Abstract
Pharmacomodulations of previously reported thiaindanones related to donepezil were achieved with the aim to enhance their AChE inhibitory activity. Condensation of the cyclopentane carbonyl group into hydrazone or cyanolefine derivatives, as well as its hydrogenation and the subsequent substitution of the resulting hydroxyl group led to new 2-(4-benzylpiperazin-1-yl)-N-(1,3-dibromo-6-hydroxy-5,6-dihydro-4H-cyclopenta[c]thien-4-yl) acetamides. The in vitro evaluation of this new series, according to the method of Ellman, shows however that it conserved only partially the biological activity. The best compound remains the alcohol 11 (IC50 = 0.40 μM, against 0.02 μM for donepezil).
Introduction
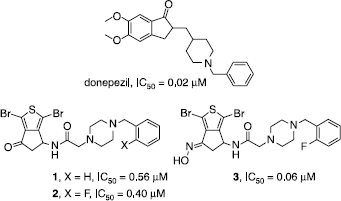
We have previously reported the synthesis and the important in vitro acetylcholinesterase (AChE) inhibitory activity of some donepezil-like thiaindanone derivatives 1-3 () [Citation1]. A molecular modelling study has further designed the interactions engaged by these inhibitors with the enzyme. In view to evaluate the role played by these interactions in the AChE inhibition and to potentially enhance the activity of our series, we have performed further pharmacomodulations whose we report herein the results which particularly concern the cyclopentane carbonyl group and the benzylpiperazine moiety.
Figure 1 Structure and anti-AChE activity of donepezil and thiaindanone derivatives 1-3.
Materials and methods
Chemistry
Instrumentation
Melting points were determined on a Kofler melting point apparatus and are uncorrected. Elemental analyses for new compounds were performed at the Institut de Recherche en Chimie Organique Fine (Rouen, France). IR spectra were recorded on a Genesis series FTIR spectrometer using KBr pellets. The 1H (400 MHz) and 13C (100 MHz) NMR spectra were obtained on a Jeol Lambda 400 spectrometer using DMSO-d6 or CDCl3 as solvent and TMS as internal standard. The chemical shifts (δ) are reported in ppm, and the coupling constants are in Hertz. Electron impact mass spectra (EIMS) were obtained using a Jeol JMS GCMate spectrometer. Reactions were monitored by thin-layer chromatography (TLC) using 0.2 mm Polygram Sil silica gel G/UV 254 precoated plates with visualization by irradiation with a short-wavelength UV light. Silica gel flash chromatography was performed using 63-200 mM Kieselgel Merck 60 silica gel.
Synthesis
2-[4-(2-Fluorobenzyl)piperazin-1-yl]-N-(1,3-dibromo-6-hydroxy-5,6-dihydro-4H-cyclopenta[c]thien-4-yl)acetamide (11)
0.21 g (5.5 mmol) of sodium borohydride was added by small portions to a solution of 2.74 g (5 mmol) of ketone 2 in 50 mL methanol at 0°C. The mixture was then stirred for 30 min at room temperature. The solvent was evaporated under reduced pressure and the residue was dissolved in 100 mL of dichloromethane and washed with 100 mL of water. The organic layer was separated, dried over anhydrous CaCl2, filtered and evaporated under reduced pressure to give 2.63 g of product as a mixture (50:50) of syn and anti isomers. Yield: 96%. MP 110 °C. IR (cm− 1): 3377, 2937, 1695, 1492, 1229, 1098, 1010, 758. MS (m/z): 544.8 (M+ − 2), 546.8 (M+), 548.8 (M++2).
Syn isomer: 1H NMR (CDCl3) δ: 7.79 (d, J = 8.0 Hz, 1H, NH), 7.27 (m, 1H, H-6′), 7.22 (m, 1H, H-4′), 7.10 (t, J = 7.0 Hz, 1H, H-5′), 7.02 (t, J = 9.2 Hz, 1H, H-3′), 4.78 (t, J = 8.0 Hz, 1H, H-6), 4.69 (dt, J = 3.2 Hz, J = 8.0 Hz, 1H, H-4), 4.61 (d, J = 8.0 Hz, 1H, OH), 3.51 (s, 2H, Hbenzyl), 3.07 (dt, J = 8.0, 19.4 Hz, 1H, H-5b), 2.93 (s, 2H, COCH2N), 2.44 (m, 8H, Hpiperazine), 2.18 (dt, J = 3.2, 19.4 Hz, 1H, H-5a). 13C NMR (CDCl3) δ: 170.33, 161.34 (d, J = 245.2 Hz), 150.21, 146.48, 131.51 (d, J = 6.5 Hz), 128.88 (d, J = 8.2 Hz), 124.24 (d, J = 14.2 Hz), 123.91 (d, J = 2.5 Hz), 115.24 (d, J = 22.3 Hz), 105.27, 103.74, 68.66, 61.32, 54.97, 53.54, 52.70, 48.89, 48.13.
