Abstract
Src family kinases (SFKs) are nonreceptor tyrosine kinases that are reported to be critical for cancer progression. Inhibiting the catalytic activity of these proteins has become one of the major therapeutic concepts in contemporary drug discovery. We report here the design and the synthesis of novel 6-substituted-5-benzyloxy-4-oxo-4H-pyran-2-carboxamides as potential inhibitors of Src kinase. The synthesis of these derivatives and the preliminary results of biological activity will be discussed.
Introduction
Src is the prototype member of the Src-family of tyrosine kinases, which comprises nine highly homologous proteins including Src, Yes, Fyn, Lyn, Lck, Hck, Blk, Fgr and Yrk [Citation1]. Src is localized to intracellular membranes of the cell and also acts as a signal transduction inhibitor that is a critical component of multiple signaling pathways that control cell growth, proliferation, invasion and apoptosis. While this protein is highly regulated and active only at low levels in most normal cells, studies have shown that Src kinase is involved in metastases and upregulated in many human tumor progression, particulary those of breast, metastatic colorectal, ovarian and pancreatic cancer Citation2Citation3Citation4.
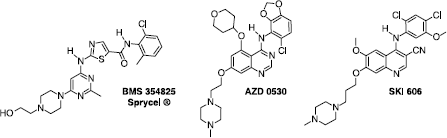
The three most advanced compounds that are marketed or are undergoing clinical evaluation are the thiazolecarboxamide BMS-354825 (Sprycel®) [Citation5], the anilinoquinazoline AZD0530 [Citation6], and the anilinoquinolinecarbonitrile SKI-606 [Citation7] ().
Figure 1 Structure of the three most advanced Src inhibitors.
In a precedent work [Citation8], we described the synthesis of various 3,6-diaryl-2,5-dihydroxy-1,4-benzoquinones I that inhibit Src kinase in a micromolar range by binding to the ATP pocket (). In continuation of our works, we focused our efforts on the design and synthesis of original O-membered rings II bearing a 5-benzyloxy-4-oxo-4H-pyran moiety. Biological evaluation of these compounds will also be reported in this paper.
Figure 2 Structure of benzoquinones I and pyranones II.
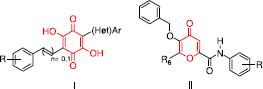
Materials and methods
Chemistry
Instrumentation
Melting points were determined using an Electrothermal IA9300 digital melting point apparatus and reported uncorrected. IR spectra were recorded on a Perkin-Elmer Paragon FTIR 1000 PC spectrometer in KBr or neat liquid films. Only the most significant absorption bands have been reported. 1H and 13C NMR spectra were recorded on a Bruker AC250 (250 MHz) or on Bruker Avance 400 spectrometer (400 MHz). Chemical shifts are expressed as δ values (ppm) relative to tetramethylsilane as internal standard (in NMR description, s = singlet, d = doublet, t = triplet, q = quartet, sept = septuplet, non = nonuplet, m = multiplet and br = broad). Coupling constants J are given in Hz. Electrospray mass spectrometric analysis was performed on a Esquire-LC Ion Trap System mass spectrometer. All reactions were monitored by TLC analytic, using 0.2 mm-thick silica gel plates 60F-254 (5735 Merck). Column chromatography was carried out using silica gel 60 (70–230 Mesh, ASTM, Merck). Chemicals and solvents used were commercially available.
Synthesis of 5-hydroxy-2-[(tetrahydro-2H-pyran-2-yloxy)methyl]-4H-pyran-4-one (2)
To a solution of kojic acid 1 (10 g, 70.4 mmol) in dichloromethane (200 mL) was added 3,4-dihydro-2H-pyran (12.9 mL, 140.8 mmol) followed by p-toluenesulfonic acid monohydrate (0.196 g, 1 mmol). After being stirred at room temperature for 45 min, the reaction mixture was concentrated to dryness by rotary evaporation. Trituration from diisopropyl ether afforded the pure product. Yield: 79%; white crystals; mp = 80-81°C; IR (KBr) ν cm− 1: 3253 (OH), 3102 (C-Har), 2948, 2865 (C-H), 1651 (C = O), 1617, 1456 (C = C), 1267, 1206, 1149 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.51-1.74 (m, 6H, 3CH2), 3.46-3.54 (m, 1H, CH2), 3.73-3.82 (m, 1H, CH2), 4.38 (d, 1H, CH2OCH, J = 13.7 Hz), 4.50 (d, 1H, CH2OCH, J = 13.7 Hz), 4.73-4.75 (m, 1H, OCHO), 6.44 (s, 1H, H3), 8.11 (s, 1H, H6), 9.20 (s, 1H, OH); NMR 13C (DMSO-d6): δ ppm 18.92 (CH2), 24.99 (CH2), 30.04 (CH2), 61.54 (CH2), 64.51 (CH2), 98.00 (CH), 111.91 (C3H), 139.85 (C6H), 146.06 (Cq), 164.32 (Cq), 173.95 (Cq); MS-ES+ (MeOH): m/z 226.
Synthesis of 3-hydroxy-2,6-bis(hydroxymethyl)-4H-pyran-4-one (3)
To a three-necked round-bottomed flask fitted with a pressure-equalized dropping funnel and containing a magnetic stirring bar were added kojic acid 1 (10 g, 70.4 mmol) and water (40 mL). The pH of the mixture was adjusted to 10.5 by treatment with 50% aqueous sodium hydroxide solution. A 37% aqueous solution of formaldehyde (5.6 mL, 75.3 mmol) was added slowly dropwise via the dropping funnel. The solution was stirred at 25°C for 4 h. The reaction mixture was acidified to pH 1 using concentrated aqueous hydrochloric acid solution, cooled, and allowed to crystallize. The product was removed by filtration. Yield: 81%; white crystals; mp = 150-151°C; IR (KBr) ν cm− 1: 3390, 3215 (OH), 2922, 2861 (C-H), 1653 (C = O), 1612 (C = C), 1248, 1093 (C-O-C); NMR 1H (DMSO-d6): δ ppm 4.34 (d, 2H, CH2OH, J = 5.6 Hz), 4.44 (d, 2H, CH2OH, J = 5.8 Hz), 5.40 (t, 1H, CH2OH, J = 5.6 Hz), 5.72 (t, 1H, CH2OH, J = 5.8 Hz), 6.34 (s, 1H, H5), 9.00 (se, 1H, OH); NMR 13C (DMSO-d6): δ ppm 55.21 (CH2), 59.71 (CH2), 109.05 (C5H), 141.86 (Cq), 149.50 (Cq), 167.77 (Cq), 174.16 (Cq); MS-ES+ (MeOH): m/z 172.
3-Hydroxy-6-hydroxymethyl-2-(1-hydroxy-2-methylpropyl)-4H-pyran-4-one (4)
was prepared as described for compound 3 using kojic acid 1 and isobutyraldehyde. Yield: 65%; white crystals; mp = 138-139°C; IR (KBr) ν cm− 1: 3220 (OH), 2957 (C-H), 1646 (C = O), 1616, 1572 (C = C), 1246, 1214, 1054 (C-O-C); NMR 1H (DMSO-d6): δ ppm 0.76 (d, 3H, CH3CH, J = 6.7 Hz), 1.02 (d, 3H, CH3CH, J = 6.7 Hz), 1.98 (septd, 1H, CH(CH3)2, J = 6.7 Hz and J = 8.5 Hz), 4.33 (d, 2H, CH2OH, J = 5.8 Hz), 4.45 (dd, 1H, CHOH, J = 4.9 Hz and J = 8.5 Hz), 5.42 (d, 1H, CHOH, J = 4.9 Hz), 5.69 (t, 1H, CH2OH, J = 5.8 Hz), 6.34 (s, 1H, H5), 8.85 (se, 1H, OH); NMR 13C (DMSO-d6): δ ppm 18.56 (CH3), 19.17 (CH3), 31.83 (CH), 59.71 (CH2), 70.02 (CH), 108.79 (C5H), 141.67 (Cq), 150.91 (Cq), 167.54 (Cq), 173.87 (Cq); MS-ES+ (MeOH): m/z 214.
3-Hydroxy-2-hydroxymethyl-6-[(tetrahydro-2H-pyran-2-yloxy)methyl]-4H-pyran-4-one (5)
was prepared as described for compound 3 using 5-hydroxy-2-[(tetrahydro-2H-pyran-2-yloxy)methyl]-4H-pyran-4-one 2, formaldehyde and methanol as a co solvent. Yield: 90%; red crystals; mp = 67-68°C; IR (KBr) ν cm− 1: 3381, 3246 (OH), 2929 (C-H), 1656 (C = O), 1621, 1585, 1462 (C = C), 1232, 1138, 1130 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.51-1.74 (m, 6H, 3CH2), 3.48-3.53 (m, 1H, CH2), 3.74-3.83 (m, 1H, CH2), 4.39 (d, 1H, CH2OCH, J = 14.0 Hz), 4.45 (s, 2H, CH2OH), 4.51 (d, 1H, CH2OCH, J = 14.0 Hz), 4.75 (se, 1H, OCHO), 5.52 (se, 1H, CH2OH), 6.42 (s, 1H, H5), 9.11 (s, 1H, OH); NMR 13C (DMSO-d6): δ ppm 18.92 (CH2), 25.01 (CH2), 30.05 (CH2), 55.06 (CH2), 61.56 (CH2), 64.46 (CH2), 97.94 (CH), 110.92 (C5H), 142.07 (Cq), 149.96 (Cq), 163.81 (Cq), 174.00 (Cq); MS-ES+ (MeOH): m/z 256.
