Abstract
Synthesis and biological evaluation of an spiro[benzoxazepine-piperidine] class of Aβ-peptide production inhibitors for treatment of Alzheimer's disease are described.
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease characterized by memory loss and cognitive decline. The amyloid peptide (Aβ) is the major component of extracellular seniles plaques found in Alzheimer's brain patients; this soluble Aβ has been found to be neurotoxic in vitro [Citation1]. Cleavages of amyloid peptide precursor protein (APP) by both β- and γ-secretases release Aβ peptide fragments. These cleavage events are thought to play a key role in the neurodegenerative pathways responsible for AD pathology. Inhibition of proteases responsible for the unfavourable cleavage of APP has been reported to be an attractive point of intervention which could alter pathophysiology of the disease rather than act as palliative treatment.
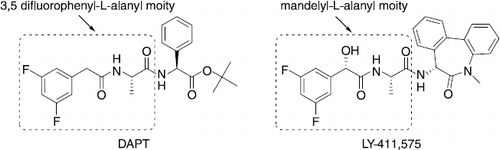
From a literature search, it was observed that N-[N-(3,5-difluorophenylacetyl-l-alanyl)]-S-phenylglycine tert-butyl ester known as DAPT [Citation2,3] (see ) and some related analogues in which the motifs 3,5-difluorophenylacetyl-l-alanyl or 3,5-difluoromandelyl-l-alanyl are coupled to bulky heterocyclic moieties [Citation4–7] are among the most potent inhibitors of Aβ peptide production through γ-secretase inhibition. For example LY-411,575 (see ) which is a potent Aβ peptide inhibitor in vitro and in vivo, includes in its structure the 3,5-difluoromandelyl-l-alanyl motif linked to a benzodiazepine moiety [Citation8]. From a synthetic point of view, this promising series of derivatives are rather complex molecules since they present three chiral centers, and appeared to present several potential liabilities that may limit their use for the treatment of AD.
Figure 1. Structure of DAPT and LY-411, 575.
Materials and methods
Herein we report the synthesis and the cellular inhibitory activity of new 4,5-dihydro-3H-spiro[1,5-benzoxazepine-2,4′-piperidine] substituted at the position R2 by a simple 3-hydroxy-3-phenyl propyl moiety on the production of Aβ-peptide (general structure shown on ).
Figure 2. General structure of spiro[benzoxazepine-piperidine].
![Figure 2. General structure of spiro[benzoxazepine-piperidine].](/cms/asset/b354054e-2e02-48bf-89e0-6dd7f60ab3f2/ienz_a_283396_f0002_b.gif)
The bulky heterocyclic moiety 4,5-dihydro-3H-spiro[1,5-benzoxazepine-2,4′-piperidine] was selected because:
– it has been observed that bioactive drugs incorporating in their structure a piperidinyl ring linked to an other cycle through a spiranyl carbon, can adopt specific conformations in which the nitrogen electron doublet can have different orientations [Citation9,10].
– this scaffold presents different positions on which substituents (R1, R2 and R3) could be introduced.
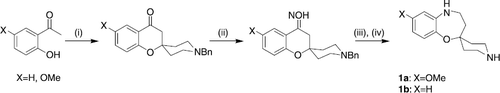
The spiro[benzoxazepine-piperidine] scaffold (see Scheme ) has been synthesized according to reported procedure [Citation15].
Scheme 1 Reagents and conditions: (i) N-benzyl piperidone, pyrrolidine, MeOH, reflux; (ii) HCl.H2N-OH, pyridine, EtOH, reflux; (iii) DIBAH, CH2Cl2, 0°C; (iv) H2, Pd(OH)2 (10%), MeOH, rt, 80%.
