection at 200 nm.
Several of the active compounds were also tested in cellular situation. In this assay cPLA2α of intact human platelets was stimulated with calcium ionophore A23187. cPLA2α-catalysed liberation of arachidonic acid from membrane phospholipids was measured with HPLC and UV-detection at 200 nm [Citation12]. To avoid metabolism of arachidonic acid via cyclooxygenase-1 and 12-lipoxygenase pathways, the dual cyclooxygenase/12-lipoxygenase inhibitor 5,8,11,14-eicosatetraynoic acid (ETYA) was added to the platelets in these experiments.
Since lysis of the platelets by a test compound may falsely indicate enzyme inhibition, we also determined the cell lytic potency of each compound by turbidimetry [Citation13]. In these experiments it was found that none of the compounds showed cell lytic properties at concentrations near its IC50 against cPLA2α.
Structure–activity relationships
Important pharmacophoric groups of the potent Shionogi cPLA2α inhibitors such as 2 () are the o-(benzoyl)benzoyl-moiety and the thiazolidinedione part of the molecules. In our effort to develop new inhibitors of cPLA2α, we wanted to synthesize compounds, in which these residues are attached to a 1-phenyl-substituted pyrrole. The pyrrole residue was chosen as central scaffold in these investigations, because structural variations of this activated heterocycle can be easily performed. During the synthesis of the concipated substances it became evident that under the reaction conditions applied the o-(benzoyl)benzoyl-residue rearranges into a 3H-isobenzofuran-1-one [Citation14–16]. Biological evaluation of the 1-phenylpyrrole 10 with 3H-isobenzofuran-1-one and thiazolidienedione bearing substituents in position 3 and 4 showed that 10 possess some inhibitory potency against cPLA2α. Its IC50-value in the isolated enzyme assay was 26 μM. In cellular situation 10 inhibited cPLA2α-mediated arachidonic acid release with an IC50 of 3.1 μM. For comparison, the known cPLA2α-inhibitor arachidonyltrifluoromethyl ketone (1) showed IC50-values of 2.3 μM and 3.3 μM in these assays ().
Table I. Inhibition of cPLA2α-activity
Since omission of the thiazolidinedione part of 10 led to a derivative with similar inhibition values (), this structurally simpler compound (11) was taken as lead in our further inhibitor development. First, we substituted the 4-fluorophenyl residue of 11 by methyl and hydrogen. In contrast to 11, which reduced cPLA2α activity at 33 μM to 40%, the obtained isobenzofuran-1-ones 12 and 13 showed no enzyme inhibition at this concentration (). Replacement of the 1-phenyl substituent of the pyrrole ring system of 11 by methyl (14) or hydrogen (15) did not change the inhibitory properties significantly. However, introduction of a benzyl moiety at the pyrrole nitrogen (16) caused a decrease of inhibitory potency, while a benzhydryl substituent (17), which is also present in several known cPLA2α inhibitors [Citation17], even led to inactivity at 33 μM.
Table II. Inhibition of cPLA2α-activity
Next, a variety of substituents were introduced into 4-position of the phenyl residue attached to the pyrrole ring of 11. All compounds synthesized were tested in the isolated enzyme assay at 33 μM. Only compounds with H-donor substituents such as COOH, CONH2, OH or NH2 in para-position showed some activity at this concentration ().
Table III. inhibition Of Cpla2α-activity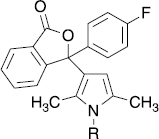
The most active of these derivatives was the 2,4-dioxothiazolidin-5-ylidenemethyl-substitued compound 35 () showing 76% inhibition at 33 μM (IC50: 7.3 μM). Interestingly, the 3-hydroxy- and the 3,4-dihydroxyphenyl substituted pyrroles 32 and 34 were less active at 33 μM against the isolated enzyme than the 4-hydroxyphenyl derivative 31.
Figure 2. Structure of 35.
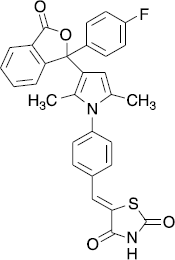
The active 3-pyrrol-3-ylisobenzofuran-1-ones with 4-hydroxyphenyl-, 4-aminophenyl and 4-(2,4-dioxothiazolidin-5-ylidenemethylphenyl)-residues (27,31,35) were also investigated in the cellular system. Here, the IC50 of hydroxy- and amino-substituted derivatives 27 and 31 was 2.8 μM each. The thiazolidinedione 35 even revealed an IC50 of 0.7 μM in this assay ().
Table IV. Inhibition of cPLA2α-activity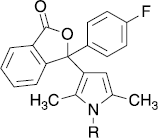
The evaluation of 3-hydroxy- and 3,4-dihydroxyphenyl-substituted compounds 32 and 34 in cellular situation led to an unexpected result. Although they were less active against the isolated enzyme, here their activity was even higher than that of the 4-hydroxy-derivative 27 (2.0 μM and 1.3 μM, respectively, vs. 2.8 μM). Taken together, the observed inhibition of cellular arachidonic acid release by the 3-pyrrol-3-ylisobenzofuran-1-ones investigated seems to be caused not only by a direct inhibition of cPLA2α. Other mechanisms may contribute to the reduction of cellular arachidonic acid liberation. Similar results have been evaluated for several indole-2-carboxylic acid derivatives before [Citation18].