Synthesis of 3-benzyloxy-2,6-bis(hydroxymethyl)-4H-pyran-4-one (7)
Sodium hydroxide (1.14 g, 28.7 mmol) dissolved in 2.4 mL distilled water was added to a solution of 3 (4.11 g, 23.9 mmol) in methanol (22 mL) and the reaction was heated to reflux. Benzyl bromide (3.41 mL, 28.7 mmol) was added dropwise and then refluxed for 5 h. The reaction mixture was concentrated to dryness by rotary evaporation, the residue taken up into dichloromethane (20 mL) and the inorganic salts filtered off. The dichloromethane layer was concentrated in vacuo to yield the crude product. Further purification by column chromatography on silica gel (eluting gradient: dichloromethane → dichloromethane/methanol 95/5) furnished 7. Yield: 71%; white crystals; mp = 76-77°C; IR (KBr) ν cm− 1: 3384 (OH), 3114 (C-Har), 2909, 2851 (C-H), 1652 (C = O), 1605, 1578 (C = C), 1246 (C-O-C); NMR 1H (DMSO-d6): δ ppm 4.31 (d, 2H, CH2OH, J = 6.1 Hz), 4.33 (dd, 2H, CH2OH, J = 6.2 Hz and J = 0.9 Hz), 5.06 (s, 2H, CH2Ph), 5.49 (t, 1H, CH2OH, J = 6.1 Hz), 5.76 (t, 1H, CH2OH, J = 6.2 Hz), 6.38 (d, 1H, H5, J = 0.9 Hz), 7.39-7.42 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 55.49 (CH2), 59.49 (CH2), 73.53 (CH2), 111.99 (C5H), 128.38 (CH), 128.54 (2CH), 128.74 (2CH), 137.00 (Cq), 142.18 (Cq), 159.31 (Cq), 168.08 (Cq), 175.20 (Cq); MS-ES+ (MeOH): m/z 262.
5-Benzyloxy-2-hydroxymethyl-4H-pyran-4-one (6)
was prepared as described for compound 7 using kojic acid 1 and benzyl bromide. Yield: 80%; white crystals; mp = 131-132°C; IR (KBr) ν cm− 1: 3329 (OH), 3114 (C-Har), 2906, 2834 (C-H), 1646 (C = O), 1608, 1578 (C = C), 1251, 1194 (C-O-C); NMR 1H (DMSO-d6): δ ppm 4.33 (d, 2H, CH2OH, J = 5.7 Hz), 4.98 (s, 2H, CH2Ph), 5.73 (t, 1H, CH2OH, J = 5.7 Hz), 6.36 (s, 1H, H3), 7.38-7.46 (m, 5H, HBn), 8.22 (s, 1H, H6); NMR 13C (DMSO-d6): δ ppm 59.50 (CH2), 70.75 (CH2), 111.35 (C3H), 128.30 (2CH), 128.36 (CH), 128.62 (2CH), 136.35 (Cq), 141.43 (C6H), 146.80 (Cq), 168.23 (Cq), 173.39 (Cq); MS-ES+ (MeOH): m/z 232.
3-Benzyloxy-6-hydroxymethyl-2-(1-hydroxy-2-methylpropyl)-4H-pyran-4-one (8)
was prepared as described for compound 7 using 3-hydroxy-6-hydroxymethyl-2-(1-hydroxy-2-methylpropyl)-4H-pyran-4-one 4 and benzyl bromide. Yield: 73%; white crystals; mp = 114-115°C; IR (KBr) ν cm− 1: 3318 (OH), 3133 (C-Har), 2947 (C-H), 1649 (C = O), 1606 (C = C), 1235, 1197 (C-O-C); NMR 1H (DMSO-d6): δ ppm 0.65 (d, 3H, CH3CH, J = 6.7 Hz), 0.99 (d, 3H, CH3CH, J = 6.7 Hz), 1.91 (septd, 1H, CH(CH3)2, J = 6.7 Hz and J = 8.6 Hz), 4.34 (d, 2H, CH2OH, J = 6.1 Hz), 4.41 (dd, 1H, CHOH, J = 5.8 Hz and J = 8.6 Hz), 5.07 (s, 2H, CH2Ph), 5.55 (d, 1H, CHOH, J = 5.8 Hz), 5.75 (t, 1H, CH2OH, J = 6.1 Hz), 6.38 (s, 1H, H5), 7.37-7.43 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 18.62 (CH3), 19.03 (CH3), 31.48 (CH), 59.51 (CH2), 70.19 (CH2), 73.19 (CH2), 111.78 (C5H), 128.26 (CH), 128.46 (2CH), 128.56 (2CH), 137.26 (Cq), 142.39 (Cq), 160.56 (Cq), 167.90 (Cq), 175.11 (Cq); MS-ES+ (MeOH): m/z 304.
3-Benzyloxy-2-hydroxymethyl-6-[(tetrahydro-2H-pyran-2-yloxy)methyl]-4H-pyran-4-one (9)
was prepared as described for compound 7 using 3-hydroxy-2-hydroxymethyl-6-[(tetrahydro-2H-pyran-2-yloxy)methyl]-4H-pyran-4-one 5 and benzyl bromide. Yield: 98%; orange oil; IR (NaCl) ν cm− 1: 3421 (OH), 3061 (C-Har), 2948, 2865 (C-H), 1651 (C = O), 1615, 1451 (C = C), 1251, 1184, 1149 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.52-1.75 (m, 6H, 3CH2), 3.46-3.54 (m, 1H, CH2), 3.74-3.83 (m, 1H, CH2), 4.33 (d, 2H, CH2OH, J = 5.9 Hz), 4.40 (d, 1H, CH2OCH, J = 14.0 Hz), 4.51 (d, 1H, CH2OCH, J = 14.0 Hz), 4.78 (se, 1H, OCHO), 5.07 (s, 2H, CH2Ph), 5.52 (t, 1H, CH2OH, J = 5.9 Hz), 6.46 (s, 1H, H5), 7.40-7.44 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 18.92 (CH2), 25.00 (CH2), 30.03 (CH2), 55.50 (CH2), 61.56 (CH2), 64.27 (CH2), 73.53 (CH2), 98.02 (CH), 113.88 (C5H), 128.38 (CH), 128.54 (2CH), 128.73 (2CH), 136.97 (Cq), 142.38 (Cq), 159.70 (Cq), 164.21 (Cq), 175.07 (Cq); MS-ES+ (MeOH): m/z 346.
Synthesis of {3-benzyloxy-4-oxo-6-[(tetrahydro-2H-pyran-2-yloxy)methyl]-4H-pyran-2-yl}methyl diethyl phosphate (10)
To a solution of 3-benzyloxy-2-hydroxymethyl-6-[(tetrahydro-2H-pyran-2-yloxy)methyl]-4H-pyran-4-one 9 (3.3 g, 9.5 mmol) in dry dichloromethane (70 mL), under nitrogen atmosphere, was added diethyl chlorophosphate (2.82 mL, 19 mmol) followed by pyridine (2.34 mL, 28.6 mmol) and DMAP (0.05 g, 0.4 mmol). After being stirred at room temperature for 24 h, the reaction mixture was diluted with dichloromethane and washed with an aqueous solution of hydrochloric acid 1 N and then with distilled water. The organic layer was dried over anhydrous sodium sulfate, filtered and concentrated to dryness by rotary evaporation. Further purification by column chromatography on silica gel (eluting gradient: dichloromethane → dichloromethane/methanol 99/1) furnished 10. Yield: 75%; yellow oil; IR (NaCl) ν cm− 1: 3061 (C-Har); 2937, 2865 (C-H), 1656 (C = O), 1630, 1446 (C = C), 1267, 1195, 1126 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.24 (t, 6H, 2CH3, J = 7.0 Hz), 1.52-1.74 (m, 6H, 3CH2), 3.37-3.54 (m, 1H, CH2), 3.70-3.82 (m, 1H, CH2), 4.04 (dq, 4H, 2CH2, J = 7.0 Hz and J = 8.2 Hz), 4.43 (d, 1H, CH2OCH, J = 14.0 Hz), 4.53 (d, 1H, CH2OCH, J = 14.0 Hz), 4.77 (m, 1H, OCHO), 4.89 (d, 2H, CH2OP, J = 8.8 Hz), 5.15 (s, 2H, CH2Ph), 6.54 (s, 1H, H5), 7.39-7.44 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 16.00 (2CH3, J = 6.4 Hz), 18.84 (CH2), 24.98 (CH2), 29.98 (CH2), 60.25 (CH2, J = 4.9 Hz), 61.50 (CH2), 63.83 (2CH2, J = 5.8 Hz), 64.20 (CH2), 73.57 (CH2), 98.00 (CH), 114.31 (C5H), 128.51 (CH), 128.59 (2CH), 128.74 (2CH), 136.61 (Cq), 143.81 (Cq), 153.91 (Cq, J = 7.1 Hz), 164.50 (Cq), 174.93 (Cq); MS-ES+ (MeOH): m/z 482.