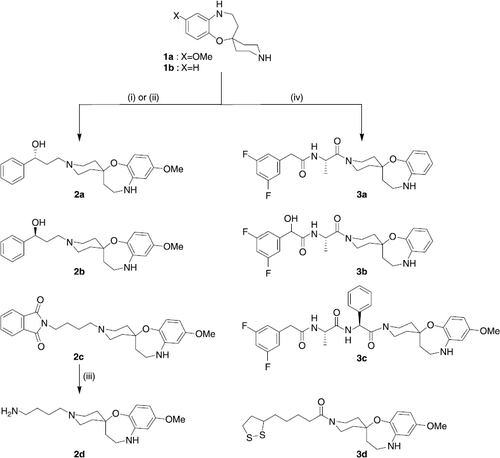
The keys intermediates 1a, 1b were subsequently functionalized with various halide derivatives or carboxylic acids (Scheme ). Compound 1a was N-alkylated [Citation16] with different alkyl halides such as (S)-( − )-3-chloro-1-phenyl-1-propanol, or (R)-(+)-3-chloro-1-phenyl-1-propanol and N-(5-bromopentyl)phthalimide. This reaction performed in the presence of potassium carbonate in acetonitrile as solvent gives respectively compounds 2a, 2b, and 2c. After removal of the phthalimide protecting group of 2c by hydrazine monohydrate in ethanol [Citation17], compound 2d was obtained. Acylation of the secondary amine 1a or 1b with acids: N-(3,5-Difluorophenylacetyl)-l-alanine, N-(3,5-Difluoromandelyl)-l-alanine [Citation18], DAPT derivative [Citation5] and (+/ − )-α-Lipoic acid [Citation19], in the presence of BOP and diisopropylethylamine afford the desired analogues 3a, 3b, 3c and 3d.
Scheme 2 Reagents and conditions: (i) ClCH2CH2CH(Ph)OH (S) or (R), CH3CN, K2CO3, 35°C, 24 h; (ii) Br(CH2)4-phthalimide, CH3CN, K2CO3, reflux, 12 h; (iii) H2NNH2, 24 h; (iv) RCO2H, BOP, DIEA, CH2Cl2, rt, 18 h.
Synthesis
General procedure for compound 2a–2c
To a stirred solution of 1a (0.16 g, 0.65 mmol) in i-PrOH (5 mL) was added K2CO3 (0.18 g, 1.29 mmol). A solution of the (R)-( − )-3-chloro-1-phenyl-1- propanol (0.11 g, 0.65 mmol) in acetonitrile (3 mL) was added slowly, and the reaction mixture was stirred at room temperature for 18 h. After concentration under reduced pressure, the residue was purified by column chromatography (silica gel, CH2Cl2/MeOH, 95:5) to give the desired compound 2a as a white solid.
2a
(Yield) 13%, Rf 0.86 (CH2Cl2/MeOH 95:5). 1H NMR (CDCl3, 250 MHz) δ: 1.61–1.79 (m 2H), 1.90–2.08 (m, 8H), 2.11–2.88 (m, 6H), 3.31 (t, J = 5.4 Hz, 2H), 3.72 (s, 3H), 4.98 (m, J = 4.6 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 6.23 (dd, J = 2.9, 8.7 Hz, 1H), 6.78 (d, J = 8.7 Hz, 1H), 7.21–7.36 (m, 5H). 13C NMR (CDCl3, 62.9 MHz) δ: 33.6, 34.4, 34.5, 40.7, 48.7, 50.1, 55.3, 56.8, 74.7, 75.7, 77.2, 103.9, 104.2, 124.6, 125.5 (2C), 126.9, 128.2 (2C), 137.2, 143.1, 144.7, 156.3. ESI-MS m/z [M + H]+ = 383. Anal. Calcd. for C23H30N2O3: C 72.22, H 7.91, N 7.32. Found: C 75.36, H 7.55, N 7.07%.
2b
(Yield) 13%, Rf 0.86 (CH2Cl2/MeOH 95:5). 1H NMR (CDCl3, 250 MHz) δ: 1.61–1.79 (m 2H), 1.90–2.08 (m, 8H), 2.11–2.88 (m, 6H), 3.31 (t, J = 5.4 Hz, 2H), 3.72 (s, 3H), 4.98 (m, J = 4.6 Hz, 1H), 6.17 (d, J = 2.8 Hz, 1H), 6.23 (dd, J = 2.9, 8.7 Hz, 1H), 6.78 (d, J = 8.7 Hz, 1H), 7.21–7.36 (m, 5H). 13C NMR (CDCl3, 62.9 MHz) δ: 33.6, 34.4, 34.5, 40.7, 48.7, 50.1, 55.3, 56.8, 74.7, 75.7, 77.2, 103.9, 104.2, 124.6, 125.5 (2C), 126.9, 128.2 (2C), 137.2, 143.1, 144.7, 156.3. ESI-MS m/z [M + H]+ = 383. Anal. Calcd. for C23H30N2O3: C 72.22, H 7.91, N 7.32. Found: C 75.52, H 7.65, N 7.15%.
