Abstract
Leishmaniasis is a protozoan vector borne disease prevalent throughout the world and present in at least 88 countries. The parasite is transmitted by infected phlebotomine sandfly bites. While conventional therapies i.e. pentavalent antimonials, amphotericin B and pentamidine continue to play a major role, it is evident that new drugs or strategies must circumvent the limitations, such as a long-term parenteral administration, toxicity, the high cost in endemic countries and the emergence of resistance, that prevail. One of the most promising drugs is miltefosine, a new oral, approved alkylphospholipid for visceral leishmaniasis with only slight adverse effects. Although we have now this recent and encouraging advance, there is still a need to develop safe, efficient and affordable new treatments for the different clinical forms that exist. This review summarises conventional therapy and the current efforts in the discovery of drugs to treat leishmaniasis with the emphasis on drug combinations to enhance efficiency and prevent the emergence of resistance, the investigation of natural products with the objective of offering new bioactive chemical structures and the development of novel antileishmanial targets.
Introduction
Despite the fact that infectious diseases have been identified as the third major cause of death in the world, many fall into the category of “neglected” diseases. Among them, leishmaniasis, which is an ancient protozoan disease affecting about 12 million people with 2 million new cases every year that constitute a serious public health problem. According to the World Health Organisation (WHO), leishmaniasis is now endemic in 88 countries, particularly in subtropical and tropical regions [Citation1]. In some countries, the persistence of this disease is explained by specific epidemiological factors such as urbanisation, armed conflict, HIV co-infection or the resistance of Leishmania and its vector. The life cycle of Leishmania involves the transmission of a flagellated promastigote by phlebotomine sandfly bites that invades macrophages and rapidly transforms into the amastigote stage which actively divides within the mononuclear phagocytes (). The resulting clinical patterns () are extremely diverse depending on the Leishmania species and the cellular immune system of the patient. Four clinical forms have been described. Cutaneous leishmaniasis (CL) is an ulcerous skin lesion localised on exposed parts of the body. Mucocutaneous leishmaniasis (MCL), also called “espundia” in South America, which leads to disfiguration. Diffuse cutaneous leishmaniasis (DCL), particularly related to a defective immune system, which manifests itself in the form of numerous nodular lesions on the face, the arms and the legs. Cutaneous lesions caused by L. major in the old-world and L. mexicana in the new world generally heal by themselves within 2-5 months while healing is slower for L. tropica, L. braziliensis and L. panamensis. Visceral leishmaniasis (VL) is the most severe form of the disease because it is lethal if not treated [Citation2]. It is important to note that in Europe, L. infantum has emerged as an opportunist pathogen in HIV patients in which the depletion of CD4 explains the tendancy to relapse if immunosuppression is not controlled by an antiretroviral therapy [Citation3,Citation4].
Figure 1 Promastigote (A) and axenic amastigotes (B) visualized in scanning electronic microscopy – original magnification × 6000 and × 15 000. Intracellular amastigotes (C) from an experimental infection of Balb/c peritoneal macrophages, arrow indicated Leishmania – original magnification × 1000. (Photographies from Laboratory of Parasitology-Nantes).
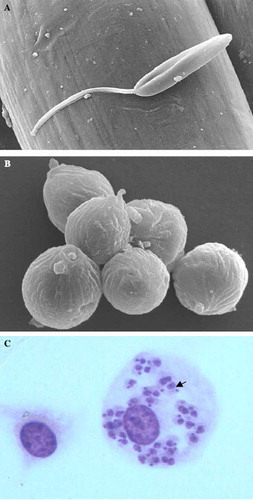
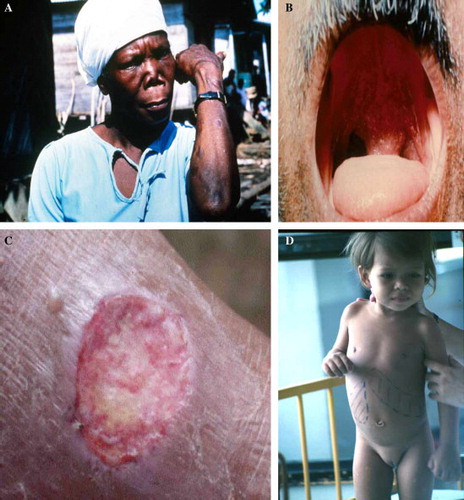
Figure 2 Some of clinical patterns of leishmaniasis. (A) Primary healed lesion and secondary destructive lesion of mucocutaneous leishmaniasis (L. panamensis). (B) Oropharyngeal lesion (L. braziliensis). (C) Cutaneous leishmaniasis with a large ulcerative lesion on the arm (L. panamensis). (D) Hepatosplenomegaly of visceral leishmaniasis (L. infantum). (Photographies from Laboratory of Parasitology-Nantes and PECET-Colombia).
Concerning therapy, pentavalent antimony compounds have remained the principal solution for nearly 75 years [Citation5]. Hovewer, lack of response to pentavalent antimonials, actually widespread in India and Sudan, led to the use of amphotericin B or pentamidine. These three drugs present, however, some limitations such as long-term parenteral administration, toxicity, high cost in endemic countries, resistance and a high rate of treatment failure in HIV co-infected patients. Among the new drugs discovered, miltefosine, a hexadecylphosphocholine, is the first promising oral drug which can be used against leishmaniasis [Citation6]. Other drugs such as paromomycin, sitamaquine, azoles and azythromicin have been reported as having variable cure rates Citation7Citation8Citation9. Consequently there is still a real need for new active compounds that can provide therapeutic benefits but with fewer side effects.
This review will summarise the drugs currently available and those which are included in clinical trials. Moreover, the current status and future perspectives in the area of combination therapy, plant product research and novel drug targets in the parasite will also be presented.