Anti isomer: 1H NMR (CDCl3) δ: 7.34 (d, J = 8.5 Hz, 1H, NH), 7.27 (m, 1H, H-6′), 7.22 (m, 1H, H-4′), 7.10 (t, J = 7.0 Hz, 1H, H-5′), 7.02 (t, J = 9.2 Hz, 1H, H-3′), 5.47 (m, 1H, H-4), 4.92 (d, J = 7.3 Hz, 1H, H-6), 3.72 (large, 1H, OH), 3.51 (s, 2H, Hbenzyl), 3.17 (ddd, J = 3.0, 7.3, 15.9 Hz, 1H, H-5a), 2.93 (s, 2H, COCH2N), 2.44 (m, 8H, Hpiperazine and H-5b). 13C NMR (CDCl3) δ: 170.22, 161.34 (d, J = 245.2 Hz), 149.73, 146.37, 131.51 (d, J = 6.5 Hz), 128.88 (d, J = 8.2 Hz), 124.24 (d, J = 14.2 Hz), 123.91 (d, J = 2.5 Hz), 115.24 (d, J = 22.3 Hz), 105.00, 103.46, 67.95, 61.25, 54.96, 53.39, 52.69, 49.38, 48.71.
2-[4-(2-Fluorobenzyl)piperazin-1-yl]-N-(1,3-dibromo-6-fluoro-5,6-dihydro-4H-cyclopenta[c]thien-4-yl)acetamide (12)
0.12 mL (1.1 mmol) of DAST in 5 mL of dry dichloromethane was added dropwise to a solution of 547 mg (1 mmol) of the alcohol 11 (syn/anti 50:50) in 20 mL of dry dichloromethane at 0°C. The mixture was stirred for further 4 hours at the same temperature. The reaction was then quenched with 100 mL of aqueous saturated NaHCO3 solution. The aqueous layer was extracted twice with 100 mL of dichloromethane. The combined organic extracts were dried over anhydrous CaCl2, filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2/MeOH 93:7) to give 300 mg of product as a mixture (50:50) of syn and anti isomers. Yield: 55%. MP 113 °C. IR (cm− 1): 3419, 1660, 1494, 1455, 1083, 777, 582. MS (m/z): 547.1 (M+ − 2), 549.1 (M+), 551.1 (M++2).
Syn isomer: 1H NMR (CDCl3) δ: 7.40 (d, J = 8.0 Hz, 1H, NH), 7.34 (m, 1H, H-6′), 7.20 (m, 1H, H-4′), 7.04 (t, J = 7.5 Hz, 1H, H-5′), 6.96 (t, J = 9.6 Hz, 1H, H-3′), 5.61 (m, 1H, H-6), 5.17 (m, 1H, H-4), 3.56 (s, 2H, Hbenzyl), 3.04 (m, 3H, COCH2N and H-5b), 2.44 (m, 8H, Hpiperazine), 2.35 (m, 1H, H-5a). 13C NMR (CDCl3) δ: 170.11, 161.40 (d, J = 255.0 Hz), 147.38, 145.51, 131.81 (d, J = 6.5 Hz), 129.25 (d, J = 8.2 Hz), 124.24 (d, J = 2.5 Hz), 124.12 (d, J = 14.2 Hz), 115.24 (d, J = 23.2 Hz), 108.37 (d, J = 5.8 Hz), 103.19, 88.03 (d, J = 176.9 Hz), 61.30, 54.90, 53.39, 52.62, 47.22 (d, J = 24.2 Hz), 46.04.