2c
(Yield) 12%, 1H NMR (CDCl3, 250 MHz) δ: 1.22–1.51 (m, 2H), 1.55–1.75 (m, 6H), 1.88 (t, J = 5.4 Hz, 2H), 1.92–2.03 (m, 2H), 2.36–2.49 (m, 4H), 2.63–2.71 (m, 2H), 3.28 (t, J = 5.4 Hz, 2H), 3.65–3.68 (m, 2H), 3.71 (s, 3H), 6.16 (d, J = 3.0 Hz, 1H), 6.24 (dd, J = 2.9, 5.7 Hz, 1H), 6.82 (d, J = 8.7 Hz, 1H), 7.68–7.72 (m, 2H), 7.81–7.87 (m, 2H). 13C NMR (CDCl3, 62.9 MHz) δ: 24.9, 26.4, 28.5, 32.4, 34.7, 38.0, 40.8 (2C), 41.8, 49.4 (2C), 55.3, 58.5, 76.1, 103.9, 104.1, 123.2 (2C), 124.8, 132.1, 133.8 (2C), 137.6, 143.2, 156.1, 168.4 (2C).ESI-MS m/z [M + H]+ = 464. Anal. Calcd. for C27H33N3O4: C 72.22, H 7.91, N 7.32. Found: C 75.52, H 7.65, N 7.15%.
2d
To a stirred solution of 2c (50 mg, 0.10 mmol) in EtOH (5 mL) was added anhydrous hydrazine (0.15 mmol). The reaction mixture was stirred at reflux for 2 h. and then allowed to cool to room temperature. After concentration under reduced pressure, the residue was purified by column chromatography (silica gel, CH2Cl2/MeOH, 95:5) to give quantitatively the desired compound 2d (33 mg) as a yellow oil. Rf 0.67 (CH2Cl2/MeOH, 95:5). 1H NMR (CDCl3, 250 MHz) δ: 1.22–1.51 (m, 2H), 1.55–1.75 (m, 6H), 1.88 (t, J = 5.4 Hz, 2H), 1.92–2.03 (m, 2H), 2.33–2.47 (m, 4H), 2.61–2.75 (m, 4H), 3.28 (t, J = 5.4 Hz, 2H), 3.71 (s, 3H), 6.16 (d, J = 3.0 Hz, 1H), 6.24 (dd, J = 2.9, 5.7 Hz, 1H), 6.84 (d, J = 8.7 Hz, 1H). 13C NMR (CDCl3, 62.9 MHz) δ: 24.9, 26.9, 26.7, 30.3, 32.8, 40.8 (2C), 41.7, 49.5 (2C), 55.3, 58.9, 76.1, 103.9, 104.1, 124.8, 137.6, 143.2, 156.1. ESI-MS m/z [M + H]+ = 334. Anal. Calcd. for C19H31N3O2: C 68.43, H 9.37, N 12.60. Found: C 68.85, H 9.34, N 12.27%.
General procedure for compound 3a–3d
The 3,5-difluorophenylacetyl alanine acid (0.11 g, 0.46 mmol) was dissolved in freshly distilled CH2Cl2 (5 mL) in the presence of BOP reagent (0.24 g, 0.55 mmol). The reaction mixture was cooled to 0°C and then DIEA (80 μL, 0.46 mmol) was added dropwise. The reaction mixture was stirred for 1 h at room temperature and then cooled once again to 0°C. A CH2Cl2 solution of the benzoxazepine piperidine derivative 1a (0.30 g, 0.46 mmol) and DIEA (240 μL, 1.38 mmol) was added dropwise. The solution was allowed to warm and stirred overnight at room temperature. The solvent was removed under reduced pressure and the residue was dissolved in EtOAc (20 mL). The organic layer was washed successively by using H2O (10 mL), brine (10 mL), 5% aqueous NaHCO3 (2 × 10 mL), and brine (10 mL), was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude residue was purified by flash chromatography (CH2Cl2/MeOH, 95:5) to give the title compound 3a (76 mg, yield: 37%) as a white solid. Rf 0.48 (CH2Cl2/MeOH, 95: 5). 1H NMR (CDCl3,250 MHz) δ: 1.32 (d, J = 6.6 Hz, 3H), 1.38–1.46 (m, 2H), 1.88–1.92 (m, 2H), 2.05–2.16 (m, 2H), 3.05–3.19 (m, 1H), 3.28–3.33 (m, 2H), 3.51 (s, 2H), 3.59–3.64 (m, 2H), 4.25–4.42 (brs, 1H), 4.85–4.90 (m, 1H), 6.63–6.92 (m, 7H), 8.05 (brs, 1H). 13C NMR (CDCl3, 62.9 MHz) δ:18.5, 34.4, 35.6, 38.4, 40.7, 41.3, 41.6, 43.0, 45.4, 73.3, 102.7 (t, J = 25.5 Hz, 1C), 112.0, 112.4, 118.7, 121.2 (d, J = 24.8 Hz, 2C), 124.4, 138.4 (t, J = 9.7 Hz, 1C), 142.2, 143.4, 163.2 (dd, J = 246.8 Hz, 2C), 168.5, 170.2. ESI-MS m/z [M + H]+ = 444. Anal. Calcd. for C24H27F2N3O3: C 65.00, H 6.14, N 9.47. Found: C 65.37, H 6.22, N 9.13%.