Conventional therapy against leishmaniasis
While conventional therapies i.e. pentavalent antimonials, amphotericin B and pentamidine () continue to play a major role, it is obvious that new drugs or strategies must circumvent limitations such as a long-term parenteral administration, toxicity (), the high cost in endemic countries, and the emergence of resistance.
Figure 3 Chemical structures of conventional antileishmanial drugs.
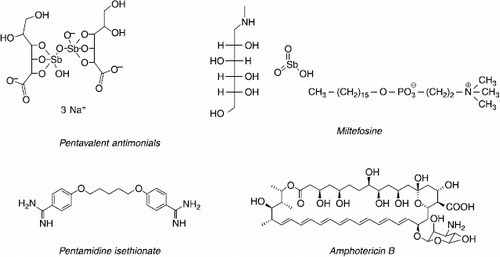
Table I. Adverse effects of conventional therapies against leishmaniasis.
Pentavalent antimonials
Despite the spread of resistance and severe cardiac, hepatic, pancreatic and renal side effects, N-methylglucamine antimoniate (Glucantime®) and sodium stibogluconate (Pentostam®), both pentavalent antimonials, remain the drugs of choice in most parts of the world [Citation5]. The intracellular reduced trivalent form is the active derivate that comes about through the alteration in parasite bioenergetic pathways and trypanothione inhibition [Citation10,Citation11]. The recommendation for the use of pentavalent antimonials is administration by intramuscular injections of 20 mg Sbv/kg/day up to a maximum of 1275 mg over 20 or 30 days. In the case of old-world cutaneous leishmaniasis, there is no significant difference between the intralesional and intramuscular route [Citation12]. For these drugs, the cure rate is generally high (85–95%), except in Bihar-India where 60% of patients with VL are now unresponsive [Citation13], as in Iran for L. tropica infected patients (CL) [Citation14] and in Peru for MCL [Citation15].
Amphotericin B
Another first line drug is amphotericin B (Fungizone®), a macrolide polyene, characterised by hydrophilic polyhydroxyl and hydrophobic polyene aspects. Amphotericin B binds to membrane ergosterol leading to the formation of pores, major constituent efflux and, finally, parasite cell lysis. Intravenous infusion (7–20 mg/kg up to 20 days) of amphotericin B is an alternative treatment in all the regions where antimonial resistance has been reported. This drug is characterised by infusion related side effects and renal toxicity [Citation16]. Lipid based formulations such as liposomal amphotericin B (Ambisome®) reduce the impairment of renal function by up to 50% with a therapeutic schedule of 6–21 mg (LV) or 2–3 mg (ML)/kg for 20 days [Citation17,Citation18]. Despite its great efficiency, the prohibitive cost of liposomal amphotericin B limits its use in developed countries.
Pentamidine isethionate
Although used less frequently, diamine pentamidine has become of special interest in CL caused by L. guyanensis, where intralesional injections have been more efficient than with pentavalent antimonials [Citation19]. Major side effects include hypotension, diabetes mellitus and renal impairment. Antileishmanial activity is based on the inhibition of polyamine biosynthesis and the disruption of mitochondrial membrane potential [Citation20].
Miltefosine
The most recently introduced drug in the armentarium is miltefosine (Impavido®), an hexadecylphosphocholine derived from cancer therapy [Citation21]. Miltefosine could alter glycosylphosphatidylinositol (GPI) anchor synthesis, ether-lipid metabolism, signal transduction and alkyl-specific acyl-coenzyme A acyl-transferase [Citation22,Citation23]. With leishmaniasis, this first oral treatment has enabled us to attain high cure rates in Indian visceral leishmaniasis (95%) and Colombian cutaneous leishmaniasis (91%) when used at 100-150 mg/day for 28 days [Citation6,Citation24,Citation25]. These clinical findings are in accordance with the laboratory results showing that L. donovani and L. panamensis are the most susceptible species [Citation26,Citation27]. A phase IV trial conducted in 2006 in India demonstrated similar efficiency of miltefosine in field conditions [Citation28]. Despite these encouraging reports, low cure rates observed in CL caused by L. braziliensis or L. major and transient cures followed by relapses in DCL or HIV/LV co-infected patients could minimise its extended use as a monotherapy Citation29Citation30Citation31. Miltefosine was first approved in India (2002), Germany (2004) and Colombia (2005).
Drugs in clinical trials
Paromomycin
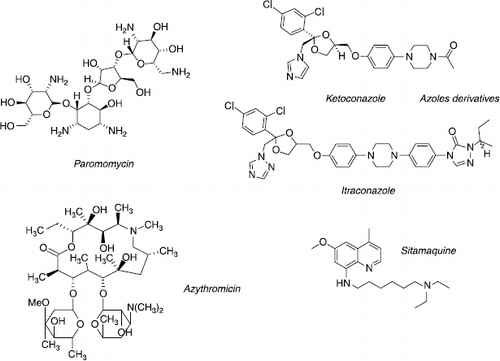
Paromomycin, an aminoglycoside antibiotic produced by Streptomyces riomosus, was developed in the 1960s as an oral therapy for intestinal protozoa. More recently, clinical formulations associating 15% paromomycin sulphate with 12% methylbenzethonium chloride (Leshcutan®) or urea in paraffin have been investigated for topical treatment of cutaneous leishmaniasis Citation32Citation33Citation34. Because of species- dependent efficiency and low tolerance (inflammation, burning sensation…), a new formulation containing 15% paromomycin and 0.5% gentamycin in a complex base (WR 279396) was evaluated in a phase II trial in patients with L. panamensis cutaneous leishmaniasis [Citation35]. No statistically significant difference was observed between cure rates after WR 279396 (61%) or placebo (55%) treatment. Recently, an Indian phase III trial was conducted to compare intramuscular paromomycin (15 mg/kg/day) and amphotericin B in patients with L. donovani visceral leishmaniasis. This trial demonstrated that the two drugs were as effective at 6 months post-treatment with cure rates of 94.6% and 98.8%, respectively [Citation7]. Since August 2006, paramomycin IM has been approved in India as a new alternative for visceral leishmaniasis treatment. The mechanism of its action is unclear. In bacteria, this antibiotic binds to the A-site on the 16S RNA in the 30S sub-units of ribosomes giving rise to nonsense proteins through a misreading during protein synthesis [Citation36]. In Leishmania, paramomycin could interfere with RNA synthesis and membrane permeability [Citation37] ().
