Abstract
A series of new ursolic and oleanolic acids derivatives was synthesized via ursolic or oleanolic acids, previously extracted from South American Ilex species. These new compounds were tested for in vitro antiparasitic activity on Leishmania amazonensis and Leishmania infantum strains. Some of these compounds showed activity against the promastigote forms of L. amazonensis or L. infantum, with IC50 ranging from 5 to 12 μM. As expected, most of the compounds showed a significant level of cytotoxicity against monocytes (IC50 = 2-50 μM). From a structure-activity relationships point of view, these pharmacological results enlightened mainly the importance of an acetylation at position 3 of the oleanolic acid skeleton in the activity against the L. amazonensis strain, and of a bis-(3-aminopropyl)piperazine moiety on the carboxylic function of ursolic acid against the L. infantum strain.
Introduction
Protozoan parasites affect 3 billion people, with malaria and trypanosomatid parasites causing the greatest morbidity. The leishmaniasis is a complex of disease syndromes caused by at least 20 species of the protozoan parasite of the genus Leishmania [Citation1,Citation2]. The disease is distributed worldwide in both the old and new worlds, but mainly in the tropics and subtropics. According to recent World Health Organization reports, 88 countries are affected, comprehending 12 million infected people worldwide, with approximately 350 million people at risk. The incidence is increasing worldwide, with 1-2 million new cases registered annually, despite all efforts being made to fight the disease Citation1Citation2Citation3Citation4Citation5. It occurs in two major forms; cutaneous/muco-cutaneous (CL) and visceral leishmaniasis (VL, or Kala-azar), depending upon parasite species. Leishmania donovani and L. infantum are major causative agents of VL, while L. major, L. tropica and L. aethiopica, L. braziliensis, L. panamensis, L. amazonensis and L. mexicana cause CL [Citation2,Citation4]. Therapy of patients with leishmaniasis is still a serious problem. Historically, leishmaniasis chemotherapy has been based on the use of toxic heavy metals, particularly antimony compounds. In fact, the drugs for leishmaniasis's treatment of all their clinical forms are meglumine antimonite (Glucantime®) and sodium stibogluconate (Pentostam®), despite the fact that they exhibit renal and cardiac toxicities [Citation2,Citation4,Citation6]. When this kind of treatment is not effective, alternative drugs include pentamidine (Pentacarinat®) and amphotericin B (Fungizone®, AmBisome®), also very toxic with serious side effects [Citation7]. Miltefosine, a phosphocholine analogue originally developed as an anticancer agent, has been found to be highly effective against leishmaniasis in vitro and in vivo. Now, this compound is the only oral agent against cutanous and visceral leishmaniasis, although presenting severe gastrointestinal problems [Citation8]. As the leishmaniasis chemotherapy is still inefficient, there is an urgent need for the development of new, efficient, and safe drugs [Citation4,Citation6,Citation9]. Traditional and ethnic medicine is often a good source for researchers looking for bioactive substances. Leishmaniasis is not an exception, and plants have been used for the treatment of people living far from modern medicine. Such compounds belong to the following groups: alkaloids, terpenes, quinines, lactones, coumarins, acetogenins of Annonaceae, chalcones, tetralones, lignans, saponins and triterpenes Citation9Citation10Citation11Citation12Citation13Citation14Citation15. For instance, some triterpenoids active against leishmaniasis were isolated from Pourouna guianensis through a bioactivy-guided fractionation process from an investigation in Brazil [Citation16]. The most active pentacyclic triterpenes isolated from P. guianensis (Moraceae) were found to be ursolic acid (3-β-hydroxy-urs-12-en-28-oic acid) and its isomer oleanolic acid (3-β-hydroxy-olean-12-en-28-oic acid), two common phytochemicals, which are naturally found in various plants such as sea-weeds, the wax-like coatings of fruits and many medicinal herbs. In a previous study, ursolic acid, isolated from Jacaranda copaia (Aublet) D. Don in Bolivia, already showed an interesting in vitro antileishmanial activity [Citation17]. These two triterpenoid acids exist in plants in the form of free acids or aglycones for triterpenoid saponins. The traditional uses of plants containing ursolic and oleanolic acids in folk medicine are multiple; they have been investigated as anti-inflammatory, hepatoprotective, gastroprotective, anti-HIV, antitumor, hypolipidemic agents Citation10Citation17Citation18Citation19Citation20Citation21Citation22Citation23.