3b
(Yield) 38%,. Rf 0.47 (CH2Cl2/MeOH, 95:5). 1H NMR (CDCl3, 250 MHz) δ: 1.25–1.60 (m, 5H), 1.88–1.93 (m, 2H), 2.04–2.12 (m, 2H), 3.07–3.19 (m, 2H), 3.25–3.35 (m, 2H), 3.56–3.81 (m, 3H), 4.31–4.42 (m, 1H), 4.83–4.89 (m, 1H), 5.04 (brs, 1H), 6.64–7.41 (m, 7H), 7.95 (brs, 1H). 13C NMR (CDCl3, 62.9 MHz) δ: 18.5, 29.7, 34.4, 35.6, 38.4, 40.7, 41.6, 45.2, 72.9, 76.32, 103.5 (t, J = 25.5 Hz, 1C), 109.4, 109.9, 118.8, 121.3 (d, J = 24.8 Hz, 2C), 124.4, 138.3 (t, J = 9.7 Hz, 1C), 142.2, 143.4, 163.2 (dd, J = 246.8 Hz, 2C), 170.3, 170.8. ESI-MS m/z [M + H]+ = 460. Anal. Calcd. for C24H27F2N3O4: C 62.73, H 5.92, N 9.15. Found: C 62.45, H 6.08, N 9.12%.
3c
(Yield) 45%, Rf 0.71 (EtOAc). 1H NMR (CDCl3, 250 MHz) δ: 1.30 (d, J = 6.8 Hz, 3H), 1.58–1.70 (m, 2H), 1.80–1.97 (m, 4H), 2.48–2.71 (m, 4H), 3.21 (t, J = 5.4 Hz, 2H), 3.61–3.69 (m, 5H), 4.40–4.49 (m, 1H), 5.75 (s, 1H), 6.13–6.42 (m, 3H), 6.74–6.78 (m, 1H), 6.89–7.08 (m, 2H), 7.29–7.50 (m, 5H). ESI-MS m/z [M + H]+ = 607. Anal. Calcd. for C33H36F2N4O5: C 65.33, H 5.98, N 9.24. Found: C 65.17, H 5.63, N 9.51%.
3d
(Yield) 52%, Rf 0.51 (CH2Cl2/MeOH 95:5). 1H NMR (CDCl3, 250 MHz) δ: 1.39–1.50 (m, 4H), 1.57–1.73 (m, 6H), 1.84–2.05 (m, 3H), 2.29–2.49 (m, 5H), 2.97–3.18 (m, 2H), 3.26–3.31 (m, 2H), 3.48–3.58 (m, 3H), 3.69 (s, 3H), 6.16 (d, J = 2.8 Hz, 1H), 6.22 (dd, J = 2.9, 5.7 Hz, 1H), 6.78 (d, J = 8.5 Hz, 1H). 13C NMR (CDCl3, 62.9 MHz) δ: 25.1, 29.2, 33.0, 34.3, 34.7, 35.7, 37.7, 38.5, 40.3, 40.6, 41.5, 41.7, 55.4, 56.5, 76.4, 104.0, 104.3, 124.5, 137.3, 143.0, 156.4, 171.2. ESI-MS m/z [M + H]+ = 437. Anal. Calcd. for C22H32N2O3S2: C 60.52, H 7.39, N 6.42. Found: C 60.37, H 7.45, N 6.01%.