Figure 4 Chemical structures of antileishmanial drugs on clinical trials.
Azole derivatives
The last example of development in new anti-infectious drugs is therapeutic switching also called “piggy-back therapy”. As in fungal cells, ergosterol is one of the most important sterols of the Leishmania membrane. Ergosterol biosynthesis requires 14a-lanosterol demethylase, making this enzyme a target in antifungal therapy, as illustrated by the success of azole derivatives (fluconazole, voriconazole). Because of this similarity in biochemical pathway, azoles are also effective in leishmaniasis therapy (ketoconazole, itraconazole) [Citation9,Citation38]. However, azoles have been the most controversial of these drugs, since the conclusions on efficiency come from few registered patients or poorly-conceived trials. Hence, a wide range of cure rates was reported for ketoconazole and itraconazole whereas promising results were obtained with fluconazole that reduced the time of healing of cutaneous lesions caused by L. major [Citation39] or for posaconazole against experimental L. amazonensis [Citation40].
Sitamaquine
The intracellular targets of the 4-methyl-6-methoxy-8-aminoquinoline, sitamaquine, are mitochondria and acidocalcisomes. This compound has been used in clinical trials against new and old-world LV with 67–92% cure rates after oral delivery (1.7–2 mg/kg/day) for 28 days [Citation8,Citation41,Citation42]. A phase II trial is ongoing to study safety and tolerance and to investigate the efficiency of a 21-day course of treatment in American visceral leishmaniasis. Further studies must be conducted in order to explore the clinical value in other forms of leishmaniasis.
Azithromycin
Azithromycin, an azalide antibiotic has demonstrated activity against various protozoa i.e. Toxoplasma gondii, Plasmodium spp, Cryptosporidium parvum. The advantages of this macrolide are its high concentration in tissues, especially in macrophages, oral administration and safety. Its antiprotozoal action is due to the inhibition of protein synthesis but stimulation of phagocytosis, chemotaxis and its enhancement of immune response cannot be excluded Citation43Citation44Citation45. Encouraging results were confirmed in American cutaneous leishmaniasis caused by L. braziliensis with high response rates (85%) at 60–120 days after varying periods of taking a 500–1000 mg daily dose Citation46Citation47. However, trials conducted in endemic foci of L. major in Iran, revealed no apparent role for azithromycin in old-world cutaneous leishmaniasis with 11.8% [Citation48] and 10.3% [Citation49] cure rates.
Resistance to antileishmanial drugs
Therapeutic failures resulting from treatment schedule modifications, such as sub-therapeutic doses or reduced treatment periods, are a typical situation in non-hospitalised patients. However variations in response to current antileishmanial drugs have indicated other aspects of resistance such as parasite intrinsic factors, drug pharmacokinetic and host immune status. The diversity of infection sites, which are specific to each clinical form, could contribute to the variations in efficiency of these drugs depending on their respective distribution in the host i.e. bone marrow, spleen, liver or dermis [Citation50,Citation51]. HIV/LV co-infected patient suffer from frequent relapses until the CD4 lymphocyte level increases, thanks to new antiretroviral therapy [Citation52]. Concerning the parasite counterpart, recent studies on the Leishmania genomic suggest a possible role of thiol metabolism and intracellular ABC transporter MRPAp in antimony resistance [Citation53]. Parasite resistance to amphotericin B could result from the replacement of the membrane ergosterol target by one of its precursor cholesta 5,7,24-trien-3β-ol after the alteration of transcripts of the C-24-D-sterol-methyltransferase [Citation54,Citation55]. Resistance to pentamidine could be related to the loss of the drug transporter PRP1 in the plasmatic membrane Citation56Citation57Citation58 or to lower accumulation in the mitochondrion thus facilitating its efflux [Citation59]. Data on the mechanisms of miltefosine resistance come from laboratory manipulated strains showing that the over-expression of the P-glycoprotein efflux pump or the dysfunction of two proteins LdMT/LdRos3 that are necessary for its uptake, have been involved [Citation60,Citation61].
Reports of lack of response to pentavalent antimony appeared in the 1970s and now this situation is well-rooted in Bihar-India and south-east Nepal. From the early 1980s to the 1990s, the cure rate after pentamidine administration decreased, demonstrating the increasing clinical resistance of Leishmania to this drug. In the light of these considerations, the monitoring of this resistance phenomenon is of crucial importance as regards the medication available as first line treatment or recently introduced into an anthroponotic situation. Instead, the larger use of amphotericin B or miltefosine in endemic regions where Glucantime® or pentamidine resistance is now established could rapidly generate wide resistance to these other first line drugs. A Fungizone® refractory case of L. infantum visceral leishmaniasis has yet been reported in an HIV patient [Citation62].
Drug combination strategy
As in other infectious diseases, combination therapy should be one of the most important approaches to leishmaniasis. Drug combination could lead to reduced conventional treatment length, higher patient compliance and to the prevention of treatment failure, antileishmanial drug resistance or high toxicity. Furthermore, the combination of topical therapies could replace the need for parenteral injections of antimonials for LC.