Anti isomer: 1H NMR (CDCl3) δ: 7.40 (d, J = 8.0 Hz, 1H, NH), 7.34 (m, 1H, H-6′), 7.20 (m, 1H, H-4′), 7.04 (t, J = 7.5 Hz, 1H, H-5′), 6.96 (t, J = 9.6 Hz, 1H, H-3′), 5.47 (m, 1H, H-6), 5.40 (m, 1H, H-4), 3.56 (s, 2H, Hbenzyl), 3.04 (m, 3H, COCH2N and H-5b), 2.44 (m, 9H, Hpiperazine and H-5a). 13C NMR (CDCl3) δ: 169.54, 161.40 (d, J = 255.0 Hz), 146.40, 145.51, 131.81 (d, J = 6.5 Hz), 129.25 (d, J = 8.2 Hz), 124.24 (d, J = 2.5 Hz), 124.12 (d, J = 14.2 Hz), 115.24 (d, J = 23.2 Hz), 108.37 (d, J = 5.8 Hz), 103.19, 86.97 (d, J = 177.3 Hz), 61.30, 54.90, 53.16, 52.62, 47.02 (d, J = 24.2 Hz), 42.36.
2-[4-(2-Fluorobenzyl)piperazin-1-yl]-N-(1,3-dibromo-6-chloro-5,6-dihydro-4H-cyclopenta[c]thien-4-yl)acetamide (13)
0.15 mL (2.0 mmol) of thionyl chloride in 5 mL of dry dichloromethane was added dropwise to a solution of 547 mg (1 mmol) of the alcohol 11 (syn/anti 50:50) in 20 mL of dry dichloromethane at 0°C. The mixture was then refluxed for 2 h. The reaction was quenched with 100 mL of aqueous saturated NaHCO3 solution. The aqueous layer was extracted twice with 100 mL of dichloromethane. The combined organic extracts were dried over anhydrous CaCl2, filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2/MeOH 93:7) to give 170 mg of amorphous solid also as a mixture (50:50) of syn and anti isomers. Yield: 30%. IR (cm− 1): 3428, 1651, 1495, 1457, 1235, 962, 763. HRMS: calculated (564.9424); found (564.9419).
Syn isomer: 1H NMR (CDCl3) δ: 7.86 (d, J = 7.1 Hz, 1H, NH), 7.39 (m, 1H, H-6′), 7.23 (m, 2H, H-4′ and H-5′), 7.08 (t, J = 9.0 Hz, 1H, H-3′), 5.51 (m, 1H, H-4), 5.02 (m, 1H, H-6), 4.16 (s, 2H, Hbenzyl), 3.33 (m, 2H, and COCH2N), 2.84 (m, 9H, H-5b and Hpiperazine), 2.42 (m, 1H, H-5a). 13C NMR (CDCl3) δ: 170.11, 161.14 (d, J = 245.2 Hz), 149.81, 147.47, 131.51 (d, J = 6.5 Hz), 128.88 (d, J = 8.2 Hz), 124.22 (d, J = 14.2 Hz), 123.90 (d, J = 2.5 Hz), 115.24 (d, J = 22.2 Hz), 105.97, 104.64, 61.66, 61.32, 54.97, 53.54, 52.70, 48.81, 48.13.
Anti isomer: 1H NMR (CDCl3) δ: 7.83 (d, J = 7.1 Hz, 1H, NH), 7.39 (m, 1H, H-6′), 7.23 (m, 2H, H-4′ and H-5′), 7.08 (t, J = 9.0 Hz, 1H, H-3′), 5.26 (m, 1H, H-4), 5.06 (m, 1H, H-6), 4.16 (s, 2H, Hbenzyl), 3.33 (m, 2H, and COCH2N), 2.84 (m, 9H, H-5b and Hpiperazine), 2.62 (m, 1H, H-5a). 13C NMR (CDCl3) δ: 170.01, 161.14 (d, J = 245.2 Hz), 149.81, 147.47, 131.51 (d, J = 6.5 Hz), 128.88 (d, J = 8.2 Hz), 124.22 (d, J = 14.2 Hz), 123.90 (d, J = 2.5 Hz), 115.24 (d, J = 22.2 Hz), 105.97, 104.64, 62.96, 61.32, 54.97, 53.54, 52.70, 49.94, 48.13.
