Abstract
A new series of fluorinated and non-fluorinated 2-phenylbenzimidazoles bearing oxygenated substituents on the phenyl ring has been synthesized. Synthesis of the new series was based on our previous discovery of 2-(3,4-dimethoxyphenyl)-5-fluorobenzothiazole (PMX 610) as a potent and selective antitumour agent in vitro (sub-nanomolar GI50 in sensitive human cancer cell lines), but with poor aqueous solubility and lack of a definitive cellular target limiting further development. In this study we test the hypothesis that 2-phenylbenzimidazoles with similar substitution patterns to PMX 610 would retain potent antitumour activity but with potentially superior pharmaceutical properties. In general the new compounds were less active than the former benzothiazole series in vitro when tested against the breast cancer cell lines MCF-7 and MDA 468; however the two most active compounds in the present series (3j and 3k) exhibit low micromolar GI50 values in both cell lines and provide the opportunity for further chemical derivatization with a view to target identification.
Introduction
The benzimidazole nucleus provides a versatile scaffold in medicinal chemistry for the design of new therapeutic agents, and a number of substituted benzimidazoles possessing a range of biological activities have been reported. Amongst the range of biological activities ascribed to benzimidazole derivatives have been antitumour, antiparasitic, antiviral and antimicrobial activity; this area of research has been recently reviewed [Citation1]. Of particular interest to us have been reports of antitumour benzimidazoles with activity against cancer drug targets of current interest, as exemplified for example by a benzimidazole-based inhibitor of polo-like kinase 1 [Citation2], 2-arylbenzimidazole-4-carboxamides as PARP-1 inhibitors [Citation3], and our own work on thioredoxin-inhibitory antitumour quinols including benzimidazole derivatives [Citation4].
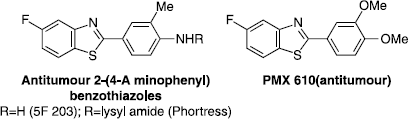
In recent years we have employed the 2-phenyl benzothiazole scaffold as inspiration for several anticancer drug discovery projects to provide novel agents acting on unusual mechanistic targets. The development of the antitumour benzothiazole prodrug Phortress (), currently in Phase 1 clinical development, illustrates a case in point [Citation5]. Phortress evolved from the discovery (through cell-based screening) of a series of 2-(4-aminophenyl)benzothiazoles with potent and selective in vitro and in vivo antitumour activity [Citation6]. Strategic installation of fluorine to circumvent deactivating metabolism led to the development of 2-(4-amino-3-methylphenyl)-5-fluorobenzothiazole (5F 203, ) as a lead compound from which the lysyl amide prodrug Phortress emerged as the clinical development candidate Citation7Citation8Citation9Citation10. More recently a further series of substituted 2-phenylbenzothiazoles has been synthesized and tested for cellular antitumour activity, leading to the identification of 2-(3,4-dimethoxyphenyl)-5-fluorobenzothiazole (PMX 610, ) as a new potent and selective antitumour agent with a selectivity profile reminiscent of the (aminophenyl)benzothiazoles, but with a distinct intracellular mechanism of action Citation11Citation12Citation13. Unfortunately the high lipophilicity associated with PMX 610 (estimated clogP 4.21; Crippen fragmentation [Citation14]) compromised further in vivo development through poor solubility in standard aqueous formulations.
Figure 1 Antitumour benzothiazoles.
In this paper we report the synthesis and in vitro antitumour evaluation of a new series of 2-phenylbenzimidazole derivatives bearing oxygenated substituents on the phenyl ring, based on the structure of PMX 610. Substituted 2-phenylbenzimidazoles are commonly synthesized via two well-established types of condensation reaction [Citation15]. The first method involves the thermal condensation of (substituted) o-phenylenediamine with carboxylic acids or their derivatives (e.g. nitriles, imidates or orthoesters). Alternatively aldehydes can be employed as reaction partners for o-phenylenediamines under oxidative conditions, since the initial product from the condensation reaction is formally a dihydrobenzimidazole.
The new analogues were synthesised to explore the following hypotheses:
Whether potent antitumour activity could be maintained for benzimidazole congeners of PMX 610, with an accompanying increase in hydrophilicity/water solubility (estimated clogP for the direct benzimidazole analogue of PMX 610 = 2.8 [Citation14]).