Results and discussion
The new synthesized compounds were first assayed for their inhibitory properties on Aβ peptide production using a conventional cellular model [Citation20]. Inhibitory potencies for each compound was determined using: HEK 293 cells over expressing APP, according to known literature procedure [Citation20–22]. Among the tested compounds, 2a and 2b were the most potent to inhibit Aβ peptide production with IC50 values ranging between 0.2 and 0.6μM ().
Table I. Inhibitory activities of spiro[benzoxazepine-piperidine] derivatives.
Since Aβ-peptide production can result from cleavages of the amyloid peptide precursor protein (APP) by both β- and γ-secretases, compounds were also assayed as β-secretase inhibitors, using a fluorescence resonance energy transfer (FRET) assay [Citation18]. In this enzymatic assay, no one of the compounds found active on Aβ peptide production, were found β-secretase inhibitors at least up to 100μM. These results suggest that the observed inhibition of Aβ-peptide production, by analogues 2a and 2b did not result from β-secretase inhibition but more likely from γ-secretase inhibition.
These results, suggest that substitution on the nitrogen atom of the piperidine ring greatly influences Aβ-peptide inhibitory activity of the resulting derivatives. Inhibitors 2a and 2b, substituted on the nitrogen atom of the piperidinyl ring, by a chiral moiety 3-hydroxy-3-phenyl-propyl group appeared equipotent to their corresponding 3a and 3b derivatives which are substituted by a difluoro phenylacetyl-l-alanyl moiety. It should be also underlined that both enantiomers 2a and 2b are equipotent in our assay. In contrast analogues bearing at the N-4 position of the piperazine ring other substituents such as N-acyl-ω-aminobutyl 2d or N-acyl-lipoyl 3d were denied of any inhibitory activity.
Conclusions
Based on the molecular structure simplicity and relatively high potency of analogues 2a and 2b, it could be of interest to investigate the effects of this 3-hydroxy-3-phenyl-propyl substituent on other positions of the 4,5-dihydro-3H-spiro[1,5-benzoxazepine-2,4′-piperidine] scaffold. These preliminary results are of interest since these new analogues bearing a heterocyclic moiety linked to a simple side chain containing only one chiral center represent new cellular Aβ-peptide inhibitor prototype, which structure can be optimized. Additional in vitro screening assays such as cell free and NICD (Notch intracellular domain) assays are needed in order to evaluate their potential for treatment of AD [Citation24].
Acknowledgements
We are grateful to Professor K. Dudley (Université de la Méditerranée, IBDML) for the preparation of the manuscript.
References
- Y Gong, L Chang, KL Viola, PN Lacor, MP Lambert, CE Finch, GA Krafft, WL Klein. Alzheimer's disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA (2003); 100(18):10417–10422.
- ER Siemers, JF Quinn, J Kaye, MR Farlow, A Porsteinsson, P Tariot, P Zoulnouni, JE Galvin, DM Holtzman, DS Knopman, et al. Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology (2006); 66(4):602–604.
- HF Dovey, V John, JP Anderson, LZ Chen, PD Andrieu, LY Fang, SB Freedman, B Folmer, E Goldbach, EJ Holsztynska, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem (2001); 76(1):173–181.
- B Schmidt. Aspartic proteases involved in Alzheimer's disease. Chembiochem (2003); 4(5):366–378.
- T Kan, Y Tominari, K Rikimaru, Y Morohashi, H Natsugari, T Tomita, T Iwatsubo, T Fukuyama. Parallel synthesis of DAPT derivatives and their g-secretase-inhibitory activity. Bioorg Med Chem Lett (2004). 14(8):1983–1985.
- CVC Prasad, S Vig, W Smith David, Q Gao, T Polson Craig, A Corsa Jason, L Guss Valerie, A Loo, M Barten Donna, M Zheng, et al. 2,3-Benzodiazepin-1,4-diones as peptidomimetic inhibitors of gamma-secretase. Bioorg Med Chem Lett (2004); 14(13):3535–3538.
- G Larbig, A Zall, B Schmidt. Inhibitors designed for presenilin 1 by means of aspartic acid activation: Preliminary communication. Helv Chim Acta (2004); 87(9):2334–2340.