2-[4-(2-Fluorobenzyl)piperazin-1-yl]-N-[(6E)-1,3-dibromo-6-hydrazono-5,6-dihydro-4H-cyclopenta[c]thien-4-yl]acetamide (14)
0.2 mL of hydrazine hydrate was added to a solution of 545 mg (1 mmol) of the ketone 12 in 20 mL of methanol. The mixture was then refluxed for three hours. The solvent was evaporated under reduced pressure and the residue was dissolved in 100 mL of dichloromethane and washed twice with 100 mL of water. The organic layer was separated, dried over anhydrous CaCl2, filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2/MeOH 95:5) to give 500 mg of the desired hydrazone. Yield: 90%. MP 115 °C. IR (cm− 1): 3382, 1652, 1494, 1489, 1456, 1229, 1096, 1011, 758. MS (m/z): 478.0 (M+ − 1 − Br), 480.0 (M++1 + Br). 1H NMR (CDCl3) δ: 7.51 (d, J = 8.4 Hz, 1H, NH), 7.36 (t, J = 6.8 Hz, 1H, H-6′), 7.23 (m, 1H, H-4′), 7.11 (t, J = 6.8 Hz, 1H, H-5′), 7.02 (t, J = 8.5 Hz, 1H, H-3′), 5.38 (dt, J = 3.4, 8.4 Hz, 1H, H-4), 4.52 (s, 2H, NH2), 3.58 (s, 2H, Hbenzyl), 3.51 (dd, J = 8.4, 17.9 Hz, 1H, H-5b), 3.04 (m, 2H, COCH2N), 2.72 (dd, J = 3.6, 17.9 Hz, 1H, H-5a), 2.51 (m, 8H, Hpiperazine). 13C NMR (CDCl3) δ: 170.20, 161.41 (d, J = 244.3 Hz), 147.62, 145.17, 142.02, 131.52 (d, J = 4.9 Hz), 128.93 (d, J = 8.2 Hz), 124.34 (d, J = 3.3 Hz), 123.91 (d, J = 14.9 Hz), 115.31 (d, J = 21.4 Hz), 104.82, 101.84, 61.35, 55.02, 53.58, 52.70, 45.46, 40.64.
2-[4-(2-Fluorobenzyl)piperazin-1-yl]-N-[(6E)-1,3-dibromo-6-cyanomethylene-5,6-dihydro-4H-cyclopenta[c]thien-4-yl]acetamide (15)
135 mg (1.2 mmol) of potassium carbonate was added to a solution of 213 mg (1.2 mmol) of diethyl cyanomethylphosphonate in 10 mL of dry THF under argon atmosphere. The mixture was refluxed for 20 min. The reaction was then allowed to return to the room temperature and 545 mg (1 mmol) of the ketone 12 was added. The mixture was then refluxed again for further three hours. The reaction was allowed to return to the room temperature and 15 mL of 10% potassium carbonate aqueous solution were then added. The mixture was extracted three times by 100 mL of AcOEt. The combined organic extracts were dried over anhydrous MgSO4, filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2/MeOH 95:5) to give 360 mg of the desired product as amorphous solid. Yield: 66%. IR (cm− 1): 3436, 2208, 1689, 1552, 1500, 1261, 1024, 762. HRMS: calculated (567.9766); found (567.9770). 1H NMR (CDCl3) δ: 7.54 (s, 1H, CHCN), 7.29 (m, 3H, NH, H-4′ and H-6′), 7.13 (t, J = 7.6 Hz, 1H, H-5′), 7.06 (t, J = 9.0 Hz, 1H, H-3′), 4.38 (m, 2H, H-4 and H-5b), 3.66 (s, 2H, Hbenzyl), 3.20 (m, 3H, COCH2N), 2.88 (dd, J = 3.6, 21.5 Hz, 1H, H-5a), 2.60 (m, 8H, Hpiperazine). 13C NMR (CDCl3) δ: 169.80, 161.63 (d, J = 244.3 Hz), 150.17, 140.81, 138.68, 131.36 (d, J = 4.1 Hz), 129.39 (d, J = 8.2 Hz), 124.11 (d, J = 3.3 Hz), 123.99 (d, J = 14.8 Hz), 116.71, 115.56 (d, J = 22.2 Hz), 108.24, 99.89, 88.60, 64.19, 64.15, 61.90, 55.14, 53.80, 52.57.