Very recently, we first reported the synthesis of a series of N-[1-(4-(3-aminopropyl)piperazinyl)propyl] ursolic acid derivatives via ursolic acid, previously isolated from the South American species Ilex paraguariensis and Ilex dumosa, and its in vitro antiparasitic activity on Plasmodium falciparum strains. In these series, seven new bis-(aminopropyl)piperazinyl analogues of ursolic acid showed significant activity in the nanomolar range suggesting the antiprotozoal activity role of this pharmacophoric group [Citation24,Citation25]. Based on these previous studies, we further examined members of these pentacyclic triterpenes family with respect to their inhibitory effects on Leishmania amazonensis and Leishmania infantum. We present here the results of our investigation in this series.
Materials and methods
Chemistry
Instrumentation
Melting points were determined with an SM-LUX-POL Leitz hot-stage microscope and reported uncorrected. Infrared (IR) spectra were determined in KBr discs on a JASCO FT/IR-4200 spectrometer. Optical rotations were measured on a Perkin-Elmer 241 polarimeter; values are given in 10− 1deg.cm2.g− 1. NMR spectra were recorded on a BRUKER AVANCE 500 spectrometer (500 MHz). Chemical shifts refer to tetramethylsilane which was used as an internal reference. Nuclear magnetic resonance (1H and 13C NMR) spectra were recorded on a BRUKER AVANCE 500 spectrometer (500 MHz) and tetramethylsilane (TMS) was used as an internal standard. 1H NMR analyses were obtained at 500 MHz (s: singlet, d: doublet, t: triplet, dd: double doublet, td: triple doublet, m: multiplet), whereas 13C NMR analyses were obtained at 125 MHz. J values are given in Hertz. The chemical shifts (δ) are given in parts per million relative to TMS (δ = 0.00). Mass spectra were recorded on a Micromass-Waters Q-TOF Ultima spectrometer. Analytical TLC was carried out on 0.25 precoated silica gel plates (POLYGRAM SIL G/UV254) with visualisation by irradiation with a UV lamp. Silica gel 60 (70-230 mesh) was used for column chromatography.
N-{3-[4-(3-Aminopropyl)piperazinyl]propyl}ursolamide (6) [Citation25,Citation26]