Synthesis of 3-benzyloxy-6-hydroxymethyl-2-methyl-4H-pyran-4-one (11)
3-Benzyloxy-2,6-bis(hydroxymethyl)-4H-pyran-4-one 7 (3.95 g, 15.1 mmol) was added to 20 mL of distilled water. Zinc powder (1.18 g, 18.1 mmol) was added followed by the dropwise addition of concentrated aqueous hydrochloric acid solution (4.25 mL) over 30 min with vigorous stirring. The reaction mixture was heated to 70°C for 3 h and extracted 3 times with ethyl acetate. The organic layers were combined, dried over anhydrous sodium sulfate, filtered and concentrated to dryness by rotary evaporation. Further purification by column chromatography on silica gel (eluting gradient: dichloromethane → dichloromethane/methanol 98/2) furnished 11. Yield: 50%; pale white oil; IR (NaCl) ν cm− 1: 3340 (OH), 2916 (C-H), 1656 (C = O), 1615 (C = C), 1195 (C-O-C); NMR 1H (DMSO-d6): δ ppm 2.15 (s, 3H, CH3), 4.30 (d, 2H, CH2OH, J = 5.6 Hz), 5.05 (s, 2H, CH2Ph), 5.72 (t, 1H, CH2OH, J = 5.6 Hz), 6.33 (s, 1H, H5), 7.39-7.42 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 14.65 (CH3), 59.46 (CH2), 72.86 (CH2), 111.90 (C5H), 128.35 (CH), 128.51 (2CH), 128.83 (2CH), 137.15 (Cq), 142.33 (Cq), 158.44 (Cq), 167.48 (Cq), 174.64 (Cq); MS-ES+ (MeOH): m/z 246.
3-Benzyloxy-6-hydroxymethyl-2-isobutyl-4H-pyran-4-one (12)
was prepared as described for compound 11 using 3-benzyloxy-6-hydroxymethyl-2-(1-hydroxy-2-methylpropyl)-4H-pyran-4-one 8 and zinc powder. Yield: 37%; pale white oil; IR (NaCl) ν cm− 1: 3397 (OH), 2957, 2865 (C-H), 1652 (C = O), 1605, 1456 (C = C), 1246, 1190, 1049 (C-O-C); NMR 1H (DMSO-d6): δ ppm 0.87 (d, 6H, 2CH3, J = 6.7 Hz), 1.91 (non, 1H, CH(CH3)2, J = 6.7 Hz), 2.41 (d, 2H, CH2CH(CH3)2, J = 7.1 Hz), 4.33 (d, 2H, CH2OH, J = 6.2 Hz), 5.07 (s, 2H, CH2Ph), 5.71 (t, 1H, CH2OH, J = 6.2 Hz), 6.35 (s, 1H, H5), 7.36-7.43 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 22.29 (2CH3), 26.64 (CH), 36.61 (CH2), 59.52 (CH2), 72.83 (CH2), 111.84 (C5H), 128.30 (CH), 128.54 (2CH), 128.63 (2CH), 137.25 (Cq), 142.83 (Cq), 160.52 (Cq), 167.59 (Cq), 174.85 (Cq); MS-ES+ (MeOH): m/z 288.
Synthesis of [3-benzyloxy-6-(hydroxymethyl)-4-oxo-4H-pyran-2-yl]methyl diethyl phosphate (13)
3-Benzyloxy-4-oxo-6-[(tetrahydro-2H-pyran-2-yloxy)methyl]-4H-pyran-2-yl}methyl diethyl phosphate 10 (2.21 g, 4.6 mmol) was added to an aqueous solution of hydrochloric acid 1 N (55 mL) and refluxed for 15 min, followed by extraction into dichloromethane. The combined organic layers were then dried over anhydrous sodium sulfate, filtered, and concentrated to dryness by rotary evaporation. Further purification by column chromatography on silica gel (eluting gradient: dichloromethane → dichloromethane/methanol 98/2) furnished 13. Yield: 78%; yellow oil; IR (NaCl) ν cm− 1: 3369 (OH), 3061 (C-Har), 2978, 2916 (C-H), 1651 (C = O), 1620, 1446 (C = C), 1256, 1195, 1026 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.24 (t, 6H, 2CH3, J = 7.0 Hz), 4.03 (qd, 4H, 2CH2, J = 7.0 Hz and J = 8.2 Hz), 4.36 (d, 2H, CH2OH, J = 6.1 Hz), 4.88 (d, 2H, CH2OP, J = 8.5 Hz), 5.14 (s, 2H, CH2Ph), 5.81 (t, 1H, CH2OH), 6.45 (s, 1H, H5), 7.39-7.43 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 16.02 (2CH3, J = 6.6 Hz), 59.40 (CH2), 60.31 (CH2, J = 4.7 Hz), 63.83 (2CH2, J = 5.7 Hz), 73.57 (CH2), 112.35 (C5H), 128.50 (CH), 128.59 (2CH), 128.73 (2CH), 136.65 (Cq), 143.70 (Cq), 153.55 (Cq, J = 7.1 Hz), 168.42 (Cq), 175.00 (Cq); MS-ES+ (MeOH): m/z 398.
Synthesis of 5-benzyloxy-6-methyl-4-oxo-4H-pyran-2-carboxylic acid (15)
3-Benzyloxy-6-hydroxymethyl-2-methyl-4H-pyran-4-one 11 (1.1 g, 4.5 mmol) was dissolved in acetone (55 mL) and cooled to − 20°C to which was added Jones reagent (3.59 mL). The resulting mixture was stirred for 1 h at this temperature. The organic material was removed by filtration and the filtrate was evaporated to dryness. The residue was then redissolved in an 1 M aqueous sodium hydroxide solution and washed with dichloromethane. Acidification of the aqueous layer with concentrated aqueous hydrochloric acid solution followed by extraction with dichloromethane gave, after drying over anhydrous sodium sulfate and concentration to dryness by rotary evaporation, the crude product. Further trituration from diethyl ether in an ice-bath furnished the pure product. Yield: 75%; green crystals; mp = 145-146°C; IR (KBr) ν cm− 1: 3421 (OH), 3089 (C-Har), 2875 (C-H), 1734 (C = O acid), 1637 (C = O), 1560, 1456 (C = C), 1257, 1194 (C-O-C); NMR 1H (DMSO-d6): δ ppm 2.21 (s, 3H, CH3), 5.09 (s, 2H, CH2Ph), 6.96 (s, 1H, H3), 7.40-7.43 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 14.88 (CH3), 72.88 (CH2), 118.25 (C3H), 128.44 (CH), 128.56 (2CH), 128.86 (2CH), 136.89 (Cq), 144.14 (Cq), 152.19 (Cq), 159.60 (Cq), 160.82 (Cq), 174.48 (Cq); MS-ES+ (MeOH): m/z 260.
5-Benzyloxy-4-oxo-4H-pyran-2-carboxylic acid (14)
was prepared as described for compound 15 using 5-benzyloxy-2-hydroxymethyl-4H-pyran-4-one 6 and Jones reagent at 0°C. Yield: 86%; white crystals; mp = 186-187°C; IR (KBr) ν cm− 1: 3460 (OH), 3088 (C-Har), 2880, 2798 (C-H), 1731 (C = O acid), 1632 (C = O), 1605, 1578 (C = C), 1260, 1214 (C-O-C); NMR 1H (DMSO-d6): δ ppm 5.02 (s, 2H, CH2Ph), 6.98 (s, 1H, H3), 7.42-7.47 (m, 5H, HBn), 8.41 (s, 1H, H6); NMR 13C (DMSO-d6): δ ppm 70.84 (CH2), 117.15 (C3H), 128.41 (2CH), 128.49 (CH), 128.66 (2CH), 136.03 (Cq), 141.66 (C6H), 148.35 (Cq), 153.24 (Cq), 160.91 (Cq), 173.28 (Cq); MS-ES+ (MeOH): m/z 246.
5-Benzyloxy-6-isobutyl-4-oxo-4H-pyran-2-carboxylic acid (16)
was prepared as described for compound 15 using 3-benzyloxy-6-hydroxymethyl-2-isobutyl-4H-pyran-4-one 12 and Jones reagent at − 20°C. Yield: 93%; green crystals; mp = 141-142°C; IR (KBr) ν cm− 1: 3342 (OH), 3081 (C-Har), 2953, 2865 (C-H), 1742 (C = O acid), 1643 (C = O), 1600, 1570 (C = C), 1244, 1185 (C-O-C); NMR 1H (DMSO-d6): δ ppm 0.88 (d, 6H, 2CH3, J = 6.4 Hz), 1.94 (non, 1H, CH(CH3)2, J = 6.4 Hz), 2.49 (d, 2H, CH2CH(CH3)2, J = 7.6 Hz), 5.11 (s, 2H, CH2Ph), 6.98 (s, 1H, H3), 7.40-7.43 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 22.24 (2CH3), 26.64 (CH), 36.60 (CH2), 72.81 (CH2), 118.20 (C3H), 128.39 (CH), 128.57 (2CH), 128.69 (2CH), 136.94 (Cq), 144.59 (Cq), 152.31 (Cq), 160.91 (Cq), 161.53 (Cq), 174.62 (Cq); MS-ES+ (MeOH): m/z 302.