Combination therapies with antimonials
A double-blind, randomised, controlled trial performed in Iran indicates that combined therapy using pentoxiffylline is more effective than Glucantime® alone, with 81.3% and 51.6% complete improvement, respectively [Citation63]. In this same country, where L. tropica is endemic, Firooz et al. reported that a combined treatment with 5% imiquimod (Aldara®), a topical immunomodulator, was no more effective than intramuscular antimonial alone. In this same region, imiquimod, however, showed a beneficial effect when meglumine antimonials were administered intralesionally [Citation64]. When evaluated for American cutaneous leishmaniasis patients resistant to Glucantime®, the combination demonstrated a cure rate of 72% in patients treated with glucantime plus imiquimod, while 35% of the patients receiving Glucantime® plus vehicle cream healed at 3 months [Citation65]. Furthermore, this combination leads to faster healing and a better aesthetic aspect [Citation66]. Considering fatal Indian visceral leishmaniasis, treatment with paramomycin 18 mg/kg on a daily basis combined with sodium stibogluconate 20 mg/kg for 21 days was statistically more effective than with sodium stibogluconate alone for producing final cure rates of 93.8% and 53.1%, respectively [Citation67]. In addition, association with parenteral paromomycin allowed the reduction of the sodium stibogluconate treatment period (17 days versus 30 days) [Citation68]. In the past, the combination of allopurinol plus sodium stibogluconate was investigated for few pentavalent resistant LV cases. Recently, in the special leishmaniasis recidivans clinical form, a chronic CL, highly resistant to current therapy, a combination with oral allopurinol (20 mg/kg for 30 days), was highly effective with 87.5% cure rates and only two relapses [Citation69].
Other combination therapies
Antileishmanial activity of other drugs could benefit from an experimental combination strategy. Amphotericin B and paromomycin increased miltefosine activity in mice infected with L. donovani [Citation70]. A combination of amphotericin B plus itraconazole did not succeed in the treatment of MCL in Peru and Bolivia [Citation71].
Plant products against Leishmania
Although there are still none present in the leishmaniasis armamentarium, interest in natural products must be maintained since chemical structure diversity could offer the basis for future drugs. Numerous papers claim the in vitro antileishmanial activity of plant extracts. Some of the more recently published genera of interest are: Portulaca spp from Brazil [Citation72] and Aphelandra spp, Byrsonima spp, Clucia spp, Tridax spp and Pentalinon spp from Mexico [Citation73,Citation74]. Some studies concern the in vitro evaluations of purified compounds. Xanthinin, a xanthanolide from Xanthium macrocarpum harbouring an α-methylene-γ-butyrolactone moiety, coumarin from Cadophyllum brasiliense, clerosterol from Cassia fistula and Jangambin from Ocotea duckei exhibited significant biological activities at promastigote or amastigote stages Citation75Citation76Citation77Citation78. A new taxoid from Taxus baccata, 10-deacethylbaccatin III, was reported to be highly active (IC50: 70 nM) against the intracellular amastigote stage. Contrary to other taxoids, this activity does not seem to be related to the interference with parasite tubulin [Citation79]. The in vitro reduction of the amastigote load was obtained after macrophage activation by propolis and garlic extracts. The latter could stimulate INFγ and nitric oxide production resulting in the reduction of footpad lesions [Citation80]. In the groups of natural products evaluated, few demonstrated in vivo antileishmanial activity. Isopropylquinolines from Bolivian Galipea longiflora exhibit parasiticidal effect in experimental VL and CL [Citation81]. Two chalcone derivatives, licochalcone A from chinese Glycyrrhiza and more recently flavokavaïn B from Piper rusbyi and a triterpenoid saponin isolated from Maesa balansae (maesabalide III) are the most promising compounds Citation82Citation83Citation84. Protoberberine related to berberine, a chalcone from Berberis aristata, and maesabalide derivatives obtained through pharmacomodulation are under investigation [Citation85,Citation86].
New targets under investigation
For a few years, scientists have been convinced that rational drug design must be carried out in conjunction with investigations into Leishmania biology in order to better understand its particularities with the objective of defining new parasite targets for new therapeutic compounds. Specific enzymes that do not exist in mammalian cells are of particular interest as relevant targets. However, although structural homology with mammalian enzymes, those supporting a specific role in the parasite, could also offer fully functionally targets for antileishmanial therapy ().
Figure 5 Chemical structures of new lead compounds.
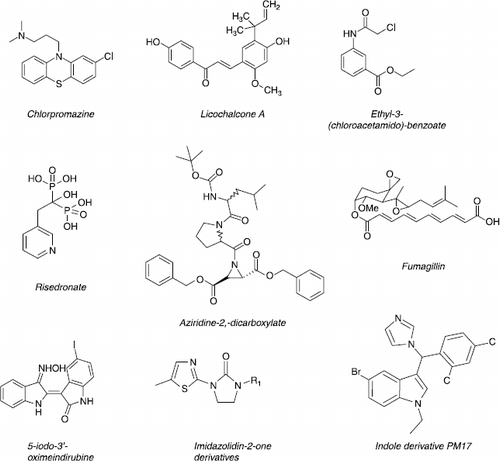
Trypanothione reductase/peroxidase
In Leishmania, glutathione is substituted by trypanothione (bis glutathionyl spermidin) to protect the cell against oxidants and xenobiotics. Specificity in the structure of trypanosomatid trypanothione reductase/peroxidase active sites rationalise the design of parasite inhibitors [Citation87]. Some 2-amino-4-chlorophenyl phenyl sulfide analogues of chlorpromazine inhibitors of trypanothione reductase from Trypanosoma cruzi were highly active against Leishmania donovani [Citation88].