To a solution of N-{3-[4-(3-aminopropyl)piperazinyl]propyl}-3-O-acetylursolamide 4 (1.48 g, 0.1 mmol) in dry ethanol (2 mL) was added a 1M aqueous solution of sodium hydroxide (1 mL). The resulting solution was refluxed for 2 h, and concentrated under vacuum. The aqueous layer was extracted with dichloromethane (3 × 1 mL). The organic layers were dried over anhydrous sodium sulfate. The solvent was removed under vacuum to afford 6. Yield: 70%, white crystals, mp = 140°C; ![]() = +56 (c 0.2, CHCl3); IR νmax (KBr)/cm− 1 3385 (NH and OH), 1700 (CO); 1H NMR δ (500 MHz, CDCl3) 0.78 (m, 1H, CH-5), 0.82 (s, 3H, CH3-24), 0.83 (d, J 7.0 Hz, 3H, CH3-30), 0.91 (d, J 6.6 Hz, 3H, CH3-29); 0.93 (m, 1H, CH2-1), 0.94 (s, 3H, CH3-25), 0.96 (m, 1H, CH-20), 0.94 (m, 1H, CH2-11), 0.98 (s, 3H, CH3-23), 1.02 (s, 3H, CH3-26), 1.14 (s, 3H, CH3-27), 1.21 (m, 1H, CH2(Hα)-7), 1.33 (m, 1H, CH-19), 1.41 (m, 2H, CH2-2), 1.55 (m, 4H, CH2(Hβ)-6, CH2(Hβ)-7, CH-9, CH2(Hα)-21), 1.62 (m, 4H, CH2(Hβ)-1, CH2(Hα)-15, CH2(Hβ)-16 and CH2(Hβ)-21), 1.70 (m, 5H, CH2-γ, CH2-γ′ and CH2(Hα)-6), 1.77 (td, J 14.0 and 6.0 Hz, 1H, CH2(Hα)-16), 1.83 (m, 2H, CH2-22), 2.00 (m, 1H, CH2(Hβ)-15), 2.52 (m, 13H, CH2-α1, CH2-α1′, CH2-α2, CH2-α2′, CH2-β, CH2-β′ and CH-18), 3.03 (m, 2H, CH2-δ′), 3,25 (dd, J 13.8 and 6.4 Hz, 1H, CH-3), 3.34 (m, 2H, CH2-δ), 5.35 (tl, 1H, CH-12), 6.46 (s, 1H, NH); 13C NMR δ (125 MHz, CDCl3) 15.8 (C-25), 15.9 (C-26), 16.8 (C-24), 17.4 (C-29), 18.7 (C-6), 21.7 (C-30), 23.8 (C-11 and C-27), 25.1 (C-16), 27.2 (C-γ), 28.3 (C-23), 28.4 (C-2), 28.5 (C-15), 31.3 (C-21), 33.2 (C-7), 31.5 (C-γ′), 37.4 (C-10), 37.8 (C-22), 38.9 (C-8), 39.0 (C-1), 39.2 (C-δ), 39.3 (C-4), 39.5 (C-19), 39.9 (C-20), 40.1 (C-δ′), 42.8 (C-14), 47.9 (C-9 and C-17), 53.4 (C-18), 53.7 (C-α1, C-α1′, C-α2, C-α2′, 55.6 (C-5), 56.5 (C-β and C-β′), 79.3 (C-3), 123.0 (C-12), 140.1 (C-13), 182.2 (C-28). HRMS (ESI-MS, m/z), (M + H)+: Calcd. for C40H71N4O2: 640.5656, Found: 640.5641.
= +56 (c 0.2, CHCl3); IR νmax (KBr)/cm− 1 3385 (NH and OH), 1700 (CO); 1H NMR δ (500 MHz, CDCl3) 0.78 (m, 1H, CH-5), 0.82 (s, 3H, CH3-24), 0.83 (d, J 7.0 Hz, 3H, CH3-30), 0.91 (d, J 6.6 Hz, 3H, CH3-29); 0.93 (m, 1H, CH2-1), 0.94 (s, 3H, CH3-25), 0.96 (m, 1H, CH-20), 0.94 (m, 1H, CH2-11), 0.98 (s, 3H, CH3-23), 1.02 (s, 3H, CH3-26), 1.14 (s, 3H, CH3-27), 1.21 (m, 1H, CH2(Hα)-7), 1.33 (m, 1H, CH-19), 1.41 (m, 2H, CH2-2), 1.55 (m, 4H, CH2(Hβ)-6, CH2(Hβ)-7, CH-9, CH2(Hα)-21), 1.62 (m, 4H, CH2(Hβ)-1, CH2(Hα)-15, CH2(Hβ)-16 and CH2(Hβ)-21), 1.70 (m, 5H, CH2-γ, CH2-γ′ and CH2(Hα)-6), 1.77 (td, J 14.0 and 6.0 Hz, 1H, CH2(Hα)-16), 1.83 (m, 2H, CH2-22), 2.00 (m, 1H, CH2(Hβ)-15), 2.52 (m, 13H, CH2-α1, CH2-α1′, CH2-α2, CH2-α2′, CH2-β, CH2-β′ and CH-18), 3.03 (m, 2H, CH2-δ′), 3,25 (dd, J 13.8 and 6.4 Hz, 1H, CH-3), 3.34 (m, 2H, CH2-δ), 5.35 (tl, 1H, CH-12), 6.46 (s, 1H, NH); 13C NMR δ (125 MHz, CDCl3) 15.8 (C-25), 15.9 (C-26), 16.8 (C-24), 17.4 (C-29), 18.7 (C-6), 21.7 (C-30), 23.8 (C-11 and C-27), 25.1 (C-16), 27.2 (C-γ), 28.3 (C-23), 28.4 (C-2), 28.5 (C-15), 31.3 (C-21), 33.2 (C-7), 31.5 (C-γ′), 37.4 (C-10), 37.8 (C-22), 38.9 (C-8), 39.0 (C-1), 39.2 (C-δ), 39.3 (C-4), 39.5 (C-19), 39.9 (C-20), 40.1 (C-δ′), 42.8 (C-14), 47.9 (C-9 and C-17), 53.4 (C-18), 53.7 (C-α1, C-α1′, C-α2, C-α2′, 55.6 (C-5), 56.5 (C-β and C-β′), 79.3 (C-3), 123.0 (C-12), 140.1 (C-13), 182.2 (C-28). HRMS (ESI-MS, m/z), (M + H)+: Calcd. for C40H71N4O2: 640.5656, Found: 640.5641.
Synthesis of 3-O-acetyloleanolic acid (8)
To a solution of oleanolic acid 7 (0.2 mmol) in dry pyridine (2.5 mL) at room temperature was slowly added acetic anhydride (2.5 mL). The reaction mixture was stirred for 24 h at room temperature and then poured in ice water. After filtration, the residue was washed with water to give 8 as a white powder. Yield: 100%, white crystals, mp = 200°C; ![]() = +119 (c 0.1, CHCl3); IR νmax (KBr)/cm− 1 3560 (OH), 1730 and 1695 (CO); 1H NMR δ (500 MHz, CDCl3) 0.80 (d, J 11.1 Hz, 1H, CH-5), 0.82 (s, 3H, CH3-25), 0.83 (m, 4H, CH3-24 and CH2(Hα)-1), 0.96 (s, 3H, CH3-26), 0.97 (s, 3H, CH3-30), 0.99 (s, 3H, CH3-23), 1.05 (s, 3H, CH3-27), 1.19 (s, 3H, CH3-29), 1.22 (m, 1H, CH2(Hα)-7), 1.33 (m, 3H, CH2-2, CH-19), 1.47 (m, 4H, CH2(Hβ)-6, CH2(Hβ)-7, CH-9 and CH2(Hα)-21), 1.58 (m, 4H, CH2(Hβ)-1, CH2(Hα)-15, CH2(Hβ)-16 and CH2(Hβ)-21), 1.67 (t, J 13.9 Hz, 1H, CH2(Hα)-6), 1.81 (t, J 13.6 Hz, 1H, CH2(Hα)-16), 1.86 (t, J 7.3 Hz, 2H, CH2-22), 1.96 (td, J 13.4 Hz, 1H, CH2(Hβ)-15), 2.10 (s, 3H, H3CCOO), 2.88 (dd, J 13.8 and 4.2 Hz, 1H, CH-18), 4.55 (t, J 13.3 and 7.5 Hz, 1H, CH-3), 5.34 (t, J 16.6 Hz, 1H, CH-12), 13C NMR δ (125 MHz, CDCl3) 15.7 (C-25), 15.9 (C-26), 17.5 (C-24), 18.7 (C-6), 21.7 (H3CCOO), 23.4 (C-11), 23.8 (C-30), 23.9 (C-27), 26.3 (C-16), 27.6 (C-15), 28.1 (C-2), 28.5 (C-23), 31.0 (C-21), 32.8 (C-29), 33.1 (C-19), 33.4 (C-7), 34.2 (C-22), 37.5 (C-10), 38.8 (C-8), 39.2 (C-1), 39.7 (C-4), 41.4 (C-20), 42.1 (C-14), 46.3 (C-9), 46.9 (C-17), 48.1 (C-18), 55.6 (C-5), 80.6 (C-3), 123.1 (C-12), 143.9 (C-13), 171.5 (H3CCOO-), 182.6(C-28). HRMS (ESI-MS, m/z), (M + H)+: Calcd. for C32H49O4: 497.3631, Found: 497,3637.