[3-Benzyloxy-6-carboxy-4-oxo-4H-pyran-2-yl]methyl diethyl phosphate (17)
was prepared as described for compound 15 using [3-benzyloxy-6-(hydroxymethyl)-4-oxo-4H-pyran-2-yl]methyl diethyl phosphate 13 and Jones reagent at − 20°C. Yield: 100%; green oil; IR (NaCl) ν cm− 1: 3401 (OH), 2987 (C-H), 1738 (C = O acid), 1652 (C = O), 1451 (C = C), 1261, 1226, 1184, 1128 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.24 (t, 6H, 2CH3, J = 7.0 Hz), 4.03 (dq, 4H, 2CH2, J = 7.0 Hz and J = 7.6 Hz), 4.94 (d, 2H, CH2OP, J = 8.5 Hz), 5.18 (s, 2H, CH2Ph), 7.07 (s, 1H, H5), 7.41-7.46 (m, 5H, HBn); NMR 13C (DMSO-d6): δ ppm 15.99 (2CH3, J = 6.4 Hz), 60.25 (CH2, J = 4.8 Hz), 63.89 (2CH2, J = 5.8 Hz), 73.60 (CH2), 118.58 (C5H), 128.58 (CH), 128.62 (2CH), 128.77 (2CH), 136.43 (Cq), 145.21 (Cq), 152.76 (Cq), 154.37 (Cq, J = 6.8 Hz), 160.63 (Cq), 175.10 (Cq); MS-ES+ (MeOH): m/z 412.
Synthesis of 5-benzyloxy-4-oxo-N-phenyl-4H-pyran-2-carboxamide (18) (Method A)
To a solution of 5-benzyloxy-4-oxo-4H-pyran-2-carboxylic acid 14 (0.4 g, 1.6 mmol) in toluene (12 mL) and acetonitrile (1.6 mL) was added triethylamine (0.91 mL, 6.5 mmol) and TBTU (1.31 g, 4.1 mmol). The mixture was stirred at room temperature for 30 min and aniline (0.22 mL, 2.5 mmol) was then added and the solution was stirred for 72 h. The solvent was removed by evaporation and the residue was dissolved into ethyl acetate. The organic layer was washed with 1 M aqueous hydrochloric acid solution, 1 M aqueous sodium hydroxide solution and with distilled water. It was then dried over anhydrous sodium sulfate, filtered, and concentrated to dryness by rotary evaporation. Further trituration from diethyl ether in an ice-bath furnished the pure product. Yield: 85%; white crystals; mp = 241-242°C; IR (KBr) ν cm− 1: 3330 (N-H), 3077 (C-Har), 2909, 2851 (C-H), 1685 (C = O amide), 1637 (C = O), 1637, 1546 (C = C), 1246, 1200 (C-O-C); NMR 1H (DMSO-d6): δ ppm 5.09 (s, 2H, CH2Ph), 7.07 (s, 1H, H3), 7.21 (td, 1H, Hc, JbC = 7.3 Hz, JaC = 1.2 Hz), 7.39- 7.51 (m, 7H, 5HBn+Hb), 7.78 (dd, 2H, Ha, Jab = 8.8 Hz, JaC = 1.2 Hz), 8.34 (s, 1H, H6), 10.69 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 70.90 (CH2), 115.03 (C3H), 121.01 (2CH), 124.97 (CH), 128.35 (2CH), 128.50 (CH), 128.70 (2CH), 128.96 (2CH), 136.06 (Cq), 137.80 (Cq), 140.95 (C6H), 148.19 (Cq), 155.32 (Cq), 157.40 (Cq), 173.19 (Cq); MS-ES+ (MeOH): m/z 321.
5-Benzyloxy-N-(2-chlorophenyl)-4-oxo-4H-pyran-2-carboxamide (19)
was prepared following Method A. Yield: 66%; white crystals; mp = 144-145°C; IR (KBr) ν cm− 1: 3386 (N-H), 3109 (C-Har), 2979, 2871 (C-H), 1702 (C = O amide), 1653 (C = O), 1589, 1530 (C = C), 1234, 1199 (C-O-C); NMR 1H (DMSO-d6): δ ppm 5.09 (s, 2H, CH2Ph), 7.06 (s, 1H, H3), 7.35-7.49 (m, 7H, 5HBn+Hb+Hc), 7.61, 7.67 (2d, 2H, Ha+Hd, Jab = 7.3 Hz, Jcd = 7.3 Hz), 8.39 (s, 1H, H6), 10.50 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 70.90 (CH2), 115.20 (C3H), 127.91 (CH), 128.18 (CH), 128.36 (2CH), 128.43 (CH), 128.50 (CH), 128.70 (2CH), 129.41 (Cq), 129.88 (CH), 133.71 (Cq), 136.03 (Cq), 141.01 (C6H), 148.29 (Cq), 154.75 (Cq), 157.72 (Cq), 173.05 (Cq); MS-ES+ (MeOH): m/z 355.
5-Benzyloxy-N-(2,6-dichlorophenyl)-4-oxo-4H-pyran-2-carboxamide (20)
was prepared following Method A. Yield: 35%; beige crystals; mp = 213-214°C; IR (KBr) ν cm− 1: 3326 (N-H), 3081 (C-Har), 2925, 2855 (C-H), 1693 (C = O amide), 1649 (C = O), 1588, 1566 (C = C), 1235, 1203 (C-O-C); NMR 1H (DMSO-d6): δ ppm 5.09 (s, 2H, CH2Ph), 7.07 (s, 1H, H3). 7.39-7.51 (m, 6H, 5HBn+Hb), 7.66 (d, 2H, Ha, Jab = 8.2 Hz), 8.40 (s, 1H, H6), 10.94 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 70.93 (CH2), 115.39 (C3H), 128.35 (2CH), 128.50 (CH), 128.70 (2CH), 128.88 (2CH), 130.31 (CH), 131.78 (Cq), 133.96 (2Cq), 136.03 (Cq), 141.11 (C6H), 148.40 (Cq), 154.21 (Cq), 157.70 (Cq), 172.93 (Cq); MS-ES+ (MeOH): m/z 390.
Synthesis of 5-benzyloxy-N-(2,6-dimethylphenyl)-4-oxo-4H-pyran-2-carboxamide (21) (Method B)
To a solution of 5-benzyloxy-4-oxo-4H-pyran-2-carboxylic acid 14 (0.2 g, 0.8 mmol) in THF (10 mL) was added DEPBT (0.49 g, 1.6 mmol). The mixture was refluxed for 15 min and 2,6-dimethylaniline (0.22 mL, 1.8 mmol) and N,N-diisopropylethylamine (0.28 mL, 1.6 mmol) were then added. The mixture was refluxed for 24 h and the solvent was removed by evaporation. The residue was dissolved into ethyl acetate and the organic layer was washed with 1 M aqueous hydrochloric acid solution, 1 M aqueous sodium hydroxide solution and with distilled water. It was then dried over anhydrous sodium sulfate, filtered, and rotary evaporated. Further purification by column chromatography on silica gel (eluting gradient: dichloromethane → dichloromethane/methanol 98/2) furnished 21. Yield: 51%; pale green crystals; mp = 175-176°C; IR (KBr) ν cm− 1: 3229 (N-H), 3066 (C-Har), 2858, 2926 (C-H), 1690 (C = O amide), 1647 (C = O), 1615, 1589 (C = C), 1248, 1197 (C-O-C); NMR 1H (DMSO-d6): δ ppm 2.19 (s, 6H, 2CH3Ph), 5.09 (s, 2H, CH2Ph), 7.05 (s, 1H, H3), 7.17 (se, 3H, HPh), 7.43-7.49 (m, 5H, HBn), 8.36 (s, 1H, H6), 10.31 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 18.10 (2CH3), 70.90 (CH2), 114.89 (C3H), 127.44 (CH), 128.04 (2CH), 128.32 (2CH), 128.48 (CH), 128.70 (2CH), 133.85(Cq), 135.56 (2Cq), 136.10 (Cq), 141.06 (C6H), 148.23 (Cq), 155.22 (Cq), 157.38 (Cq), 173.17 (Cq); MS-ES+ (MeOH): m/z 349.
5-Benzyloxy-N-(2-chloro-6-methylphenyl)-4-oxo-4H-pyran-2-carboxamide (22)
was prepared following Method B. Yield: 23%; beige crystals; mp = 193-194°C; IR (KBr) ν cm− 1: 3198 (N-H), 3081 (C-Har), 2989, 2865 (C-H), 1691 (C = O amide), 1645 (C = O), 1620, 1588 (C = C), 1251, 1200 (C-O-C); NMR 1H (DMSO-d6): δ ppm 2.25 (s, 3H, CH3), 5.09 (s, 2H, CH2Ph), 7.06 (s, 1H, H3), 7.32-7.34 (m, 2H, 2HPh), 7.41-7.52 (m, 6H, 5HBn+1HPh), 8.38 (s, 1H, H6), 10.60 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 18.28 (CH3), 70.92 (CH2), 115.14 (C3H), 127.30 (CH), 128.33 (2CH), 128.48 (CH), 128.69 (2CH), 128.95 (CH), 129.35 (CH), 132.14 (Cq), 132.54 (Cq), 136.06 (Cq), 138.58 (Cq), 141.09 (C6H), 148.31 (Cq), 154.72 (Cq), 157.57 (Cq), 173.05 (Cq); MS-ES+ (MeOH): m/z 369.