N-(1,3-Dibromo-6-oxo-5,6-dihydro-4H-cyclopenta[c]thien-4-yl)-6-phenylhexanamide (16)
To a suspension of 350 mg (1 mmol) of the salt 9 in acetontrile (15 mL) was added 0.14 mL (1 mmol) of triethylamine. The obtained solution was cooled to 0°C and added 296 mg (1 mmol) of ethyl 6-phenylhexanoyl carbonate (prepared separately from equimolar quantities of 6-phenylhexanoic acid, ethyl chloroformate and triethylamine). The reaction was stirred at room temperature for one hour. The formed precipitate was filtered, washed with petroleum spirit and dried to give 350 mg of the desired product. Yield: 72%. MP 193 °C. IR (cm− 1): 3280, 1718, 1647, 2931, 1539, 1472, 1074, 695. MS (m/z): 583.0 (M+ − 2), 585.0 (M+), 587.0 (M++2). 1H NMR (DMSO-d6) δ: 8.40 (d, J = 8.3 Hz, 1H, NH), 7.25 (t, J = 7,4 Hz, 2H, H-3′ and H-5′), 7.17 (m, 3H, H-2′, H-4′ and H-6′), 5.16 (dt, J= 3.2, 8.3 Hz, 1H, H-4), 3.34 (dd, J = 8.3, 18.6 Hz, 1H, H-5b), 2.64 (dd, J = 3.2, 18.6 Hz, 1H, H-5a), 2.53 (t, J = 7.8 Hz, 2H, H-6″), 2.05 (t, J = 7.8 Hz, 2H, H-2″), 1.53 (m, 4H, H-3″ and H-5″), 1.26 (m, 2H, H-4″). 13C NMR (DMSO-d6) δ: 193.30, 171.50, 153.41, 142.19, 142.17, 128.30, 128.18, 125.57, 109.94, 105.00, 51.44, 42.67, 35.21, 35.01, 30.69, 28.20, 24.95. Anl. CHN C19H19Br2NO2S.
N-(1,3-Dibromo-6-oxo-5,6-dihydro-4H-cyclopenta[c]thien-4-yl)-4-(1H-indol-3-yl)butanamide (17)
To a suspension of 350 mg (1 mmol) of the salt 9 in acetontrile (15 mL) was added 0.14 mL (1 mmol) of triethylamine. The obtained solution was cooled to 0°C and added 275 mg (1 mmol) of ethyl 4-(1H-indol-3-yl)butanoyl carbonate (prepared separately from equimolar quantities of 3-indolebutyric acid, ethyl chloroformate and triethylamine). The reaction was stirred at room temperature for one hour. The solvent was evaporated under reduced pressure and the residue was dissolved in 100 mL of dichloromethane and washed successively with 2 N aqueous hydrochloric acid solution, aqueous saturated NaHCO3 solution and brine. The organic layer was separated, dried over anhydrous CaCl2, filtered and evaporated under reduced pressure to give 200 mg of product. Yield: 40%. MP 180 °C. IR (cm− 1): 3375, 2926, 1718, 1652, 1548, 1472, 1066, 743. MS (m/z): 593.9 (M+ − 2), 595.9 (M+), 597.9 (M++2). 1H NMR (DMSO-d6) δ: 10.75 (s, 1H, NHindole), 8.41 (d, J = 8.3 Hz, 1H, NHamide), 7.47 (d, J = 7.8 Hz, 1H, H-7′), 7.31 (d, J = 7.8 Hz, 1H, H-4′), 7.08 (s, 1H, H-2′), 7.03 (t, J = 7.8 Hz, 1H, H-5′), 6.95 (t, J = 7.8 Hz, 1H, H-6′), 5.36 (dt, J = 3.4, 8.3 Hz, 1H, H-4), 3.36 (m, 1H, H-5b), 2.67 (m, 3H, H-4″ and H-5a), 2.13 (t, J = 7.3 Hz, 2H, H-2″), 1.88 (quintuplet, J = 7.3 Hz, 2H, H-3″). 13C NMR (DMSO-d6) δ: 193.32, 171.56, 153.48, 142.17, 136.28, 127.14, 122.27, 120.79, 118.28, 118.07, 114.00, 111.29, 109.92, 105.00, 51.41, 42.68, 35.16, 26.00, 24.26).