The necessity for fluorination on the benzimidazole ring for optimal activity, since fluorine is essential for the antitumour activity of PMX 610.
Materials and methods
Melting points were measured on a Griffin apparatus and are uncorrected. NMR spectra were recorded on a Bruker AVANCE 500 MHz instrument; coupling constants (J values) are in Hz. Thin layer chromatography (TLC) was performed using pre-coated, aluminium backed silica gel plates (F254-Merck) with UV visualisation. Merck silica gel 60 (40-60 μM) was used for column chromatography. All commercially available starting materials were used without further purification.
General procedure for the synthesis of 2-phenylbenzimidazoles 3a-f
Sodium metabisulfite (0.607 g, 3.16 mmol) was added to a stirring mixture of (substituted)-1,2-phenylenediamine 1a-c (3.12 mmol) and substituted benzaldehyde 2a-b (3.16 mmol) in DMF (10 mL), and the reaction mixture was heated under reflux for two hours. After completion of the reaction (monitored by TLC), the mixture was allowed to cool to room temperature. Addition of water (approximately 20 mL) caused precipitation of a solid, which was collected by vacuum filtration and dried. The crude product was further purified by recrystallization (aqueous methanol) to give the pure substituted 2-arylbenzimidazole as a solid in yields of 46-61%. The following compounds were prepared:
2-(3,4-Dimethoxyphenyl)-1H-benzimidazole (3a)
From 1,2-phenylenediamine and 3,4-dimethoxybenzaldehyde (55% yield), mp 231-233°C (MeOH/H2O); 1H NMR (DMSO-d6) δ 3.84 (3H, s, OMe), 3.88 (3H, s, OMe), 7.13 (1H, d, J 8.5 Hz, H-5′), 7.18 (2H, dt, J 3.7, 7.0 Hz, H-5, H-6), 7.57 (2H, dd, J 3.5, 5.0 Hz, H-4, H-7), 7.76 (1H, dd, J 2.0, 8.5 Hz, H-6′), 7.78 (1H, d, J 2.0 Hz, H-2′), 12.84 (1H, bs, NH). Anal. calcd. for C15H14N2O2: C, 70.85; H, 5.55; N, 11.01. Found: C, 70.56; H, 5.62; N, 10.75%.
2-(3,4-Dimethoxyphenyl)-5-fluoro-1H-benzimidazole (3b)
From 4-fluoro-1,2-phenylenediamine and 3,4-dimethoxybenzaldehyde (52% yield), mp 212-214°C (MeOH/H2O); 1H NMR (DMSO-d6) δ 3.83 (3H, s, OMe), 3.88 (3H, s, OMe), 7.03 (1H, dt, J 2.5, 10.5 Hz, H-6), 7.13 (1H, d, J = 8.5 Hz, H-5′) 7.36 (1H, m, ArH) 7.55 (1H, m, ArH), 7.73 (1H, dd, J 1.5, 8.5 Hz, H-6′), 7.75 (1H, d, J 1.5 Hz, H-2′), 12.78 (1H, bs, NH). Anal. calcd. for C15H13N2O2F: C, 66.17; H, 4.81; N, 10.28. Found: C, 66.00; H, 4.86; N, 10.08%.
2-(3,4-Dimethoxyphenyl)-1-methyl-1H-benzimidazole (3c)
From N-methyl-1,2-phenylenediamine and 3,4-dimethoxybenzaldehyde (61% yield), mp 107-109°C (MeOH/H2O); 1H NMR (CDCl3) δ 3.85 (3H, s, NMe), 3.95 (3H, s, OMe), 3.96 (3H, s, OMe), 6.98 (1H, d, J 8.0 Hz, H-5′), 7.24 (1H, dd, J = 1.8, 8.3 Hz, H-6′), 7.30 (2H, m, ArH), 7.36 (2H, m, ArH), 7.79 (1H, m, ArH). Anal. calcd. for C16H16N2O2: C, 71.62; H, 6.01; N, 10.44. Found: C, 71.85; H, 6.02; N, 10.39%.