Synthesis of 5-benzyloxy-6-methyl-4-oxo-N-phenyl-4H-pyran-2-carboxamide (23) (Method C)
To a solution of 5-benzyloxy-6-methyl-4-oxo-4H-pyran-2-carboxylic acid 15 (0.1 g, 0.4 mmol) in dichloromethane (5 mL) was added CMPI (0.12 g, 0.5 mmol). The mixture was refluxed for 1 h and aniline (0.042 mL, 0.5 mmol) and triethylamine (0.137 mL, 1.0 mmol) were then added. The mixture was refluxed for 14 h and the solvent was removed by evaporation. The residue was dissolved into ethyl acetate and the organic layer was washed with 1 M aqueous hydrochloric acid solution, 1 M aqueous sodium hydroxide solution and with distilled water. It was then dried over anhydrous sodium sulfate, filtered, and concentrated to dryness by rotary evaporation. Further purification by column chromatography on silica gel (eluting gradient: dichloromethane → dichloromethane/methanol 98/2) furnished 23. Yield: 62%; yellow crystals; mp = 140-141°C; IR (KBr) ν cm− 1: 3277, 3227 (N-H), 3061, 3030 (C-Har), 2957, 2916 (C-H), 1693 (C = O amide), 1644 (C = O), 1600, 1534 (C = C), 1215, 1177 (C-O-C); NMR 1H (DMSO-d6): δ ppm 2.30 (s, 3H, CH3), 5.13 (s, 2H, CH2Ph), 7.04 (s, 1H, H3), 7.21 (t, 1H, Hc, JbC = 7.3 Hz), 7.40-7.46 (m, 7H, 5HBn+Hb), 7.75 (dd, 2H, Ha, Jab = 8.5 Hz, JaC = 0.9 Hz), 10.55 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 14.89 (CH3), 72.89 (CH2), 115.96 (C3H), 121.25 (2CH), 125.07 (CH), 128.47 (CH), 128.57 (2CH), 128.92 (2CH), 128.96 (2CH), 136.91 (Cq), 137.62 (Cq), 143.93 (Cq), 154.69 (Cq), 157.30 (Cq), 159.24 (Cq), 174.35 (Cq); MS-ES+ (MeOH): m/z 235.
5-Benzyloxy-6-methyl-N-(2-chlorophenyl)-4-oxo-4H-pyran-2-carboxamide (24)
was prepared following Method C. Yield: 58%; pink crystals; mp = 121-122°C; IR (KBr) ν cm− 1: 3390 (N-H), 3030 (C-Har), 2958 (C-H), 1699 (C = O amide), 1655 (C = O), 1597, 1531 (C = C), 1220, 1183 (C-O-C); NMR 1H (DMSO-d6): δ ppm 2.30 (s, 3H, CH3), 5.13 (s, 2H, CH2Ph), 7.03 (s, 1H, H3), 7.35-7.48 (m, 7H, 5HBn+Hb+Hc), 7.60-7.65 (m, 2H, Ha+Hd), 10.54 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 14.90 (CH3), 72.93 (CH2), 116.14 (C3H), 127.93 (CH), 128.44 (CH), 128.48 (CH), 128.59 (3CH), 128.91 (2CH), 129.70 (Cq), 129.90 (CH), 133.64 (Cq), 136.92 (Cq), 144.07 (Cq), 154.12 (Cq), 157.58 (Cq), 159.21 (Cq), 174.28 (Cq); MS-ES+ (MeOH): m/z 369.
5-Benzyloxy-6-methyl-N-(2,6-dichlorophenyl)-4-oxo-4H-pyran-2-carboxamide (25)
was prepared following Method C. Yield: 25%; brown crystals; mp = 192-193°C; IR (KBr) ν cm− 1: 3411 (N-H), 3133 (C-Har), 2973 (C-H), 1697 (C = O amide), 1646 (C = O), 1585, 1501 (C = C), 1220, 1190 (C-O-C); NMR 1H (DMSO-d6): δ ppm 2.32 (s, 3H, CH3), 5.13 (s, 2H, CH2Ph), 7.05 (s, 1H, H3), 7.39-7.52 (m, 6H, 5HBn+Hb), 7.67 (d, 2H, Ha, Jab = 8.2 Hz), 10.88 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 14.95 (CH3), 72.96 (CH2), 116.37 (C3H), 128.48 (CH), 128.59 (2CH), 128.88 (2CH), 128.91 (2CH), 130.36 (CH), 131.70 (Cq), 133.96 (2Cq), 136.94 (Cq), 144.26 (Cq), 153.57 (Cq), 157.57 (Cq), 159.40 (Cq), 174.18 (Cq); MS-ES+ (MeOH): m/z 404.
5-Benzyloxy-6-methyl-N-(2,6-dimethylphenyl)-4-oxo-4H-pyran-2-carboxamide (26)
was prepared following Method C. Yield: 79%; brown crystals; mp = 158-159°C; IR (KBr) ν cm− 1: 3255 (N-H), 3076 (C-Har), 2919 (C-H), 1690 (C = O amide), 1646 (C = O), 1589, 1501 (C = C), 1235, 1188 (C-O-C); NMR 1H (DMSO-d6): δ ppm 2.20 (s, 6H, 2CH3Ph), 2.32 (s, 3H, CH3), 5.13 (s, 2H, CH2Ph), 7.03 (s, 1H, H3), 7.19-7.20 (m, 3H, HPh), 7.39-7.51 (m, 5H, HBn), 10.28 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 14.93 (CH3), 18.14 (2CH3), 72.94 (CH2), 115.78 (C3H), 127.49 (CH), 128.06 (2CH), 128.45 (CH), 128.58 (2CH), 128.85 (2CH), 133.77 (Cq), 135.59 (2Cq), 137.02 (Cq), 144.10 (Cq), 154.53 (Cq), 157.22 (Cq), 159.20 (Cq), 174.39 (Cq); MS-ES+ (MeOH): m/z 363.
5-Benzyloxy-6-methyl-N-(2-chloro-6-methylphenyl)-4-oxo-4H-pyran-2-carboxamide (27)
was prepared following Method C. Yield: 57%; brown crystals; mp = 160-161°C; IR (KBr) ν cm− 1: 3431 (N-H), 3166 (C-Har), 2968, 2927 (C-H), 1693 (C = O amide), 1645 (C = O), 1589, 1502 (C = C), 1221, 1190 (C-O-C); NMR 1H (DMSO-d6): δ ppm 2.25 (s, 3H, CH3), 2.31 (d, 3H, CH3Ph, J = 0.9 Hz), 5.13 (s, 2H, CH2Ph), 7.04 (s, 1H, H3), 7.33-7.47 (m, 8H, 5HBn+3HPh), 10.56 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 14.95 (CH3), 18.32 (CH3), 72.96 (CH2), 116.09 (C3H), 127.33 (CH), 128.48 (CH), 128.59 (2CH), 128.88 (2CH), 129.01 (CH), 129.38 (CH), 132.17 (Cq), 132.48 (Cq), 136.99 (Cq), 138.61 (Cq), 144.18 (Cq), 154.07 (Cq), 157.44 (Cq), 159.31 (Cq), 174.30 (Cq); MS-ES+ (MeOH): m/z 383.
5-Benzyloxy-6-isobutyl-4-oxo-N-phenyl-4H-pyran-2-carboxamide (28)
was prepared following Method C. Yield: 85%; brown crystals; mp = 120-121°C; IR (KBr) ν cm− 1: 3345 (N-H), 3061 (C-Har), 2957, 2865 (C-H), 1699 (C = O amide), 1636 (C = O), 1547 (C = C), 1257, 1181 (C-O-C); NMR 1H (DMSO-d6): δ ppm 0.91 (d, 6H, 2CH3, J = 6.4 Hz), 2.05 (non, 1H, CH(CH3)2, J = 6.4 Hz), 2.56 (d, 2H, CH2CH(CH3)2, J = 7.0 Hz), 5.14 (s, 2H, CH2Ph), 7.09 (s, 1H, H3), 7.22 (td, 1H, Hc, JbC = 7.3 Hz and JaC = 1.2 Hz), 7.38-7.48 (m, 7H, 5HBn+Hb), 7.74 (dd, 2H, Ha, Jab = 7.6 Hz and JaC = 1.2 Hz), 10.50 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 22.25 (2CH3), 26.66 (CH), 36.62 (CH2), 72.82 (CH2), 115.89 (C3H), 121.40 (2CH), 125.10 (CH), 128.39 (CH), 128.57 (2CH), 128.68 (2CH), 128.93 (2CH), 136.97 (Cq), 137.59 (Cq), 144.38 (Cq), 154.71 (Cq), 157.41 (Cq), 161.29 (Cq), 174.53 (Cq); MS-ES+ (MeOH): m/z 377.