= +119 (c 0.1, CHCl3); IR νmax (KBr)/cm− 1 3560 (OH), 1730 and 1695 (CO); 1H NMR δ (500 MHz, CDCl3) 0.80 (d, J 11.1 Hz, 1H, CH-5), 0.82 (s, 3H, CH3-25), 0.83 (m, 4H, CH3-24 and CH2(Hα)-1), 0.96 (s, 3H, CH3-26), 0.97 (s, 3H, CH3-30), 0.99 (s, 3H, CH3-23), 1.05 (s, 3H, CH3-27), 1.19 (s, 3H, CH3-29), 1.22 (m, 1H, CH2(Hα)-7), 1.33 (m, 3H, CH2-2, CH-19), 1.47 (m, 4H, CH2(Hβ)-6, CH2(Hβ)-7, CH-9 and CH2(Hα)-21), 1.58 (m, 4H, CH2(Hβ)-1, CH2(Hα)-15, CH2(Hβ)-16 and CH2(Hβ)-21), 1.67 (t, J 13.9 Hz, 1H, CH2(Hα)-6), 1.81 (t, J 13.6 Hz, 1H, CH2(Hα)-16), 1.86 (t, J 7.3 Hz, 2H, CH2-22), 1.96 (td, J 13.4 Hz, 1H, CH2(Hβ)-15), 2.10 (s, 3H, H3CCOO), 2.88 (dd, J 13.8 and 4.2 Hz, 1H, CH-18), 4.55 (t, J 13.3 and 7.5 Hz, 1H, CH-3), 5.34 (t, J 16.6 Hz, 1H, CH-12), 13C NMR δ (125 MHz, CDCl3) 15.7 (C-25), 15.9 (C-26), 17.5 (C-24), 18.7 (C-6), 21.7 (H3CCOO), 23.4 (C-11), 23.8 (C-30), 23.9 (C-27), 26.3 (C-16), 27.6 (C-15), 28.1 (C-2), 28.5 (C-23), 31.0 (C-21), 32.8 (C-29), 33.1 (C-19), 33.4 (C-7), 34.2 (C-22), 37.5 (C-10), 38.8 (C-8), 39.2 (C-1), 39.7 (C-4), 41.4 (C-20), 42.1 (C-14), 46.3 (C-9), 46.9 (C-17), 48.1 (C-18), 55.6 (C-5), 80.6 (C-3), 123.1 (C-12), 143.9 (C-13), 171.5 (H3CCOO-), 182.6(C-28). HRMS (ESI-MS, m/z), (M + H)+: Calcd. for C32H49O4: 497.3631, Found: 497,3637.
Pharmacology
In vitro L. amazonensis and L. infantum culture and drug assays
Promastigotes of the L. infantum (clone MCAN/GR/82/LEM497) and L. amazonensis (MHOM/BR/1987/BA) were maintained at 26°C in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum (FCS), 200 U/mL penicillin, 100 μg/mL streptomycin, sodium bicarbonate and non-essential amino acids (all from Gibco, Peisley, UK). At stationary growth phase parasites (106/mL) were harvested, washed and incubated in culture media with tested molecules. The viability of promastigotes was checked using the MTS tetrazolium colorimetric assay CellTiter 96 Aqueous (Promega, USA). The MTS cell proliferation assay is a colorimetric assay system, which measures the reduction of a tetrazolium component (MTS) into formazan produced by the mitochondria of viable cells. Cells were plated in triplicate into microtiter-plate wells in 100 μM culture media with our compounds at increasing concentrations (0, 1, 5, 10 and 25 μM). After 3 h of incubation with 20 μL MTS/well, the samples were read using an ELISA plate reader at 490 nm wavelength. The amount of colour produced was directly proportional to the number of viable cells. The results are expressed as the concentrations inhibiting parasite growth by 50% (IC50) after a 1-2 day incubation period.