Topoisomerases
DNA topoisomerases are a class of enzyme involved in the regulation of DNA supercoiling and they are crucial during DNA transcription and replication. These enzymes are leishmanial targets for some new fluoroquinolones and pentamidine [Citation89].
Sphingolipids synthesis
While the biosynthetic pathway for the formation of membrane sphingolipids of mammalian cells requires sphingomyeline synthase, in kinetoplastids inositol phosphorylceramide synthase carries out the same role, indicating that it could be specifically targeted [Citation90].
Fumarate reductase
In the Leishmania respiratory chain, succinate is converted into fumarate by fumarate reductase [Citation91]. As described before, this enzyme might be the specific target for the antileishmanial chalcones [Citation92].
Microtubule associated protein (MAP2)
In the past, parasite microtubules have been identified as a potential target for therapy but the high toxicity of antimicrotubule drugs rapidly prevented their use in practice. An ethyl 3-(chloroacetamido)-benzoate, efficient on various Leishmania species, alters parasite microtubule organisation through interaction with a microtubule associated protein MAP2. Its in vivo activity was demonstrated in a L. major mice model by the important reduction of the parasite burden in the lymph node, spleen and liver [Citation93].
Squalene synthase
In addition to lanosterol 14α-demethylase, squalene synthase involved in squalene formation by dimerisation of two molecules of farnesyl pyrophosphate, was identified as a new, valuable antileishmanial target [Citation94]. Indeed, a 3-hydroxyquinuclidine (squalene synthase inhibitor) and its derivatives induced Leishmania growth arrest after treatment with submicromolar concentrations [Citation95]. Recently, a recombinant L. major squalene synthase was produced which permitted the screening of selective inhibitors [Citation96]. Moreover, these compounds lead to major alterations in flagella, mitochondrion membranes and nuclear chromatin suggesting a possible apoptotic phenomenon [Citation97]. Located downstream from this enzyme, farnesyl pyrophosphate synthase was successfully targeted by biphosphonates such as risedronate [Citation98].
Cysteine proteases
Cysteine proteases identified in the amastigote stage actively participate in differentiation, pathogenicity and virulence. Moreover, these enzymes are involved in the survival of Leishmania within the macrophage cells [Citation99]. The reduction of macrophage infection by cysteine protease inhibitors of aziridine-2,-dicarboxylate series could be mediated through the inhibition of parasite replication and the increase of nitric oxide production [Citation100].
Methionine aminopeptidase 2 (MetAP-2)
MetAP-2 is a cellular metallo-exopeptidase that takes part in late hydrolysis of the initiator methionine of protein synthesis [Citation101]. A MetAP-2 inhibitor, fumagillin, blocked the replication of Leishmania donavani [Citation102]. The three-dimensional structure of this enzyme has been modelled for Plasmodium falciparum showing difference in the binding pocket between parasite and human MetAP-2.
Protein kinases
A cdc2-related protein kinase encoding by the CRK3 gene is essential for controlling cell cycle progression at the G2/M-phase transition [Citation103]. Included among the potential inhibitors of CRK3 cyclin-dependent kinase of Leishmania, indirubin derivatives exhibited growth arrest and change in the DNA content [Citation104]. Evidence that PKC plays a critical role in the invasion process is highlighted by a study demonstrating that pre-treatment of intact parasites by imidazolidinone compounds inhibited PKC activity and the parasite-host cell invasion process [Citation105].
IL4 production
Indole derivatives have been reported as provoking the inhibition of interleukine-4 (IL-4) secretion. So, it was considered that pharmacomodulation associating azole and indole fragments in the same compound could be of great interest in leishmaniasis treatment. In the series of 3-(α-azolylbenzyl)indoles, one compound exhibited high in vitro and in vivo antileishmanial activity. Concerning the mechanism action study, as anticipated, it was highlighted that it decreases ergosterol biosynthesis leading to membrane fungal cell alteration. Moreover it was proved that this imidazole antifungal agent induces a parasite burden-correlated decrease in IL-4 production, both in the splenocytes and the lymph node [Citation106]. The 3D-QSAR CoMSIA study offered a new model for further design of more promising inhibitors in the 3-(imidazol-1-ylmethyl)indole series [Citation107].
Conclusions
Although pentavalent antimonials are still the first choice of drugs in most countries, several trials have undertaken the hunt for less toxic and less expensive drugs and for orally available treatment. The recent availability of oral miltefosine for visceral leishmaniasis has been the most significant development in the past few years. Nevertheless, the spectrum of resistance is always present, indicating the need for further research in order to assess, in greater depth, the efficiency and safety of drug combinations. Efforts to find new lead compounds and to identify new targets will also contribute to the fight against leishmanial diseases and the preparation of additional resources for the drug discovery pipeline.
Declaration of interest: The authors reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
References
- World Health Organization. The leishmaniases and Leishmania/HIV co-infections Fact sheet 116 2004.
- BL Herwaldt. (1999). Lancet 354:1191–1199.
- J Alvar, C Canavate, B Gutierrez-Solar, M Jiminez, F Laguna, R Lopez-Velez, R Molina, and J Moreno. (1997). Clin Microbiol Rev 10:298–319.
- MJ Fernández Cotarelo, J Abellán Martínez, JM Guerra Vales, P Martínez Sánchez, M Rodrigo Gómez De La Bárcena, and E Salto Fernández. (2003). Clin Infect Dis 37:973–977.
- PD Deps, MC Viana, A Falqueto, and R Dietze. (2000). Rev Soc Bras Med Trop 33:535–543.
- S Sundar, and HW Murray. (2005). Bull World Health Organ 83:394–395.