In conclusion, we have found some 3-pyrrol-3-ylisobenzofuran-1-ones to be cPLA2α inhibitors. Since these isobenzofuranes show higher cPLA2α inhibitory potency in cellular situation than against the isolated enzyme, it seems to be possible that they exert their cellular activities not only by a direct interaction with the enzyme but also by other as yet unknown mechanisms.
References
- I Kudo, and M Murakami. (2002). Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat 68–69:3–58.
- CN Serhan, JZ Haeggström, and CC Leslie. (1996). Lipid mediator networks in cell signaling: Update and impact of cytokines. FASEB J 10:1147–1158.
- S Connolly, and DH Robinson. (1995). The search for inhibitors of the phospholipases A2. Expert Opin Ther Pat 5:673–683.
- JD Clark, and S Tam. (2004). Potential therapeutic uses of phospholipase A2 inhibitors. Expert Opin Ther Pat 14:937–950.
- M Lehr. (2006). Inhibitors of phospholipase A2α as potential anti-inflammatory drugs. Anti-Inflammatory Anti-Allergy Agents Med Chem 5:149–161.
- K Seno, T Okuno, K Nishi, Y Murakami, F Watanabe, T Matsuura, M Wada, Y Fujii, M Yamada, T Ogawa, T Okada, H Hashizume, M Kii, S Hara, S Hagishita, S Nakamoto, K Yamada, Y Chikazawa, M Ueno, I Teshirogi, T Ono, and M Ohtani. (2000). Pyrrolidine inhibitors of human cytosolic phospholipase A2. J Med Chem 43:1041–1044.
- S Connolly, C Bennion, S Botterell, PJ Croshaw, C Hallam, K Hardy, P Hartopp, CG Jackson, SJ King, L Lawrence, A Mete, D Murray, DH Robinson, GM Smith, L Stein, I Walters, E Wells, and WJ Withnall. (2002). Design and synthesis of a novel and potent series of inhibitors of cytosolic phospholipase A2 based on a 1,3-disubstituted propan-2-one skeleton. J Med Chem 45:1348–1362.
- R Rips, and NP Buu-Hoi. (1959). Aldehydes derived from 1,2,5-trisubstituted pyrroles. J Org Chem 24:372–374.
- K Seno, T Okuno, K Nishi, Y Murakami, K Yamada, S Nakamoto, and T Ono. (2001). Pyrrolidine inhibitors of human cytosolic phospholipase A2 part 2: Synthesis of potent and crystallized 4-triphenylmethylthio derivative “pyrrophenone”. Bioorg Med Chem Lett 11:587–590.
- M Schmitt, and M Lehr. (2004). HPLC assay with UV spectrometric detection for the evaluation of inhibitors of cytosolic phospholipase A2. J Pharm Biomed Anal 35:135–142.
- F Ghomashchi, R Loo, J Balsinde, F Bartoli, R Apitz-Castro, JD Clark, EA Dennis, and MH Gelb. (1999). Trifluoromethyl ketones and methyl fluorophosphonates as inhibitors of group IV and VI phospholipases A2: Structure-function studies with vesicle, micelle, and membrane assays. Biochim Biophys Acta 1420:45–56.
- M Lehr, and A Schulze Elfringhoff. (2000). Comparison of the inhibition of the cytosolic phospholipase A2-mediated arachidonic acid release by several indole-2-carboxylic acids and 3-(pyrrol-2-yl)propionic acids in bovine and in human platelets. Arch Pharm Pharm Med Chem 333:312–314.
- M Lehr, and K Griessbach. (1999). Cell lytic and cPLA2-inhibitory properties in bovine platelets of the commercially available cPLA2 inhibitors, arachidonyltrifluoromethyl ketone, methyl arachidonylfluorophosphonate and palmityltrifluoromethyl ketone. Pharm Pharmacol Commun 5:389–393.
- A Haller, and A Guyot. (1901). Investigations on the tautomerism of o-benzoylbenzoic acid. Bull Soc Chim Paris 25:49–56.
- W Graf, E Girod, E Schmid, and WG Stoll. (1959). The structure of benzophenone-2-carboxylic acid derivatives. Helv Chim Acta 42:1085–1101.
- DM Knauss, and JT Bender. (2002). Phthalide as an activating group for the synthesis of poly(aryl ether phthalide)s by nucleophilic aromatic substitution. J Polym Sci Part A: Polym Chem 40:3046–3054.
- JC McKew, MA Foley, P Thakker, ML Behnke, FE Lovering, FW Sum, S Tam, K Wu, MW Shen, W Zhang, M Gonzalez, S Liu, A Mahadevan, H Sard, SP Khor, and JD Clark. (2006). Inhibition of cytosolic phospholipase A2α: hit to lead optimization. J Med Chem 49:135–158.
- A Ghasemi, A Schulze Elfringhoff, and M Lehr. (2005). Structure–activity relationship studies of 3-dodecanoylindole-2-carboxylic acid inhibitors of cytosolic phospholipase A2α-mediated arachidonic acid release in intact platelets: variation of the keto moiety. J Enz Inhib Med Chem 20:429–437.