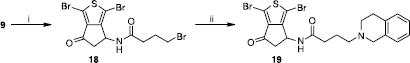
4-(3,4-Dihydro-1H-isoquinolin-2-yl)-N-(1,3-dibromo-6-oxo-5,6-dihydro-4H-cyclopenta[c]thiophen-4-yl)butyramide (19)
To a solution of 460 mg (1 mmol) of the bromobutyramide 18 in dichloromethane (20 mL) wad added 160 mg (1.2 mmol) of 1,2,3,4-tetrahydroisoquinoline and 135 mg (1.2 mmol) of potassium carbonate. The reaction mixture was stirred at room temperature over night. 50 mL of dichloromethane was then added and the mixture was washed several times with water. The organic layer was separated, dried over anhydrous CaCl2, filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (CH2Cl2/MeOH 99.5:0.5) to give 395 mg of the desired product. Yield: 77%. MP 181 °C. IR (cm− 1): 3330, 1720, 1657, 1548, 1470, 1129, 742. MS (m/z): 510.9 (M+ − 2), 512.9 (M+), 514.9 (M++2). 1H NMR (DMSO-d6) δ: 9.24 (d, J = 8.2 Hz, 1H, NH), 7.09 (t, J = 7.3 Hz, 1H, H-7′), 7.03 (t, J = 7.3 Hz, 1H, H-6′), 6.92 (d, J = 7.3 Hz, 1H, H-5′), 6.85 (d, J = 7.3 Hz, 1H, H-8′), 5.24 (dt, J = 4.4 Hz, J = 8.2 Hz, 1H, H-4), 3.61 (d, J = 16.2 Hz, 2H, H-1′), 3.19 (dd, J = 8.2, 19.1 Hz, 1H, H-5b), 2.70 (m, 7H, H-5a, H-3′, H-4′, and COCH2CH2CH2), 2.42 (m, 2H, COCH2CH2CH2), 1.88 (m, 2H, COCH2CH2CH2). 13C NMR (DMSO-d6) δ: 192.09, 172.72, 151.81, 141.33, 132.83, 132.52, 128.58, 126.84, 126.24, 126.21, 111.37, 105.52, 58.57, 55.40, 51.80, 51.03, 42.78, 36.40, 28.25, 21.65.
In vitro tests of AChE biological activity
Inhibitory ability of our compounds on AChE activity was evaluated through the use of the spectrometric method of Ellman et al. [Citation2]. Lyophilized electric eel AChE (Type III, electric eel, Sigma Chemical Co.) was dissolved in 0.2 M phosphate buffer pH 7.4 so as to have an enzyme solution stock with 2.5 units/mL AChE activity. Acetylthiocholine iodide (Sigma Chemical Co.) was used as a substrate of the enzymatic reaction and 5,5-dithiobis-(2-nitrobenzoic) acid (DTNB, Sigma Chemical Co.) as marker for the measurement of cholinesterase activity. In the procedure, 1880 μL of 60 mg / 500 mL DTNB dissolved in phosphate buffer pH 7.4 were mixed with 40 μL of test compound solution and 40 μL of enzyme stock solution were mixed. After 5 minutes of preincubation, 40 μL of 10 mM acetylthio-choline iodide solution was added to the assay solution. The change in absorbance at 412 nm was recorded (AGILENT 8453 UV-VIS Spectroscopy System) during 10 min. First screening of AChE activity was carried out at a 10− 5 M concentration of compounds under study. For the compounds with significant inhibition after 4 minutes of reaction, IC50 values were determined graphically from log concentration – inhibition curves, using a range of 10− 9 M–10− 3 M concentrations of the test compounds.
Results and discussion
Chemistry
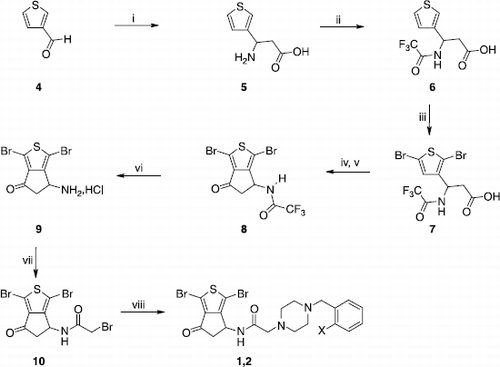
We had previously observed a significant augmentation of the anti-AChE activity of 2 by the replacement of its cyclopentane carbonyl group by a hydroximine one. In order to first determine the biological importance of this carbonyl, we decided to reduce it into the corresponding alcohol, and subsequently replace the resulting hydroxyl by an atom of fluorine or chlorine. The multi-step synthesis of compounds 1-3 has been previously reported by us [Citation1] using thiophene-3-carboxaldehyde 4 as starting material that involve a Rodionov-Jonhson reaction [Citation3,Citation4] to give the β-aryl-β-aminoacid 5 which after N-protection, bromination of the α-thiophene positions and Friedel-Crafts cyclisation led to the trifluoroacetylamino-cyclopenta[c]thiophenone 8 (Scheme ). The hydrolysis of the latter with aqueous concentrated HCl afforded the hydrochloric salt 9 that was then treated in alkaline medium to liberate the free base. The latter was reacted immediately with the bromoacetyl bromide to yield the bromoacetamide 10. The bromine atom of 10 was finally substituted by suitable benzylpiperazines to provide the titled derivatives 1 and 2.