2-(3,4,5-Trimethoxyphenyl)-1H-benzimidazole (3d)
From 1,2-phenylenediamine and 3,4,5-trimethoxybenzaldehyde (46% yield), mp 254-255°C (MeOH/H2O); 1H NMR (DMSO-d6) δ 3.74 (3H, s, OMe), 3.91 (6H, s, 2 × OMe), 7.20 (2H, dd, J 3.0, 5.5 Hz, H-4, H-7), 7.53 (2H, s, H-2′, H-6′), 7.60 (2H, m, H-5, H-6), 12.83 (1H, bs, NH). Anal. calcd. for C16H16N2O3: C, 67.59; H, 5.67; N, 9.85. Found: C, 67.65; H, 5.71; N, 9.73%.
5-Fluoro-2-(3,4,5-trimethoxyphenyl)-1H-benzimidazole (3e)
From 4-fluoro-1,2-phenylenediamine and 3,4,5-trimethoxybenzaldehyde (57% yield), mp 243-245°C (MeOH/H2O); 1H NMR (DMSO-d6) δ 3.74 (3H, s, OMe), 3.91 (6H, s, 2 × OMe), 7.06 (1H, dt, J 2.5, 9.5 Hz, H-6), 7.40 (1H, d, J 9.0 Hz, ArH), 7.51 (2H, s, H-2′, H-6′), 7.59 (1H, m, Ar-H), 12.83 (1H, bs, NH). Anal. calcd. for C16H15N2O3F: C, 63.57; H, 5.00; N, 9.26. Found: C, 63.51; H, 5.03; N, 9.14%.
1-Methyl-2-(3,4,5-trimethoxyphenyl)-1H-benzimidazole (3f)
From N-methyl-1,2-phenylenediamine and 3,4,5-trimethoxybenzaldehyde (59% yield), mp 114-116°C (MeOH/H2O); 1H NMR (CDCl3) δ 3.89 (3H, s, NMe), 3.93 (3H, s, 4′-OMe), 3.94 (6H, s, 2 × OMe), 6.96 (2H, s, H-2′, H-6′), 7.31 (2H, m, ArH), 7.39 (1H, m, ArH), 7.82 (1H, m, ArH). Anal. calcd. for C17H18N2O3: C, 68.44; H, 6.08; N, 9.39. Found: C, 68.85; H, 6.19; N, 9.48%.
General method for the synthesis of 2-phenylbenzimidazoles 3g-i
Triethylamine (5.8 mL, 4 mmol) was added to a solution of (substituted)-1,2-phenylenediamine (0.291 g, 2.7 mmol) and piperonyloyl chloride (0.5 g, 2.7 mmol) in THF (10 mL) at 0°C with stirring. The reaction mixture was stirred at 0°C for around one hour (until reaction was complete by TLC), then the solution was concentrated in vacuo. The resulting residue was further heated under reflux in acetic acid (100°C) for 12 h. The solution was then neutralised (aqueous sodium hydroxide), and the product extracted using ethyl acetate (2 × 20 mL), then dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (60% CH2Cl2-40% EtOAc) afforded the title compound as a white solid.
2-(Benzo[1,3]dioxol-5-yl)-1H-benzimidazole (3g)
From 1,2-phenylenediamine and piperonyloyl chloride (46% yield), m.p. 242-244°C (MeOH/H2O); 1H NMR (DMSO-d6) δ 6.13 (2H, s, OCH2), 7.10 (1H, d, J 8.8 Hz, H-5′), 7.18 (2H, m, Ar-H), 7.56 (1H, m, Ar-H) 7.65 (1H, m, Ar-H), 7.69 (1H, d, J 1.5 Hz, H-2′), 7.72 (1H, dd, J 1.5, 8.8 Hz, H-6′), 12.79 (1H, bs, NH). Anal. calcd. for C14H10N2O2: C, 70.58; H, 4.23; N, 11.75. Found: C, 70.31; H, 4.21; N, 11.66%.