- S Sundar, TK Jha, CP Thakur, PK Sinha, and SK.N Bhattacharya. (2007). Engl J Med 356:2571–2581.
- R Dietze, SF Carvalho, LC Valli, J Berman, T Brewer, W Milhous, J Sanchez, B Schuster, and M Grogl. (2001). Am J Trop Med Hyg 65:685–689.
- DH Beach, LJ Goad, and GG HolzJr. (1988). Mol Biochem Parasitol 31:149–162.
- M Ephros, A Bitnum, P Shaked, A Waldman, and D Zilberstein. (1999). Antimicrob Agents Chemother 43:278–282.
- S Wyllie, ML Cunningham, and AH Fairlamb. (2004). J Biol Chem 279:39925–39932.
- I Esfandiarpour, and A Alavi. (2002). Int J Dermatol 41:521–524.
- S Sundar. (2001). Trop Med Int Health 6:849–854.
- R Hadighi, M Mohebali, P Boucher, H Hajjaran, A Khamesipour, and M Ouellette. (2006). PLoS Med 3:e162.
- JP Guthmann, D Arlt, LM Garcia, M Rosales, J de Jesus Sanchez, E Alvarez, S Lonlas, M Conte, G Bertoletti, C Fournier, R Huari, E Torreele, and A Llanos-Cuentas. (2005). Trop Med Int Health 10:856–862.
- JL Burges, and R Birchall. (1972). Am J Med 53:77–84.
- R Nonata, R Sampaio, and PD Marsden. (1997). Trans R Soc Trop Med Hyg 91:77.
- JD Berman, R Badaro, CP Thakur, KM Wasunna, K Behbehani, R Davidson, F Kuzoe, L Pang, K Weerasuriya, and AD Bryceson. (1998). Bull World Health Organ 76:25–32.
- EM Andersen, M Cruz-Saldarriaga, A Llanos-Cuentas, M Luz-Cjuno, J Echevarria, C Miranda-Verastegui, O Colina, and JD Berma. (2005). Am J Trop Med Hyg 72:133–137.
- M Basselin, MA Badet-Denisot, F Lawrence, and M Robert-Gero. (1997). Exp Parasitol 85:274–282.
- SL Croft, and GH Coombs. (2003). Trends Parasitol 19:502–508.
- H Lux, DT Hart, PJ Parker, and T Klenner. (1996). Adv Exp Med Biol 416:201–211.
- H Lux, N Heise, T Klenner, D Hart, and FR Opperdoes. (2000). Mol Biochem Parasitol 111:1–14.
- TK Jha, S Sundar, CP Thakur, P Bachmann, J Karbwang, C Fischer, A Voss, and J Berman. (1999). N Engl J Med 341:1795–1800.
- J Soto, BA Arana, J Toledo, N Rizzo, JC Vega, A Diaz, M Luz, P Gutierrez, M Arboleda, JD Berman, K Junge, J Engel, and H Sindermann. (2004). Clin Infect Dis 38:1266–1272.
- P Escobar, S Matu, C Marques, and SL Corft. (2002). Acta Trop 81:151–157.
- V Yardley, SL Croft, S de Doncker, JC Dujardin, S Koirala, S Rijal, C Miranda, A Llanos-Cuentas, and F Chappuis. (2005). Am J Trop Med Hyg 73:272–275.
- SK Bhattacharya, PK Sinha, S Sundar, CP Thakur, TK Jha, K Pandey, VR Das, N Kumar, C Lal, N Verma, VP Singh, A Ranjan, RB Verma, G Anders, H Sindermann, and NK Ganguly. (2007). J Infect Dis 196:591–598.
- J Soto, J Toledo, P Gutierrez, RS Nicholls, J Padilla, J Engel, C Fischer, A Voss, and J Berman. (2001). Clin Infect Dis 33:57–61.
- O Zerpa, M Ulrich, B Blanco, M Polegre, A Avila, N Matos, I Mendoza, F Pratlong, C Ravel, and J Convit. (2007). Br J Dermatol 156:1328–1335.
- J Troya, A Casquero, E Refoyo, ML Fernández-Guerrero, M Górgolas, and J Scand. (2008). Infect Dis 40:78–80.
- J El-On, R Livshin, Z Evan-Paz, D Hamburger, and L Weinrauch. (1986). J Invest Dermatol 87:284–288.
- A BenSalah, H Zakraoui, A Zaatour, A Ftaiti, B Zaafouri, A Garaoui, PL Olliaro, K Dellagi, and R Ben Ismail. (1995). Am J Trop Med Hyg 53:162–166.
- A Asilain, T Jalayer, JAG Whitworth, RL Ghasemi, M Nilforooshzadeh, and P Ollivaro. (1995). Am J Trop Med Hyg 53:648–651.
- JM Soto, JT Tolédo, P Gutierrez, M Arboleda, RS Nicholls, JR Padilla, JD Berman, CK English, and M Grogl. (2002). Acta Trop Med Hyg 66:147–151.
- L Kotra, J Haddad, and S Mobashery. (2000). Antimicrob Agents Chemother 44:3249–3256.
- M Maarouf, Y de Kouchkoxsky, S Brown, PX Petit, and M Robert-Gero. (1997). Exp Cell Res 232:339–348.
- M Kumaresan, P Kumar, and J Ind. (2007). Dermatol Venereol Leprol 73:361–362.
- M Raffa, S Ingen-Housz-Oro, L Méry, F Le Turdu, J Wendling, C Pauwels, and M Sigal-Grinberg. (2007). Ann Dermatol Venereol 134:682–683.
- HM Al-Abdely, JR Graybill, D Loebenberg, and PC Melby. (1999). Antimicrob Agents Chemother 43:2910–2914.