Scheme 1 Synthesis of compounds 1 and 2. Reagents: (i) AcONH4, CH2(COOH)2, EtOH; (ii) TFA2O, Et2O; (iii) Br2, CH2Cl2; (iv) SOCl2; (v) AlCl3, CH2Cl2; (vi) HCl, H2O, EtOH; (vii) TEA, BrCOCH2Br, CH2Cl2; (viii) N-substituted benzylpiperazine, K2CO3, CH2Cl2.
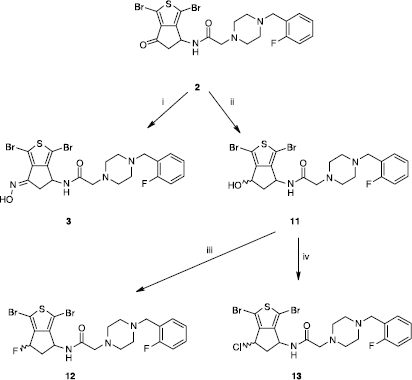
While the condensation of the carbonyl group of 2 with the hydroxylamine led selectively to the E-oxime 3, its reduction by sodium borohydride resulted, with excellent yield (97%), in the alcohol 11 as a mixture (50:50) of syn and anti isomers (Scheme ). This alcohol was then submitted to the action of (diethylamino)sulphur trifluoride (DAST) to produce with 55% yield the fluorinated compound 12 also as a mixture (50:50) of syn and anti isomers. In the same way, the hydroxyl group of 11 was replaced by a chlorine atom by means of treatment with two equivalents of thionyl chloride. Compound 13 (50:50 syn and anti) was then isolated with a moderate yield (30%).
Scheme 2 Synthesis of compounds 3,11-13. Reagents: (i) NH2OH·HCl, AcONa, EtOH, H2O; (ii) NaBH4, MeOH; (iii) DAST, CH2Cl2; (iv) SOCl2, CH2Cl2.
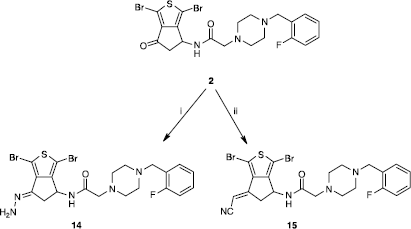
Furthermore, the carbonyl group of 2 was condensed into the corresponding hydrazone 14 (Scheme ); the reaction was accomplished by refluxing compound 2 in methanol with excess of hydrazine hydrate and hydrazone 14 was then isolated with good yied (90%) as a unique isomer (E). The cyanomethylene group of derivative 15 was also installed stereoselectively with 66% yield via a reaction of Wadsworth-Emmons [Citation5].
Scheme 3 Synthesis of compounds 14 and 15. Reagents: (i) NH2·NH2·xH20, MeOH; (ii) NCCH2PO(OEt)2, K2CO3, THF.
On the other hand, to explore the importance of the benzylpiperazine moiety, we decided to suppress the piperazine cycle of our system and to replace it by other linkers. The first modification tried concerned a phenyl ring connected to the carboxamide group by a simple carbon chain. Thus, we obtained with 72% yield the compound 16 by the reaction of the hydrochloric salt 9 in alkaline medium with 6-phenylhexanoic acid activated as a mixed anhydride (Scheme ).
Scheme 4 Synthesis of compounds 16 and 17. Reagents: (i) a- Ph(CH2)5CO2H, TEA, ClCO2Et, CH2Cl2. b- 9, TEA, MeCN; (ii) a- 3-indolebutyric acid, TEA, ClCO2Et, CH2Cl2. b- 9, TEA, MeCN.