2-(Benzo[1,3]dioxol-5-yl)-5-fluoro-1H-benzimidazole (3h)
From 4-fluoro-1,2-phenylenediamine and piperonyloyl chloride (46% yield), mp 205-207°C (MeOH/H2O); 1H NMR (DMSO-d6) δ 6.13 (2H, s, OCH2), 7.04 (1H, dt, J 2.5, 10.0 Hz, H-6), 7.10 (1H, d, J 8.0 Hz, H-5′), 7.36 (1H, m, Ar-H), 7.55 (1H, m, Ar-H) 7.66 (1H, d, J 2.0 Hz, H-2′), 7.70 (1H, dd, J 2.0, 8.0 Hz, H-6′), 12.86 (1H, bs, NH). Anal. calcd. for C14H9N2O2F: C, 65.62; H, 3.54; N, 10.93. Found: C, 65.38; H, 3.56; N, 10.74%.
2-(Benzo[1,3]dioxol-5-yl)-1-methyl-1H-benzimidazole (3i)
From N-methyl-1,2- phenylenediamine and piperonyloyl chloride (59% yield), mp 149-151°C (MeOH/H2O); 1H NMR (CDCl3) δ 3.84 (3H, s, NCH3), 6.05 (2H, s, OCH2), 6.94 (1H, d, J 8.0 Hz, H-5′), 7.23 (2H, m, Ar-H), 7.29 (2H, m, Ar-H) 7.36 (1H, m, Ar-H), 7.77 (1H, m, Ar-H). Anal. calcd. for C15H12N2O2: C, 71.42; H, 4.79; N, 11.10. Found: C, 71.02; H, 4.75; N, 11.00%.
General method for the synthesis of 2-phenylbenzimidazoles 3j-k
p-Toluenesulfonylchloride (1.1 mmol) was added in small portions to a stirring solution of 2-(3,4-dimethoxyphenyl)-5-fluoro-1H-benzimidazole 3b (0.73 mmol) in pyridine (10 mL) under nitrogen, and the reaction mixture was stirred for 24 h. The reaction mixture was then diluted with H2O and extracted with dichloromethane (3 × 20 mL), dried over MgSO4 and concentrated in vacuo. The two regioisomeric products (3j and 3k) were separated by column chromatography (DCM/EtOAc) to give the pure title compounds as solids.
2-(3,4-Dimethoxyphenyl)-5-fluoro-1-toluenesulfonyl-1H-benzimidazole (3j)
(21% yield), m.p.123-125°C (MeOH/H2O); 1H NMR (CDCl3) δ 2.43 (3H, s, CH3), 3.81 (3H, s, OCH3), 3.91 (3H, s, OCH3), 6.88 (1H, d, J 8.0 Hz, H-5′), 7.01 (2H, d, J 8.5 Hz, H-3″, H-5″), 7.03 (1H, d, J 2.0 Hz, H-2′), 7.08 (1H, dt, J 2.5 Hz, 9.0 Hz, H-6), 7.17 (1H, dd, J 2.0 Hz, 8.0 Hz, H-6′), 7.18 (2H, d, J 8.5 Hz, H-2″,H-6″), 7.29 (1H, dd, J 2.5 Hz, 9.5 Hz, H-4), 8.08 (1H, dd, J 5.0 Hz, 9.5 Hz, H-7). Anal. calcd. for C22H19N2O4FS: C, 61.96; H, 4.49; N, 6.57. Found: C, 61.64; H, 4.46; N, 6.38%.
2-(3,4-Dimethoxyphenyl)-6-fluoro-1-toluenesulfonyl-1H-benzimidazole (3k)
(16% yield), m.p.129-131°C (MeOH/H2O); 1H NMR (CDCl3) δ 2.43 (3H, s, CH3), 3.81 (3H, s, OCH3), 3.91 (3H, s, OCH3), 6.87 (1H, d, J 8.5 Hz, H-5′), 7.00 (1H, d, J 2.0 Hz, H-2′), 7.04 (2H, d, J 8.5 Hz, H-3″, H-5″), 7.07 (1H, dt, J 2.5 Hz, 9.0 Hz, H-5), 7.15 (1H, dd, J 2.0 Hz, 8.5 Hz, H-6′), 7.22 (2H, d, J 8.5 Hz, H-2″, H-6″), 7.56 (1H, dd, J 5.0 Hz, 9.5 Hz, H-4), 7.88 (1H, dd, J 2.5 Hz, 9.5 Hz, H-7). Anal. calcd. for C22H19N2O4FS: C, 61.96; H, 4.49; N, 6.57. Found: C, 61.65; H, 4.48; N, 6.28%.