5-Benzyloxy-6-isobutyl-N-(2-chlorophenyl)-4-oxo-4H-pyran-2-carboxamide (29)
was prepared following Method C. Yield: 80%; brown crystals; mp = 99-100°C; IR (KBr) ν cm− 1: 3401 (N-H), 3030 (C-Har), 2958 (C-H), 1702 (C = O amide), 1656 (C = O), 1595, 1528 (C = C), 1241, 1179 (C-O-C); NMR 1H (DMSO-d6): δ ppm 0.92 (d, 6H, 2CH3, J = 6.7 Hz), 2.09 (non, 1H, CH(CH3)2, J = 6.7 Hz), 2.56 (d, 2H, CH2CH(CH3)2, J = 7.3 Hz), 5.14 (s, 2H, CH2Ph), 7.06 (s, 1H, H3), 7.35-7.50 (m, 7H, 5HBn+Hb+Hc), 7.66 (dd, 1H, Hd, Jcd = 7.6 Hz and Jbd = 1.8 Hz), 7.69 (dd, 1H, Ha, Jab = 7.9 Hz and JaC = 1.5 Hz), 10.37 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 22.27 (2CH3), 26.66 (CH), 36.66 (CH2), 72.86 (CH2), 116.04 (C3H), 127.95 (CH), 128.02 (CH), 128.40 (CH), 128.58 (3CH), 128.68 (2CH), 129.20 (Cq), 129.86 (CH), 133.63 (Cq), 136.96 (Cq), 144.52 (Cq), 154.11 (Cq), 157.53 (Cq), 161.16 (Cq), 174.46 (Cq); MS-ES+ (MeOH): m/z 411.
5-Benzyloxy-6-isobutyl-N-(2,6-dichlorophenyl)-4-oxo-4H-pyran-2-carboxamide (30)
was prepared following Method C. Yield: 37%; brown crystals; mp = 147-148°C; IR (KBr) ν cm− 1: 3442 (N-H), 3081 (C-Har), 2958, 2927 (C-H), 1697 (C = O amide), 1646 (C = O), 1585, 1497 (C = C), 1246, 1185 (C-O-C); NMR 1H (DMSO-d6): δ ppm 0.91 (d, 6H, 2CH3, J = 6.7 Hz), 2.09 (non, 1H, CH(CH3)2, J = 6.7 Hz), 2.56 (d, 2H, CH2CH(CH3)2, J = 7.3 Hz), 5.15 (s, 2H, CH2Ph), 7.06 (s, 1H, H3), 7.39-7.52 (m, 6H, 5HBn+Hb), 7.67 (d, 2H, Ha, Jab = 7.9 Hz), 10.77 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 22.20 (2CH3), 26.69 (CH), 36.66 (CH2), 72.89 (CH2), 116.25 (C3H), 128.41 (CH), 128.59 (2CH), 128.67 (2CH), 128.88 (2CH), 130.38 (CH), 131.73 (Cq), 134.01 (2Cq), 136.99 (Cq), 144.66 (Cq), 153.61 (Cq), 157.54 (Cq), 161.36 (Cq), 174.36 (Cq); MS-ES+ (MeOH): m/z 446.
5-Benzyloxy-6-isobutyl-N-(2,6-dimethylphenyl)-4-oxo-4H-pyran-2-carboxamide (31)
was prepared following Method C. Yield: 84%; brown crystals; mp = 131-132°C; IR (KBr) ν cm− 1: 3277 (N-H), 3020 (C-Har), 2963 (C-H), 1672 (C = O amide), 1648 (C = O), 1590, 1499 (C = C), 1241, 1180 (C-O-C); NMR 1H (DMSO-d6): δ ppm 0.92 (d, 6H, 2CH3, J = 6.7 Hz), 2.09 (non, 1H, CH(CH3)2, J = 6.7 Hz), 2.22 (s, 6H, 2CH3), 2.56 (d, 2H, CH2CH(CH3)2, J = 7.3 Hz), 5.14 (s, 2H, CH2Ph), 7.03 (s, 1H, H3), 7.15-7.21 (m, 3H, HPh), 7.39-7.49 (m, 5H, HBn), 10.22 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 18.06 (2CH3), 22.21 (2CH3), 26.78 (CH), 36.65 (CH2), 72.87 (CH2), 115.65 (C3H), 127.52 (CH), 128.03 (2CH), 128.38 (CH), 128.58 (2CH), 128.64 (2CH), 133.79 (Cq), 135.63 (2Cq), 137.06 (Cq), 144.50 (Cq), 154.56 (Cq), 157.15 (Cq), 161.19 (Cq), 174.58 (Cq); MS-ES+ (MeOH): m/z 405.
5-Benzyloxy-6-isobutyl-N-(2-chloro-6-methylphenyl)-4-oxo-4H-pyran-2-carboxamide (32)
was prepared following Method C. Yield: 65%; brown crystals; mp = 120-121°C; IR (KBr) ν cm− 1: 3298 (N-H), 3071 (C-Har), 2959 (C-H), 1699 (C = O amide), 1645 (C = O), 1610, 1585, 1498 (C = C), 1246, 1185 (C-O-C); NMR 1H (DMSO-d6): δ ppm 0.92 (d, 6H, 2CH3, J = 6.7 Hz), 2.10 (non, 1H, CH(CH3)2, J = 6.7 Hz), 2.25 (s, 3H, CH3), 2.56 (d, 2H, CH2CH(CH3)2, J = 8.2 Hz), 5.14 (s, 2H, CH2Ph), 7.05 (s, 1H, H3), 7.34-7.49 (m, 8H, 5HBn+3HPh), 10.47 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 18.26 (CH3), 22.21 (2CH3), 26.74 (CH), 36.66 (CH2), 72.89 (CH2), 115.95 (C3H), 127.30 (CH), 128.40 (CH), 128.59 (2CH), 128.66 (2CH), 129.04 (CH), 129.36 (CH), 132.21 (Cq), 132.49 (Cq), 137.03 (Cq), 138.65 (Cq), 144.58 (Cq), 154.10 (Cq), 157.40 (Cq), 161.28 (Cq), 174.49 (Cq); MS-ES+ (MeOH): m/z 425.
[6-(Anilinocarbonyl)-3-benzyloxy-4-oxo-4H-pyran-2-yl]methyl diethyl phosphate (33)
was prepared following Method C. Yield: 75%; brown crystals; mp = 101-102°C; IR (KBr) ν cm− 1: 3257 (N-H), 3071 (C-Har), 2978 (C-H), 1687 (C = O amide), 1656 (C = O), 1551 (C = C), 1255, 1179 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.25 (t, 6H, 2CH3, J = 7.0 Hz), 4.08 (quint, 4H, 2CH2, J = 7.0 Hz), 5.03 (d, 2H, CH2OP, J = 10.7 Hz), 5.21 (s, 2H, CH2Ph), 7.14 (s, 1H, H5), 7.19 (t, 1H, Hc, JbC = 7.3 Hz), 7.42-7.50 (m, 7H, 5HBn+Hb), 7.80 (d, 2H, Ha, Jab = 7.6 Hz), 10.66 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 16.00 (2CH3, J = 6.4 Hz), 60.29 (CH2, J = 5.0 Hz), 64.22 (2CH2, J = 6.1 Hz), 73.63 (CH2), 116.05 (C5H), 120.68 (2CH), 125.11 (CH), 128.63 (CH), 128.64 (2CH), 128.92 (2CH), 129.10 (2CH), 136.35 (Cq), 137.68 (Cq), 144.86 (Cq), 153.67 (Cq, J = 4.9 Hz), 154.85 (Cq), 156.81 (Cq), 175.08 (Cq); MS-ES+ (MeOH): m/z 487.
{6-[(2-Chloroanilino)carbonyl]-3-benzyloxy-4-oxo-4H-pyran-2-yl}methyl diethyl phosphate (34)
was prepared following Method C. Yield: 78%; yellow oil; IR (NaCl) ν cm− 1: 3390 (N-H), 3071 (C-Har), 2978 (C-H), 1697 (C = O amide), 1651 (C = O), 1589, 1528 (C = C), 1261, 1174 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.23 (td, 6H, 2CH3, J = 7.0 Hz and J = 0.9 Hz), 4.06 (dq, 4H, 2CH2, J = 7.0 Hz and J = 8.2 Hz), 5.02 (d, 2H, CH2OP, J = 8.9 Hz), 5.21 (s, 2H, CH2Ph), 7.15 (s, 1H, H5), 7.35-7.52 (m, 7H, 5HBn+Hb+Hc), 7.63 (dd, 2H, Ha+Hd, Jab = Jcd = 7.6 Hz, JaC = Jbd = 1.7 Hz), 10.57 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 16.00 (2CH3, J = 6.4 Hz), 60.40 (CH2, J = 4.8 Hz), 63.99 (2CH2, J = 5.9 Hz), 73.65 (CH2), 116.40 (C5H), 127.98 (CH), 128.13 (CH), 128.57 (CH), 128.60 (CH), 128.63 (2CH), 128.82 (2CH), 129.36 (Cq), 129.94 (CH), 133.50 (Cq), 136.43 (Cq), 145.27 (Cq), 153.88 (Cq, J = 6.0 Hz), 154.44 (Cq), 157.33 (Cq), 174.97 (Cq); MS-ES+ (MeOH): m/z 521.