- MK Wasunna, JR Rachid, J Mbui, G Kirigi, D Kinoti, H Lodenyo, JM Felton, AJ Sabin, MJ Albert, and J Horton. (2005). Am J Trop Med Hyg 73:871–876.
- TK Jha, S Sundar, CP Thakur, MJ Felton, AJ Sabin, and J Horton. (2005). Am J Trop Med Hyg 73:1005–1011.
- CJ Beckers, DS Roos, RG Donald, BJ Luft, JC Schwab, Y Cao, and KA Joiner. (1995). J Clin Invest 95:367–376.
- A Ianaro, A Ialenti, P Maffia, L Sautebin, L Rombolà, R Carnuccio, T Iuvone, F D'Acquisto, and M Di Rosa. (2000). J Pharmacol Exp Ther 292:156–163.
- G Xu, J Fujita, K Negayama, K Yuube, S Hojo, Y Yamaji, K Kawanishi, and J Takahara. (1996). Microbiol Immunol 40:473–479.
- AJ Krolewiecki, P Scott, and D Abraham. (1999). Rev Soc Bras Med Trop 32:137.
- A Prata, ML Silva-Vergara, L Costa, A Rocha, A Krolewiecki, JC Silva, EV de Paula, FG Pimenta Junior, and LE Giraldo. (2003). Rev Soc Bras Med Trop 36:65–69.
- AZ Momeni, A Shafiei, M Emamjoeh, M Aminjavaheri, and A Momeni. (2006). Eur J Dermatol 16:701–702.
- P Layegh, MJ Yazdanpanah, EM Vosugh, F Pezeshkpoor, MT Shakeri, and T Moghiman. (2007). Am J Trop Med Hyg 77:99–101.
- PE Carson. W Peters, and WHG Richards. Springer-Verlag, Berlin Germany, (1984) Vol. 83 121.
- J Leyden. (1998). J Am Acad Dermatol 38:S42–S47.
- R De La Rosa, JA Pineda, J Delgado, J Macías, F Morillas, J Martín-Sánchez, M Leal, A Sánchez-Quijano, and E Lissen. (2001). Clin Infect Dis 32:633–635.
- MK Mittal, S Rai, Ashutosh, Ravinder, S Gupta, S Sundar, and N Goyal. (2007). Am J Trop Med Hyg 76:681–688.
- N Mbongo, PM Loiseau, MA Billion, and M Robert-Gero. (1998). Antimicrob Agents Chemother 42:352–357.
- M Pourshafie, S Morand, A Virion, M Rakotomanga, C Dupuy, and PM Loiseau. (2004). Antimicrob Agents Chemother 48:2409–2414.
- MP Barrett, and AH Fairlam. (1999). Parasitol Today 15:136–140.
- PG Bray, MP Barrett, SA Ward, and HP de Koninqg. (2003). Trends Parasitol 19:232–239.
- AC Coelho, SM Beverly, and PC Cotrim. (2003). Mol Biochem Parasitol 130:83–90.
- M Basselin, H Denise, GH Coombs, and MP Barrett. (2002). Antimicrob Agents Chemother 46:3731–3738.
- JF Perez-Victoria, F Gamarro, M Ouellette, and S Castanys. (2003). J Biol Chem 278:49965–49971.
- JM Perez-Victoria, A Di Pietro, D Barron, AG Ravelo, S Castanys, and F Gamarro. (2002). Curr Drug Targets 3:311–333.
- GC Di Giorgio, F Faraut-Gambarelli, A Imbert, P Minodier, M Gasquet, and H Dumon. (1999). J Antimicrob Chemother 44:71–76.
- G Sadeghian, and MA Nilforoushzadeh. (2006). Int J Dermatol 45:819–821.
- A Firooz, A Khamesipour, MH Ghoorchi, M Nassiri-Kashani, SE Eskandari, A Khatami, B Hooshmand, F Gorouhi, M Rashighi-Firoozabadi, and Y Dowlati. (2006). Arch Dermatol 142:1575–1579.
- C Miranda-Verástegui, A Llanos-Cuentas, I Arévalo, BJ Ward, and G Matlashewski. (2005). Clin Infect Dis 40:1395–1403.
- I Arevalo, G Tulliano, A Quispe, G Spaeth, G Matlashewski, A Llanos-Cuentas, and H Pollack. (2007). Clin Infect Dis 44:1549–1554.
- CP Thakur, TP Kanyok, AK Pandey, A Sinha, E Zaniewski, HH Houlihan, and P Olliaro. (2000). Trans R Soc Trop Med Hyg 94:429–431.
- Y Melaku, SM Collin, K Keus, F Gatluak, K Ritmeijer, and RN Davidson. (2007). Am J Trop Med Hyg 77:89–94.
- I Esfandiarpour, and SH Dabiri. (2007). Int J Dermatol 46:848–852.
- K Seifert, and SL Croft. (2006). Antimicrob Agents Chemother 50:73–79.
- LV Rodriguez, JP Dedet, V Paredes, C Mendoza, and F Cardenas. (1995). Mem Inst Oswaldo Cruz 90:525–528.
- JF Costa, AC Kiperstok, JP David, JM David, AM Giulietti, LP de Queiroz, RR Dos Santos, and MB Soares. (2007). Fitoterapia 78:510–514.
- SR Peraza-Sánchez, F Cen-Pacheco, A Noh-Chimal, F May-Pat, P Simá-Polanco, E Dumonteil, MR García-Miss, and M Mut-Martín. (2007). Fitoterapia 78:315–318.
- CM Lezama-Dávila, AP Isaac-Márquez, P Zamora-Crescencio, R Uc-Encalada M del, SY Justiniano-Apolinar, L del Angel-Robles, A Satoskar, and L Hernández-Rivero. (2007). Fitoterapia 78:255–257.