In vitro antitumour evaluation in human tumour cell lines
Compounds were prepared as 10 mM top stock solutions, dissolved in DMSO, and stored at 4°C, protected from light for a maximum period of 4 weeks. Human derived cell lines (MCF-7 (ER+), MDA 468 (ER-) breast carcinoma) were routinely cultivated at 37°C in an atmosphere of 5% CO2 in RPMI 1640 medium supplemented with 2 mM L-glutamine and 10% fetal calf serum and subcultured twice weekly to maintain continuous logarithmic growth. Cells were seeded into 96-well microtiter plates at a density of 5 × 103 per well and allowed 24 h to adhere before drugs were introduced (final concentration 0.1 nM-100 μM, n = 8). Serial drug dilutions were prepared in medium immediately prior to each assay. At the time of drug addition and following 72 h exposure, 1-(4,5-dimethylthiazol-2-yl)-3,5-diphenylformazan (MTT) was added to each well (final concentration 400 μg/mL). Incubation at 37°C for 4 h allowed reduction of MTT by viable cells to an insoluble formazan product. Well contents were aspirated and formazan solubilized by addition of DMSO. Absorbance was read on an Anthos Labtec systems plate reader at 550 nm as a measure of cell viability; thus cell growth or drug toxicity was determined.
Results
Benzimidazole synthesis
The synthesis of substituted 2-phenylbenzimidazoles 3a-f was accomplished in a single synthetic step from commercially available starting materials, as illustrated in Scheme . Following heating of (substituted) benzene-1,2-diamine and substituted benzaldehyde under reflux for two hours in the presence of sodium metabisulfite, addition of water produced a precipitate that was collected and recrystallized to give the pure 2-phenylbenzimidazole product in moderate yield (46-61%) without the need for chromatographic purification. A different route was employed to access the 2-phenylbenzimidazoles 3g-i containing a methylenedioxy bridge, due to the restricted commercial availability of piperonal. In these cases the benzenediamine 1a-c was treated with piperonoyl chloride 4 in the presence of triethylamine at 0°C followed by heating in acetic acid. Neutralisation, extraction and purification using column chromatography gave the required pure 2-phenylbenzimidazole product 3g-i in moderate yield (46-59%).
Scheme 1 Synthesis of 2-phenylbenzimidazole products 3a-k.
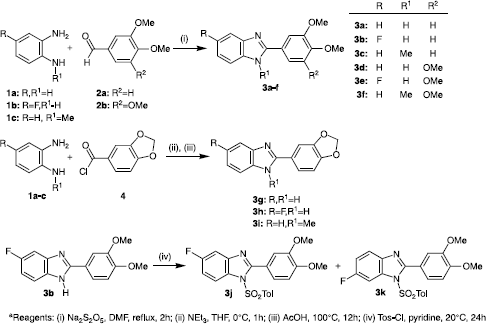
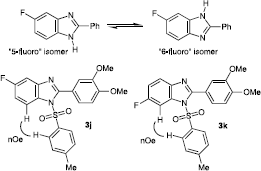
The fluorinated 2-phenylbenzimidazole products (3b, 3e, 3h) resulting from reaction of 4-fluoro-1,2-benzenediamine 1b, although represented here as 5-fluorobenzimidazole products, are actually a rapidly equilibrating mixture of the formal 5- and 6-substituted products due to tautomerism of the benzimidazole N-proton on the NMR timescale (). Deprotonation and substitution of the benzimidazole N-proton would prevent this rapid tautomerism and lead to a separable equimolar mixture of 5- and 6-substituted products. Hence the fluorinated 2-phenylbenzimidazole product 3b was treated with p-toluenesulfonyl chloride (Tos-Cl) in pyridine, leading to the isolation of an equimolar mixture of products, which were separated using column chromatography to give the pure 5-fluoro- and 6-fluoro-benzimidazole products 3j and 3k respectively. Characterisation of the individual isomeric products was carried out by observation of a 1H NMR nuclear Overhauser enhancements (nOe) between tolyl protons and the benzimidazole H-7, allowing the H-7 proton to be characterised and identified for each particular fluorinated isomer on the basis of its 1H NMR splitting pattern (, see Materials and Methods for details).