{6-[(2,6-Dichloroanilino)carbonyl]-3-benzyloxy-4-oxo-4H-pyran-2-yl}methyl diethyl phosphate (35)
was prepared following Method C. Yield: 31%; yellow oil; IR (NaCl) ν cm− 1: 3185 (N-H), 3067 (C-Har), 2978 (C-H), 1697 (C = O amide), 1654 (C = O), 1518 (C = C), 1264, 1179 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.23 (t, 6H, 2CH3, J = 7.0 Hz), 4.06 (dq, 4H, 2CH2, J = 7.0 Hz and J = 7.9 Hz), 5.04 (d, 2H, CH2OP, J = 9.1 Hz), 5.21 (s, 2H, CH2Ph), 7.18 (s, 1H, H5), 7.42-7.54 (m, 6H, 5HBn+Hb), 7.68 (d, 2H, Ha, Jab = 7.9 Hz), 10.97 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 16.01 (2CH3, J = 6.4 Hz), 60.47 (CH2, J = 4.9 Hz), 64.00 (2CH2, J = 5.8 Hz), 73.68 (CH2), 116.64 (C5H), 128.58 (CH), 128.62 (2CH), 128.78 (2CH), 128.95 (2CH), 130.43 (CH), 131.53 (Cq), 133.85 (2Cq), 136.48 (Cq), 145.52 (Cq), 153.88 (Cq), 154.11 (Cq, J = 6.6 Hz), 157.33 (Cq), 174.90 (Cq); MS-ES+ (MeOH): m/z 556.
{6-[(2,6-Dimethylanilino)carbonyl]-3-benzyloxy-4-oxo-4H-pyran-2-yl}methyl diethyl phosphate (36)
was prepared following Method C. Yield: 83%; yellow oil; IR (NaCl) ν cm− 1: 3225 (N-H), 3050 (C-Har), 2978 (C-H), 1687 (C = O amide), 1646 (C = O), 1517 (C = C), 1261, 1174 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.23 (t, 6H, 2CH3, J = 7.0 Hz), 2.21 (s, 6H, 2CH3), 4.05 (dq, 4H, 2CH2, J = 7.0 Hz and J = 8.2 Hz), 5.04 (d, 2H, CH2OP, J = 9.5 Hz), 5.21 (s, 2H, CH2Ph), 7.14 (s, 1H, H5), 7.19-7.20 (m, 3H, HPh), 7.41-7.54 (m, 5H, HBn), 10.35 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 15.99 (2CH3, J = 6.3 Hz), 18.05 (2CH3), 60.48 (CH2, J = 4.9 Hz), 64.00 (2CH2, J = 5.8 Hz), 73.66 (CH2), 116.04 (C5H), 127.53 (CH), 128.10 (2CH), 128.57 (CH), 128.62 (2CH), 128.79 (2CH), 133.61 (Cq), 135.46 (2Cq), 136.52 (Cq), 145.30 (Cq), 153.89 (Cq, J = 6.6 Hz), 154.85 (Cq), 156.93 (Cq), 175.10 (Cq); MS-ES+ (MeOH): m/z 515.
{6-[(2-Chloro-6-methylanilino)carbonyl]-3-benzyloxy-4-oxo-4H-pyran-2-yl}methyl diethyl phosphate (37)
was prepared following Method C. Yield: 74%; brown oil; IR (NaCl) ν cm− 1: 3411 (N-H), 2978 (C-H), 1692 (C = O amide), 1651 (C = O), 1523 (C = C), 1256, 1174 (C-O-C); NMR 1H (DMSO-d6): δ ppm 1.23 (td, 6H, 2CH3, J = 7.0 Hz and J = 0.9 Hz), 2.26 (s, 3H, CH3), 4.05 (dq, 4H, 2CH2, J = 7.0 Hz and J = 8.2 Hz), 5.04 (d, 2H, CH2OP, J = 9.2 Hz), 5.21 (s, 2H, CH2Ph), 7.16 (s, 1H, H5), 7.34-7.53 (m, 8H, 5HBn+3HPh), 10.64 (s, 1H, NH); NMR 13C (DMSO-d6): δ ppm 16.00 (2CH3, J = 6.3 Hz), 18.24 (CH3), 60.47 (CH2, J = 5.0 Hz), 64.02 (2CH2, J = 5.9 Hz), 73.68 (CH2), 116.35 (C5H), 127.38 (CH), 128.58 (CH), 128.63 (2CH), 128.79 (2CH), 129.07 (CH), 129.43 (CH), 132.05 (Cq), 132.31 (Cq), 136.51 (Cq), 138.48 (Cq), 145.41 (Cq), 154.00 (Cq, J = 6.4 Hz), 154.38 (Cq), 157.17 (Cq), 175.01 (Cq); MS-ES+ (MeOH): m/z 535.
Pharmacology
Inhibitors were diluted on a robotic Tecan Evo150 platform. The kinase assay was performed with 4 μL of diluted inhibitor (10% DMSO), 10 μL of kinase assay buffer 4x concentrated (80 mM MgCl2 - 200 mM Hepes – 0.4 mM EDTA – 2 mM dTT), 10 μL substrate peptide (KVEKIGEGYYGVVYK – 370 nM) and 6 μL Src kinase (stock GTP purified diluted with 1x kinase assay buffer to 200 mM). 10 μL co-substrate (40 μM ATP with 0.2 μCi P33-γ-ATP) was added with a robotic Precision 2000 (Biotek) platform. The assay was incubated 20 minutes at 30°C then stopped by adding 200 μL 0.85% ortho-phosphoric acid, then transferred to a phosphocellulose filter microplate (Whatman – P81). Three washes were performed with 200 μL aqueous 0.85% ortho-phosphoric acid solution and the filter plate was dried with 200 μL acetone. The remaining activity was measured on a topcount with 25 μL Scintillation solution (Packard Ultima Gold).
Results and discussion
Lead identification
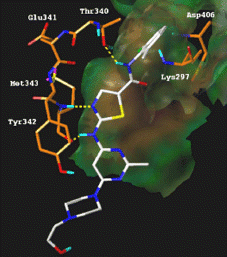
The three most studied Src inhibitors described in the literature (BMS-354825 (Sprycel®), AZD0530, SKI-606), share common properties: they act by competition with ATP in its binding site by creating hydrogen bonds and hydrophobic interactions with the protein. Based on the crystal structure of an active Src kinase domain in complex with the inhibitor CGP77675 (1YOL.pdb) [Citation9], we have docked BMS-354825 into the ATP site following a flexible ligand/rigid receptor docking protocol using GOLD software [Citation10]. As described by Louis J. Lombardo in 2004 [Citation11], this inhibitor makes critical hydrogen bonds with residue Met343 in the hinge segment which are essential for the recognition and also with the Src specific gate keeper residue Thr340. In addition, the 2-chloro-6-methylphenyl substituent, which is directed perpendicularly to the plan of the molecule, targets the specific hydrophobic backpocket and plays a key role in enhancing the binding of the moiety deep within this pocket ().
Figure 3 Illustration of a possible binding mode of BMS-354825 in the ATP binding site of active Src kinase.
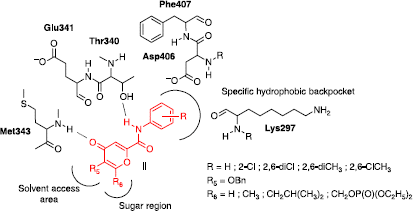
Based on this binding mode, we decided to design original compounds II which could interact with the protein in the same way. We planned to replace benzoquinone ring from I by a pyranone moiety as a H-bond acceptor group in the hinge segment. As described for BMS-354825, this scaffold could be substituted in position 2 by a carboxamide group and a phenyl substituent Citation12Citation13Citation14 orthogonal to the pyranone core, positioning this ring deep within the hydrophobic pocket. In order to target the sugar region which can often be exploited to design selective inhibitors [Citation6], various alkyl or phosphate substituents were also introduced in position 6. Finally, substitution by a 5-benzyloxy group oriented toward the solvent access area, was chosen because it was easy to prepare and to modulate ().
Figure 4 Possible interactions of pyranones II with Src.
Chemistry
The general methodology adopted for the synthesis of pyranones II is summarised in Scheme . Starting from commercially available kojic acid 1, the α-position to the ring hydroxyl was functionalised [Citation15] in an analogous fashion to the aldol condensation whereby an enolate, in this case the pyranone anion, attacks a carbonyl compound (formaldehyde or isobutyraldehyde), under alkaline aqueous conditions to furnish, on acidic work up, 3 and 4 in good yields. The 5-hydroxyl group was then selectively benzylated using benzyl bromide and aqueous sodium hydroxide in methanol. In order to obtain phosphate derivative 10, primary alcohol of kojic acid 1 was firstly selectively protected [Citation16] using 3,4-dihydro-2H-pyran before aldol condensation with formaldehyde. Subsequent benzylation and condensation of diethyl chlorophosphate with alcohol 9 afforded the desired product. Further deprotection resulted in the corresponding primary alcohol 13 in good yield. In addition, compounds 11 and 12, with an alkyl substituent in position 6, can be prepared by direct reduction of alcohols 7 and 8 using Clemmensen-type reduction with zinc powder in concentrated aqueous hydrochloric acid solution.