Cytotoxicity test upon human cells
The toxicity of various molecules was evaluated on non-activated, freshly isolated normal human peripheral blood mononuclear cells (PBMNC), as well as phytohemagglutinin (PHA)-induced cells. PBMNC from healthy volunteers were obtained following centrifugation on Ficoll gradient. Cells were then incubated in medium alone or induced to enter cell cycle by the addition of PHA (5 μg/mL, Murex Biotech Limited, Dartford, UK). Tested molecules were added as described under results. Following cell cultures during 3-4 days, cells were harvested, washed, and counted with trypan blue exclusion. In some experiments, the proliferation of PBMNC was checked using the MTS dye colorimetric method as described previously. The 50% inhibitory concentrations (IC50) were determined by linear regression analysis, expressed in μM ± SD and the selectivity index was calculated for each compound.
Results and discussion
Chemistry
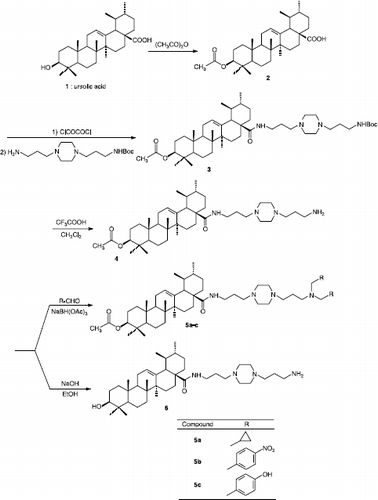
The preparation of compounds 2-6 was outlined in Scheme . 3-Acetylursolic acid 2 was synthesized by stirring ursolic acid 1 previously extracted from the south American species Ilex paraguariensis [Citation24,Citation25], with acetic anhydride in dry pyridine at room temperature. Compound 3 was prepared by treating 3-acetylursolic acid 2 with oxalyl chloride in dry dichloromethane, following by the treatment of this intermediate acid chloride with N-Boc-bis(aminopropyl)piperazine [Citation25,Citation26]. The N-Boc-amino protecting group was removed by treatment of 3 with a TFA (10%)/CH2Cl2 mixture to yield the deprotected analogue 4 [Citation25,Citation26]. Reductive amination of 4 with various aldehydes, and using sodium triacetoxyborohydride as a reducing agent, gave the N,N-disubstituted compounds 5a-c [Citation24,Citation25]. In order to investigate the importance of the acetyl substitution at the C-3 position for the antileishmanial activity of this series, derivative 4 was also submitted to a reaction of deacetylation leading to 6 [Citation25,Citation26].
Scheme 1 Synthesis of new ursolic acid derivatives 2-6.
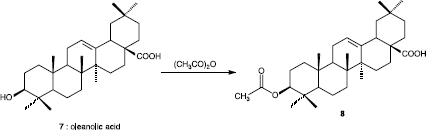
The preparation of compound 8 was outlined in Scheme . 3-Acetyloleanolic acid 8 was synthesized by stirring oleanolic acid 7 previously extracted from the south American species Ilex dumosa [Citation24], with acetic anhydride in dry pyridine at room temperature.
Scheme 2 Synthesis of 3-O-acetyloleanolic acid 8.
Pharmacology
Antileishmanial activity
Compounds 1-2 and 4-8 were tested for their in vitro antileishmanial activity upon the L. amazonensis and L. infantum strains Citation27Citation28 with Amphotericin B as the reference standard drug (). Among the seven compounds tested, ursolamide 5c was found the most active compound against the promastigote forms of L. amazonensis with IC50 of 10 μM. However, this new active hemi-synthetic compound remains ten times less active than the reference drug, amphotericin B (1 μM). No probative antipromastigote activity was observed for the natural product 1 (IC50 = 20 μM). Interestingly, in the ursolic acid amides 5a, 5b, and 5c in which the bis-(3-aminopropyl)piperazine moiety has been variously modulated, the bisalkylation of the lateral chain by substituted benzyl groups seems to increase the activity; the replacement of the para-nitro group on the benzyl moieties by a para-hydroxy one in compound 5c was found to induce interesting antileishmanial activity against L. amazonensis (IC50 of 25 μM for 5b versus 10 μM for 5c).