Figure 2 N-1/N-3 proton tautomerisation in compounds 3b, 3e and 3h, and nOe effects in products 3j and 3k.
In vitro antitumour activity
New compounds were tested for in vitro antitumour activity in the human breast carcinoma cell lines MCF-7 (ER + ve) and MDA 468 (ER-ve) using the well-established MTT assay [Citation11] following drug incubation over 72 h (see Materials and Methods for details). MCF-7 and MDA 468 cell lines were chosen here since they were previously shown to be exquisitely sensitive to the lead benzothiazole PMX 610. lists the results (GI50 values) of our antitumour evaluation studies and includes the data previously obtained using the same method for PMX 610 [Citation11].
Table I. In vitro antitumour activity against the MCF-7 and MDA 468 breast cancer cell lines.
Discussion
Inspection of reveals that the new 2-phenylbenzimidazole derivatives prepared in this study do not display antitumour activity at the sub-nanomolar GI50 level seen for PMX 610, for the two breast cancer cell lines examined. In most cases growth inhibitory values were in the micromolar range. This point is illustrated by comparison of the lead benzothiazole (PMX 610; MCF-7 GI50 = < 0.0001 μM) with its direct benzimidazole analogue (3b MCF-7 GI50 = 85.7 μM). Notable observations were that the presence of a fluorine atom in the benzimidazole ring did not lead to substantial enhancement of antitumour activity (compare 3b versus 3a, or 3e versus 3d for example). Also noteworthy was that the N-methylated compounds 3c, 3f and 3i were essentially inactive.
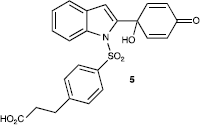
The most potent and interesting compounds from this new series were the 5-fluoro- and 6-fluoro-benzimidazoles 3j and 3k where the benzimidazole nitrogen bears an electron-withdrawing group (p-tolylsulfonyl). In these cases low micromolar GI50 values were obtained, suggesting that there is space for further drug design and potential for the installation of other electron-withdrawing groups in this position in order to further optimise activity. Clearly the synthesis of further agents of this type would be greatly assisted by identification of a molecular target responsible for the observed antitumour activity. Fortunately the tolylsulfonyl-substituted benzimidazole offers the potential for further derivatisation with a view to linking to a solid support followed by challenge with cellular lysate and identification of potential molecular targets using mass spectrometry. An analogous approach has been previously tested in our group to identify molecular targets of the arylsulfonyl-derived indole quinol 5 (), through derivatisation with amino-terminal agarose beads [Citation16]. This type of proteomic approach should also be achievable here provided that a carboxylic acid terminal group attached to the arylsulfonyl moiety does not compromise the antitumour activity.
Figure 3 Structure of carboxylic acid linked (arylsulfonyl) indole-substituted antitumour quinol 5.
Conclusions
The new series of substituted 2-phenylbenzimidazoles 3a-k did not recapitulate the potent activity previously observed for the corresponding benzothiazole PMX 610; however the tolylsulfonyl-derived benzimidazoles 3j and 3k gave low micromolar GI50 values in both MCF-7 and MDA 468 human cancer cell lines and will be the focus of future studies. The search for a molecular target on which to base a rational drug design effort to optimise antitumour activity may be aided to the presence of the arylsulfonyl group, through installation of a synthetic handle for proteomic target identification studies.
Acknowledgements
We thank the Algerian Government for a PhD studentship (to HK), and Pharminox and Cancer Research UK for providing funding to support the biological studies.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- M Boiani, and M Gonzalez. (2005). Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini Rev Med Chem 5:409–424.
- TJ Lansing, RT McConnell, DR Duckett, GM Spehar, VB Knick, DF Hassler, N Noro, M Furuta, KA Emmitte, TM Gilmer, RA MookJr, and M Cheung. (2007). In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol Cancer Therap 6:450–459.
- AW White, NJ Curtin, BW Eastman, BT Golding, Z Hostomsky, S Kyle, J Li, KA Maegley, DJ Skalitzky, SE Webber, XH Yu, and RJ Griffin. (2004). Potentiation of cytotoxic drug activity in human tumour cell lines, by amino-substituted 2-arylbenzimidazole-4-carboxamide PARP-1 inhibitors. Bioorg Med Chem Lett 14:2433–2437.