Scheme 1 Synthesis of 5-benzyloxy-4-oxo-4H-pyran-2-carboxamide and [6-aminocarbonyl-3-benzyloxy-4-oxo-4H-pyran-2-yl]methyl diethyl phosphate derivatives 18-37. Reagents and conditions: (i) DHP, PTSA·H2O, CH2Cl2, rt, 2; (ii) HCHO, NaOH/H2O, rt, 3; (CH3)2CHCHO, NaOH/H2O, rt, 4; HCHO, NaOH/H2O, MeOH, rt, 5; (iii) BnBr, NaOH/H2O, MeOH, Δ, 6-9; (iv) ClP(O)(OC2H5)2, pyridine, DMAP, CH2Cl2, rt, 10; (v) Zn/HCl, H2O, 70°C, 11, 12; (vi) HCl 1 N, Δ, 13; (vii) CrO3, H2SO4, acetone, 0°C or − 20°C, 14-17; (viii) Method A: TBTU, Et3N, toluene/acetonitrile, rt, 18-20; Method B: DEPBT, DIEA, THF, Δ, 21, 22; Method C: CMPI, Et3N, CH2Cl2, Δ, 23-37.
![Scheme 1 Synthesis of 5-benzyloxy-4-oxo-4H-pyran-2-carboxamide and [6-aminocarbonyl-3-benzyloxy-4-oxo-4H-pyran-2-yl]methyl diethyl phosphate derivatives 18-37. Reagents and conditions: (i) DHP, PTSA·H2O, CH2Cl2, rt, 2; (ii) HCHO, NaOH/H2O, rt, 3; (CH3)2CHCHO, NaOH/H2O, rt, 4; HCHO, NaOH/H2O, MeOH, rt, 5; (iii) BnBr, NaOH/H2O, MeOH, Δ, 6-9; (iv) ClP(O)(OC2H5)2, pyridine, DMAP, CH2Cl2, rt, 10; (v) Zn/HCl, H2O, 70°C, 11, 12; (vi) HCl 1 N, Δ, 13; (vii) CrO3, H2SO4, acetone, 0°C or − 20°C, 14-17; (viii) Method A: TBTU, Et3N, toluene/acetonitrile, rt, 18-20; Method B: DEPBT, DIEA, THF, Δ, 21, 22; Method C: CMPI, Et3N, CH2Cl2, Δ, 23-37.](/cms/asset/fb4f753c-0e92-428e-906c-062059c252b9/ienz_a_320696_f0005_b.gif)
Thereafter, direct conversion of primary alcohol to a carboxylic acid was attempted using Jones reagent at low temperature, in order to prevent extensive degradation of the pyranone moiety.
Finally, amide bond formation was first attempted with various substituted anilines by activation of acid function with 2-mercaptothiazoline [Citation17] using DDCI as the coupling reagent and DMAP as an acylation catalyst. However this method, described on this scaffold, was found to be unsuccessful in the synthesis of desired compounds. Thus, we had to find and optimize other conditions, depending on the nature of the substrate, to achieve this coupling. First carboxy components can be activated as acyl chlorides by using thionyl chloride or oxalyl chloride but we didn't observe the formation of desired compounds. We also decided to generate the acylating agent in situ from the acid by the addition of an activating or coupling agent. Among all reagents available in our laboratory and tested: EDCI, NHS, DCCI and CMPI, only Mukaiyama's reagent [Citation18] gave us good results for all pyranones susbtituted by alkyl or phosphate groups in R6 (compounds 23-37). However, if the heterocycle wasn't substituted in this position, only traces of the amide was obtained.
Aytemir et al. [Citation19] described a second method using TBTU in pyranone series, as activating agent for the reaction between an acid and a less reactive amine. In the same time, reactions with DEPBT [Citation20], a recently used coupling reagent in the laboratory, were attempted. This two methods were optimized and afforded the desired compounds 18-22 in various yields (see ).
Table I. Amide bond formation.
Biological assays
The biological activities of synthesized compounds were estimated by an inhibition test of the non-receptor tyrosine kinase Src. The inhibitory activity rate was determinated in vitro at two concentrations (10 μM and 1 μM). Unfortunately all compounds tested were inactive towards this kinase even if the key structural seems to be reached.
Indeed, comparison between BMS-354825 structure and compound 22 sharing a common core in the hydrophobic pocket seems to imply the pyranone moiety substituted by a 5-benzyloxy group as the main cause of inactivity. In addition, exploration of the sugar region did not improve the potency of this series. One explanation could involved the binding mode with the protein (angles and H-bonds distances) which might not be optimal. Therefore, an additional key H-bond interaction with the carbonyl oxygen of Met343 at the hinge region could be envisaged. Replacement of the 5-benzyloxy group by a 5-arylamino substituent in the ligand would lead to an essential donor-acceptor system, frequently observed in Src inhibitors.
Conclusion
In summary, we have designed and developed an efficient synthesis of new 6-substituted-5-benzyloxy-4-oxo-4H-pyran-2-carboxamide derivatives using the crystal structure of an active Src kinase domain. All the pharmacomodulations achieved on the pyranone core led to inactive compounds even if the key structural requirements were well identified. Structural modifications on position 5 could be carried out to improve the potency of this series.
Acknowledgements
We wish to thank Les Laboratoires Servier for their financial support and biological evalutations.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- AP Belsches-Jablonski, ML Demory, JT Parsons, and SJ Parsons. (2005). Drug Discov Today: Therap Strat 2:313–321.
- M Susa, M Missbach, and J Green. (2000). Trends Pharmacol Sci 21:489–495.
- RB Irby, and TJ Yeatman. (2000). Oncogene 19:5636–5642.
- JM Summy, and GE Gallick. (2003). Cancer Metastasis 22:337–358.
- J Das, P Chen, D Norris, R Padmanabha, J Lin, RV Moquin, Z Shen, LS Cook, AM Doweyko, S Pitt, S Pang, DR Shen, Q Fang, HF De Fex, KW McIntyre, DJ Shuster, KM Gillooly, K Behnia, GL Schieven, J Wityak, and JC Barrish. (2006). J Med Chem 49:6819–6832.
- LF Hennequin, J Allen, J Breed, J Curwen, M Fennell, TP Green, C Lambert-van der Brempt, R Morgentin, RA Norman, A Olivier, L Otterbein, PA Ple, N Warin, and G Costello. (2006). J Med Chem 49:6465–6488.
- R Thaimattam, PR Daga, R Banerjee, and J Iqbal. (2005). Bioorg Med Chem 13:4704–4712.
- Lardic M. 2004., Ph. D. Thesis, University of Nantes, November 24.
- CB Breitenlechner, NA Kairies, K Honold, S Scheiblich, H Koll, E Greiter, S Koch, W Schafer, R Huber, and RA Engh. (2005). J Mol Biol 353:222–231.
- G Jones, P Willett, RC Glen, AR Leach, and RJ Taylor. (1997). J Mol Biol 267:727–748.
- LJ Lombardo, FY Lee, P Chen, D Norris, JC Barrish, K Behnia, S Castaneda, LAM Cornelius, J Das, AM Doweyko, C Fairchild, JT Hunt, I Inigo, K Johnston, A Kamath, D Kan, H Klei, P Marathe, S Pang, R Peterson, S Pitt, RJ Schmidt, J Tokarski, ML Wen, J Wityak, and RM Borzilleri. (2004). J Med Chem 47:6658–6661.
- G Noronha, K Barrett, A Boccia, T Brodhag, J Cao, CP Chow, E Dneprovskaia, J Doukas, R Fine, X Gong, C Gritzen, H Gu, E Hanna, JD Hood, S Hu, X Kang, J Key, B Klebansky, A Kousba, G Li, D Lohse, CC Mak, A McPherson, MSS Palanki, VP Pathak, J Renick, F Shi, R Soll, U Splittgerber, S Stoughton, S Tang, S Yee, B Zeng, N Zhao, and H Zhu. (2007). Bioorg Med Chem Lett 17:602–608.
- G Noronha, K Barrett, J Cao, E Dneprovskaia, R Fine, X Gong, C Gritzen, C Gritzen, JD Hood, X Kang, B Klebansky, G Li, C Liao, D Lohse, CC Mak, A McPherson, MSS Palanki, VP Pathak, J Renick, R Soll, U Splittgerber, W Wrasidlo, B Zeng, N Zhao, and Y Zhouc. (2006). Bioorg Med Chem Lett 16:5546–5550.
- H Mukaiyama, T Nishimura, S Kobayashi, T Ozawa, N Kamada, Y Komatsu, S Kikuchi, H Oonota, and H Kusama. (2007). Bioorg Med Chem 15:868–885.
- BL Ellis, AK Duhme, RC Hider, MB Hossain, S Rizvi, and D Van der Helm. (1996). J Med Chem 39:3659–3670.
- Y Ma, W Luo, PJ Quinn, ZD Liu, and RC Hider. (2004). J Med Chem 47:6349–6362.
- ZD Liu, S Piyamongkol, DY Liu, HH Khodr, SL Lu, and RC Hider. (2001). Bioorg Med Chem 9:563–573.
- T Mukaiyama. (1979). Angew Chem Int Ed Engl 18:707–721.
- MD Aytemir, RC Hider, DD Erol, M Özalp, and M Ekizoglu. (2003). Turk J Chem 27:445–452.
- Y Ye, H Li, and H Jiang. (2005). Peptide Sci 80:172–178.