Table I. In vitro sensitivity of compounds 1-2 and 4-8 on L. amazonensis and L infantum strains, and cytotoxicity on human peripheral blood mononuclear cells PBMNC + PHA.
A second pharmacological evaluation was achieved against the promastigote forms of L. infantum in an experimental visceral leishmaniasis model, with amphotericin B as the reference (). While the natural triterpenoid acid 1 and its acetyl derivative 2 were found inactive on L. infantum, the introduction of a lateral bis-(3-aminopropyl)piperazine moiety on the carboxylic function (compound 4) resulted in the increase of the antileishmanial activity (IC50 = 5 μM). The bis-substitution of the primary amino group of this lateral chain (compounds 5a-c) seems to slightly or totally decrease the activity in comparison with their unsubstituted derivative 4 with IC50 = 10, 12 and >25 μM, respectively. In order to evaluate the influence of the acetyl function at position C-3 of the ursolic acid skeleton, compound 4 was submitted to deacetylation leading to 6. This structural modification induced a notable decrease in the activity (IC50 >25 μM compared with 5 μM for 4). As a general rule, the introduction of substituents on the bis-(3-aminopropyl)piperazine moiety seemed to decrease the antileishmanial activity as well as the deacetylation of the C-3 position of the N-{3-[4-(3-amino)propyl)piperazinyl]propyl}-ursolamide. Lastly we tried to experiment against the same Leishmania strains, oleanolic acid 7 and its O-acetyl derivative 8. Although 7 exhibited a lower activity than 1 against L. amazonensis, 8 exerted a fair activity (IC50 = 5 ± 0.1 μM), contrarily to its isomer 2. This preliminary pharmacological result enlightens the favourable incidence of the O-acetylation of the oleanolic acid skeleton.
Cytotoxicity
All compounds 1-2 and 4-8 were tested on activated human peripheral blood mononuclear cells (PBMNC + PHA) () [Citation28]. As expected, most of the compounds showed a significant level of cytotoxicity against monocytes (IC50 = 2-50 μM). Moreover, among these last compounds, the hemi-synthetic derivatives, substituted by a bis-(3-aminopropyl)piperazine moiety on the carboxylic acid function, showed high cytotoxicity (IC50 = 5-30 μM). Otherwise, ursolic acid 1 was found more toxic than its acetylated analogue 2 (IC50 = 20 μM for 1 versus 50 μM for 2). Index of selectivity (IS) was defined as the ratio of the IC50 value on the human mononuclear cells to the IC50 value on the L. amazonensis or L. infantum strains (promastigotes). Examination of the IS values () brings to the fore that the most active compound, 4, in the ursolic acid substituted series, is very cytotoxic (IS ≤ 1) compared to amphotericin B (IS # 100). The fact that the O-acetyloleanolic acid 8 (IC50 >50 μM; IS > 10) appeared to be at least ten-fold less toxic than 4 constitutes an encouraging result which prompt us to carry out further pharmacomodulation with this pentacyclic triterpene.
Conclusion
In the present report, we described the hemi-synthesis of new ursolic and oleanolic acid derivatives and presented their anti-leishmanial activity. These preliminary pharmacological results mainly enlightened the importance of an acetylation at position 3 of the oleanolic acid skeleton in the activity against the L. amazonensis strain, and of a bis-(3-aminopropyl)piperazine moiety on the carboxylic function of ursolic acid against the L. infantum strain. Further pharmacological studies should be now investigated to determine the intracellular target(s) of these new ursolic and oleanolic acid derivatives.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- World Health Organization. Division of control of tropical diseases, Available: http://www.who.int/leishmaniasis/en/ via the INTERNET. Accessed 2007 Sept 4.