- JM Berry, TD Bradshaw, I Fichtner, R Ren, CH Schwalbe, G Wells, E-H Chew, MFG Stevens, and AD Westwell. (2005). Quinols as novel therapeutic agents. 2. 4-(1-arylsulfonylindol-2-yl)-4-hydroxycyclohexa-2,5-dien-1-ones and related agents as potent and selective antitumor agents. J Med Chem 48:639–644.
- TD Bradshaw, and AD Westwell. (2004). The development of the antitumour benzothiazole prodrug, Phortress, as a clinical candidate. Curr Med Chem 11:1241–1253.
- TD Bradshaw, MFG Stevens, and AD Westwell. (2001). The discovery of the potent and selective antitumour agent 2-(4-amino-3-methylphenyl)benzothiazole (DF 203) and related compounds. Curr Med Chem 8:203–210.
- P Shah, and AD Westwell. (2007). The role of fluorine in medicinal chemistry. J Enz Inhib Med Chem 22:527–540.
- I Hutchinson, M-S Chua, H Browne, V Trapani, I Fichtner, TD Bradshaw, AD Westwell, and MFG Stevens. (2001). Antitumor benzothiazoles. 14. The synthesis and in vitro biological properties of fluorinated 2-(4-aminophenyl)benzothiazoles. J Med Chem 44:1446–1455.
- E Kashiyama, I Hutchinson, M-S Chua, SF Stinson, LR Phillips, G Kaur, EA Sausville, TD Bradshaw, AD Westwell, and MFG Stevens. (1999). Antitumor benzothiazoles. 8. Synthesis, metabolic formation, and biological properties of the C- and N- oxidation products of antitumor 2-(4-aminophenyl)benzothiazoles. J Med Chem 42:4172–4184.
- I Hutchinson, SA Jennings, BR Vishnuvajjala, AD Westwell, and MFG Stevens. (2002). Antitumor benzothiazoles. 16. Synthesis and pharmaceutical properties of antitumor 2-(4-aminophenyl)benzothiazole amino acid prodrugs. J Med Chem 45:744–747.
- CG Mortimer, G Wells, J-P Crochard, EL Stone, TD Bradshaw, MFG Stevens, and AD Westwell. (2006). Antitumor benzothiazoles. 26. 2-(3,4-Dimethoxyphenyl)-5-fluorobenzothiazole (GW 610, NSC 721648), a simple fluorinated 2-arylbenzothiazole, shows potent and selective inhibitory activity against lung, colon, and breast cancer cell lines. J Med Chem 49:179–185.
- E Brantley, V Trapani, MC Alley, CD Hose, TD Bradshaw, MFG Stevens, EA Sausville, and SF Stinson. (2004). Fluorinated 2-(4-amino-3-methylphenyl)benzothiazoles induce CYP1A1 expression, become metabolized, and bind to macromolecules in sensitive human cancer cells. Drug Metab Dispos 32:1392–1401.
- M-S Chua, E Kashiyama, TD Bradshaw, SF Stinson, E Brantley, EA Sausville, and MFG Stevens. (2000). Role of CYP1A1 in modulation of antitumor properties of the novel agent 2-(4-amino-3-methylphenyl)benzothiazole (DF 203, NSC 674495) in human breast cancer cells. Cancer Res 60:5196–5203.
- AK Ghose, and GM Crippen. (1987). Atomic physicochemical parameters for 3-dimensional structure-activity-directed quantitative structure-activity relationships. 2. Modeling dispersive and hydrophobic interactions. J Chem Inf Comp Sci 27:21–35.
- MR Grimmet. Imidazoles and their benzo derivativesComprehensive heterocyclic chemistryAR Katritzky, and CW Rees. Pergamon, Oxford, (1984) Vol. 5 457.
- E-H Chew, CS Matthews, J Zhang, AJ McCarroll, T Hagen, MFG Stevens, AD Westwell, and TD Bradshaw. (2006). Antitumor quinols: Role of glutathione in modulating quinol-induced apoptosis and identification of putative cellular protein targets. Biochem Biophys Res Comm 346:242–251.