- M Lavault, A Landreau, G Larcher, JP Bouchara, F Pagniez, P Le Pape, and P Richomme. (2005). Fitoterapia 76:363–366.
- MA Brenzan, CV Nakamura, B Prado Dias Filho, T Ueda-Nakamura, MC Young, and D Aparício Garcia Cortez. (2007). Parasitol Res 101:715–722.
- P Sartorelli, SP Andrade, MS Melhem, FO Prado, and AG Tempone. (2007). Phytother Res 21:644–647.
- RL Monte, JM Barbosa, LM Sousa, PF Athayde, CS Dias, and MR Oliveira. (2007). Z Naturforsch 62:348–352.
- K Georgopoulou, D Smirlis, S Bisti, E Xingi, L Skaltsounis, and K Soteriadou. (2007). Planta Med 73:1081–1088.
- MR Gamboa-León, I Aranda-González, M Mut-Martín, MR García-Miss, and E Dumonteil. (2007). Scand J Immunol 66:508–514.
- A Fournet, ME Ferreira, A Rojas De Arias, S Torres De Ortiz, S Fuentes, H Nakayama, A Schinini, and R Hocquemiller. (1996). Antimicrob Agents Chemother 40:2447–2451.
- M Chen, SB Christiensen, TG Theander, and A Kharasmi. (1994). Antimicrob Agents Chemother 38:1339–1344.
- N Flores, G Cabrera, IA Jiménez, J Piñero, A Giménez, G Bourdy, F Cortés-Selva, and IL Bazzocchi. (2007). Planta Med 73:206–211.
- L Maes, N Germonprez, L Quirijnen, L Van Puyvelde, P Cos, and D Vanden Berghe. (2004). Antimicrob Agents Chemother 48:2056–2060.
- JF Marquis, D Makhey, EJ LaVoie, and M Olivier. (2003). J Parasitol 89:1048–1052.
- N Germonprez, L Maes, L Van Puyvelde, M Van Tri, DA Tuan, and N De Kimpe. (2005). J Med Chem 48:32–37.
- A Ariza, TJ Vickers, N Greig, KA Armour, MJ Dixon, IM Eggleston, AH Fairlamb, and CS Bond. (2006). Mol Microbiol 59:1239–1248.
- S Parveen, MO Khan, SE Austin, SL Croft, V Yardley, P Rock, and KT Douglas. (2005). J Med Chem 48:8087–8097.
- G Singh, and CS Dey. (2007). Acta Trop 103:172–185.
- PW Denny, H Shams-Eldin, HP Price, DF Smith, and RT Schwarz. (2006). J Biol Chem 281:28200–28209.
- KR Santhamma, and A Bhaduri. (1995). Mol Biochem Parasitol 75:43–53.
- M Chen, L Zhai, SB Christensen, TG Theander, and A Kharazmi. (2001). Antimicrob Agents Chemother 45:2023–2029.
- H Abdalah, D Sebastien, B George, F Arlette, J Kalil, and P Le Pape. (2006). J Enz Inhib Med Chem 21:305–312.
- JA Urbina, JL Concepcion, S Rangel, G Visbal, and R Lira. (2002). Mol Biochem Parasitol 125:35–45.
- JCF Rodrigues, JA Urbina, and W de Souza. (2005). Exp Parasitol 111:230–238.
- SB Cammerer, C Jimenez, S Jones, L Gros, SO Lorente, C Rodrigues, JC Rodrigues, A Caldera, LM Ruiz Perez, W da Souza, M Kaiser, R Brun, JA Urbina, D Gonzalez Pacanowska, and IH Gilbert. (2007). Antimicrob Agents Chemother 51:4049–4061.
- AC Granthon, MV Braga, JC Rodrigues, S Cammerer, SO Lorente, IH Gilbert, JA Urbina, and W de Souza. (2007). Vet Parasitol 146:25–34.
- V Yardley, AA Khan, MB Martin, TR Slifer, FG Araujo, SN Moreno, R Docampo, SL Croft, and E Oldfield. (2002). Antimicrob Agents Chemother 46:929–931.
- V Mundodi, AS Kucknoor, and L Gedamu. (2005). BMC Mol Biol 6:3.
- A Ponte-Sucre, R Vicik, M Schultheis, T Schirmeister, and H Moll. (2006). Antimicrob Agents Chemother 50:2439–2447.
- Handbook of proteolytic enzymesKF Walker, SM Arfin, and RA Bradshaw. Academic Press, London, (1998) 1394–1399.
- P Zhang, DE Nicholson, JM Bujnicki, X Su, JJ Brendle, M Ferdig, DE Kyle, WK Milhous, and PK Chiang. (2002). J Biomed Sci 9:34–40.
- P Hassan, D Fergusson, KM Grant, and JC Mottram. (2001). Mol Biochem Parasitol 113:189–198.
- KM Grant, MH Dunion, V Yardley, AL Skaltsounis, D Marko, G Eisenbrand, SL Croft, L Meijer, and JC Mottram. (2004). Antimicrob Agents Chemother 48:3033–3042.
- N Alvarez, S Robledo, ID Velez, JM Robert, G Le Baut, and P Le Pape. (2002). J Enz Inhib Med Chem 17:443–447.
- F Pagniez, H Abdala-Valencia, P Marchand, M Le Borgne, G Le Baut, S Robert-Piessard, and P Le Pape. (2006). J Enz Inhib Med Chem 21:277–283.
- F Giraud, C Loge, M Le Borgne, F Pagniez, YM Na, P Le Pape, and QSAR SAR. (2006). Environ Res 17:299–309.