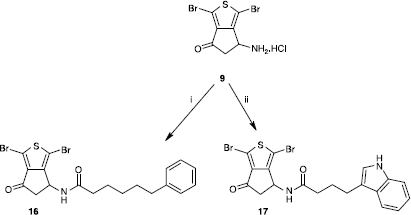
In a similar manner, the activated 3-indolebutyric acid was reacted with the hydrochloric salt 9 to give with low yield (40%) the indole derivative 17. The isoquinoline derivative 19 was finally easily obtained (77% yield) by a nucleophilic substitution of the already described ω-bromobutyryllamide 18 [Citation1] using commercially available tetrahydroisoquinoline (Scheme ).
Scheme 5 Synthesis of compound 19. Reagents: (i) TEA, BrCO(CH2)3Br, CH2Cl2; (ii) 1,2,3,4-tetrahydroisoquinoline, K2CO3, CH2Cl2.
Biology
All the compounds synthesized in this work were tested for in vitro inhibition of AChE on the commercially available electric eel enzyme according to the Ellman method [Citation2].
The docking studies previously carried out in our laboratory showed that our best compounds 2,3 took a position in the binding site of the enzyme very similar to that of donepezil [Citation1]. On one hand their upper part exhibited a π–π stacking with the indole ring of Trp279. This interaction appeared better preserved with the oxime group of 3 than with the carbonyl group of 2. On the other hand, we have also shown that the bottom part of our compounds 2,3 conserved the interactions that donepezil engaged with the enzyme: the benzyl moiety stacked against the indole ring of Trp84 and the charged nitrogen of the piperazine ring was oriented in a suitable position for a π-cation interaction with the phenyl ring Phe330. Furthermore, the docking results suggested that substitution of the benzyl at the ortho position by a fluorine atom can modify the orientation of the benzyl ring in a manner that its π-stacking with the Trp84 can be amplified.
Concerning the present study, whereas the reduction of the carbonyl group of 2 conserves the anti-AChE activity (), the replacement of the hydroxyl function of 11 by a chlorine atom slightly reduced the biological activity. Meanwhile the fluorinated derivative 12 dramatically lost such activity. Contrarily to the oxime 3, the hydrazone 14 is four times less active than the ketone, and the cyanomethylene compound 15 is totally inactive.
Table I. Anti-AChE activity of compounds 2, 3, 11-15. .
.
This work confirms also the crucial role for the benzylpiperazine system in our series since the replacement of the piperazine ring with other moieties resulted in compounds 16, 17 and 19, totally devoid of anti-AChE activity ().
Table II. Anti-AChE activity of compounds 1, 17, 18, 20. .
.
In conclusion, the previously experimented oxime 3 remains the most active compound in the thiaindanone series (IC50 = 0.06 μM against 0.02 μM for donepezil). It appears today that to enhance the biological activity of the latter, some interactions with AChE must be preserved by the test compounds. In this field, a particular interest will have to be brought in those which concern the Peripheral Anionic Site (PAS) of the enzyme. The latter has been recently identified to be responsible of promoting beta-amyloid protein assembly and this non-classical function of AChE is of renewing interest in the search of new AChE inhibitors directed towards the PAS Citation6Citation7Citation8Citation9.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Z Omran, T Cailly, E Lescot, J Sopkova-de Oliveira Santos, JH Agondanou, V Lisowski, F Fabis, AM Godard, S Stiebing, G Le Flem, M Boulouard, F Dauphin, P Dallemagne, and S Rault. (2005). Eur J Med Chem 40:1222–1245.
- JL Ellman, KD Courtney, V Andres, and RM Featherstone. (1961). Biochem Pharmacol 7:88–95.
- VM Rodionow, and ET Malwinskaja. (1926). Chem Ber 59:2952–2958.
- TB Johnson, and JE Livak. (1936). J Amer Chem Soc 58:299–303.
- WS Wadsworth, and WD Emmons. (1961). J Amer Chem Soc 83:1733–1738.
- I Silman, and JL Sussman. (2005). Curr Opin Pharmacol 5:293–302.
- U Holzgrabe, P Kapkova, V Alptuzun, J Scheiber, and E Kugelmann. (2007). Expert Opin Ther Targets 11:161–179.
- M Recanatini, and P Valenti. (2004). Curr Pharm Des 10:3157–3166.
- A Castro, and A Martinez. (2006). Curr Pharm Des 12:4377–4387.