- SL Croft, and V Yardley. (2002). Curr Pharm Des 8:319–342.
- P Desjeux. (2004). Comp Immunol Microbiol Infect Dis 27:305–318.
- SL Croft, and GH Coombs. (2003). Trends Parasitol 19:502–508.
- MH Gelb, and WGJ Hol. (2002). Science 297:343–344.
- AJ Davis, and L Kedzierski. (2005). Curr Opin Investig Drugs 6:163–169.
- A Mcgregor. (1998). Lancet 351:575.
- H Sangraula, KK Sharma, S Rijal, S Dwivedi, and S Koirala. (2003). J Assoc Physicians India 51:686–690.
- M Ouellette, J Drummelsmith, and B Papadopoulou. (2004). Drug Resist Updat 7:257–266.
- MJ Chan-Bacab, and LM Pena-Rodriguez. (2001). Nat Prod Rep 18:674–688.
- A Fournet, and V Munoz. (2002). Curr Top Med Chem 2:1215–1237.
- F Franca, EL Lago, and PD Marsden. (1996). Brazil Rev Soc Bras Med Trop 29:229–232.
- B Akendengue, E Ngou-Milama, A Laurens, and R Hocquemiller. (1999). Parasite 6:3–8.
- PB De Carvalho, and EI Ferreira. (2001). Fitoterapia 72:599–618.
- O Kayser, AF Kiderlen, and SL Croft. (2003). Parasitol Res 90:S55–S62.
- EC Torres-Santos, D Lopes, R Rodrigues Oliveira, JPP Carauta, M Bandeira Falcao, MAC Kaplan, and B Rossi-Bergmann. (2004). Phytomedicine 11:114–120.
- M Sauvain, J-P Dedet, N Kunesch, J Poisson, J-C Gantier, P Gayral, and G Kunesch. (1993). Phytother Res 7:167–171.
- C Farina, M Pinza, and G Pifferi. (1998). Il Farmaco 53:22–32.
- Z Ovesna, K Kozics, and D Slamenova. (2006). Mutat Res 600:131–137.
- EM Giner-Larza, S Manez, MC Recio, RM Giner, JM Prieto, M Cerda-Nicolas, and JL Rios. (2001). Eur J Pharmacol 428:137–143.
- HY Hsu, JJ Yang, and CC Lin. (1997). Can Lett 111:7–13.
- YM Zhu, JK Shen, HK Wang, LM Cosentino, and KH Lee. (2001). Bioorg Med Chem Lett 11:3115–3118.
- HJ Finlay, T Honda, GW Gribble, D Danielpour, NE Benoit, N Suh, C Williams, and MB Sporn. (1997). Biorg Med Chem Lett 7:1769–1772.
- Gnoatto SCB. Conception et synthèse de nouveaux dérivés de génines triterpéniques d'Ilex à visée antipaludique [thesis] 2007. p 219, University of Amiens.
- SCB Gnoatto, S Susplugas, LD Vechia, TB Ferreira, A Dassonville-Klimpt, KR Zimmer, C Demailly, S Da Nascimento, J Guillon, P Grellier, H Verli, G Gosmann, and P Sonnet. (2008). Bioorg Med Chem 16:771–782.
- SCB Gnoatto, A Dassonville-Klimpt, S Da Nascimento, P Galera, K Boumediene, G Gosmann, P Sonnet, and S Moslemi. (2008). Eur J Med Chem in press
- J Guillon, I Forfar, M Mamani-Matsuda, V Desplat, M Saliège, D Thiolat, S Massip, A Tabourier, J-M Léger, B Dufaure, G Haumont, C Jarry, and D Mossalayi. (2007). Bioorg Med Chem 15:194–210.
- J Guillon, I Forfar, V Desplat, S Belisle Fabre, D Thiolat, S Massip, H Carrie, D Mossalayi, and C Jarry. (2007). J Enz Inhib Med Chem 22:541–549.