- GT Wong, D Manfra, FM Poulet, Q Zhang, H Josien, T Bara, L Engstrom, M Pinzon-Ortiz, JS Fine, HJ Lee, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem (2004); 279(13):12876–12882.
- MJ Mokrosz, B Duszynska, AJ Bojarski, JL Mokrosz. Structure-activity relationship studies of CNS agents–XVII. Spiro[piperidine-4′,1-(1,2,3,4-tetrahydro-beta-carboline)] as a probe defining the extended topographic model of 5-HT1A receptors. Bioorg Med Chem (1995); 3(5):533–538.
- L Yang, G Morriello, K Prendergast, K Cheng, T Jacks, WW Chan, KD Schleim, RG Smith, AA Patchett. Potent 3-spiropiperidine growth hormone secretagogues. Bioorg Med Chem Lett (1998); 8(1):107–112.
- M Rosini, V Andrisano, M Bartolini, ML Bolognesi, P Hrelia, A Minarini, A Tarozzi, C Melchiorre. Rational approach to discover multipotent anti-Alzheimer drugs. J Med Chem (2005); 48(2):360–363.
- L Packer, EH Witt, HJ Tritschler. Alpha-lipoic acid as a biological antioxidant. Free Radic Biol Med (1995); 19(2):227–250.
- L Packer. Alpha-lipoic acid: A metabolic antioxidant which regulates NF-kappa B signal transduction and protects against oxidative injury. Drug Metab Rev (1998). 30(2):245–275.
- K Hager, A Marahrens, M Kenklies, P Riederer, G Munch. Alpha-lipoic acid as a new treatment option for Azheimer type dementia. Arch Gerontol Geriatr (2001); 32(3):275–282.
- Y Laras, N Pietrancosta, V Moret, S Marc, C Garino, A Rolland, V Monnier, J Kraus. Synthesis of new substituted 4,5-dihydro-3h-spiro[1,5]-benzoxazepine-2,4′-piperidine and biological properties. Aust J Chem (2006); 59(11):812–818.
- G Campiani, S Butini, F Trotta, C Fattorusso, B Catalanotti, F Aiello, S Gemma, V Nacci, E Novellino, JA Stark, et al. Synthesis and pharmacological evaluation of potent and highly selective D3 receptor ligands: Inhibition of cocaine-seeking behavior and the role of dopamine D3/D2 receptors. J Med Chem (2003); 46(18):3822–3839.
- M Govoni, RA Bakker, I van de Wetering, MJ Smit, WM Menge, H Timmerman, S Elz, W Schunack, R Leurs. Synthesis and pharmacological identification of neutral histamine H1-receptor antagonists. J Med Chem (2003); 46(26):5812–5824.
- N Pietrancosta, G Quelever, Y Laras, C Garino, S Burlet, JL Kraus. Design of beta-secretase inhibitors by introduction of a mandelyl moiety in DAPT analogues. Aust J Chem (2005); 58(8):585–594.
- PR Andres, R Lunkwitz, GR Pabst, K Bohn, D Wouters, S Schmatloch, US Schubert. New 4′-functionalized 2,2′: 6′,2″-terpyridines for applications in macromolecular chemistry and nanoscience. Eur J Org Chem (2003); (19):3769–3776.
- T Tomita, K Maruyama, TC Saido, H Kume, K Shinozaki, S Tokuhiro, A Capell, J Walter, J Grunberg, C Haass, et al. The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid beta protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci USA (1997); 94(5):2025–2030.
- A Asami-Odaka, Y Ishibashi, T Kikuchi, C Kitada, N Suzuki. Long amyloid b-protein secreted from wild-type human neuroblastoma IMR-32 cells. Biochemistry (1995); 34(32):10272–10278.
- M Ito, S Yamamoto, K Nimura, K Hiraoka, K Tamai, Y Kaneda. Rad51 siRNA delivered by HVJ envelope vector enhances the anti-cancer effect of cisplatin. J Gene Med (2005). 7(8):1044–1052.
- LD Plant, JP Boyle, IF Smith, C Peers, HA Pearson. The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J Neurosci (2003); 23(13):5531–5535.
- M Hellstrom, LK Phng, JJ Hofmann, E Wallgard, L Coultas, P Lindblom, J Alva, AK Nilsson, L Karlsson, N Gaiano, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature (2007); 445(7129):776–780.