Abstract
A new series of 1-benzyl-3-(imidazol-1-ylmethyl)indoles were synthesized according to a previous 3D-QSAR predictive model and assayed for their antiparasitic activity upon Leishmania mexicana promastigotes. The biological results obtained for these twelve molecules showed an IC50 ranging from 2.3 to 32 μM and mainly illustrated the importance of the hydrophobic parameter the para-position of the benzyl group. In order to improve the activities of these compounds and to check the potential influence of the electronic parameter on this particular position, a Craig diagram was used to select original electro-donating and lipophilic substituents. Synthesis and biological evaluation of ten new compounds (IC50 between 2.5 and 5.4 μM) confirmed that only the hydrophobic field is essential for a high level of activity.
Introduction
Leishmaniasis is a frequently observed disease in developed countries with more than two million new cases per year. It is transmitted by the bite of a sandfly which inoculates the parasite under its promastigote form to the human body [Citation1]. After a differentiation process, promastigotes are converted to a non-flagellated form, amastigotes. Multiplication and invasion of the different organs sign the first clinical aspects of the disease which can be considered as a major public health problem. Leishmaniasis occurs in mainly three different forms, depending on the parasite species and the immune response of the patients [Citation2]. In some cases, a co-infection with AIDS is observed and makes a therapeutic approach more difficult to treat these patients. Development of international tourism in infected regions is also a cause for the predominance of the disease in European countries [Citation3].
Several efforts have been made to find appropriate treatments to reduce the impact of the disease. The first class of compounds was the antimonials, but due to their high toxicity, pentamidine or amphotericine B have been developed [Citation4–5]. Some resistance cases have been reported, reducing their use and showing the importance to find more relevant drugs to treat this disease [Citation6–7].
In our interest for the research of new antifungal agents, we formerly isolated a series of 3-(imidazol-1-ylmethyl)-1H-indoles which showed an interesting level of activity on Leishmania mexicana promastigotes [Citation8–10]. They probably act on the cytochrome P450 lanosterol 14α-demethylase (CYP51) enzyme. This key enzyme is involved in the ergosterol biosynthetic pathway, one of the major constituent of the parasite membrane [Citation11]. A major class of compounds acting as CYP51 inhibitors is represented by the azole superfamily. This class of compounds has been studied for several years for its antifungal properties. All of them bear an azole ring (imidazole or triazole) which enables them to coordinate with the iron atom of the heme substructure.
Unfortunately no crystallographic data of the targeted enzyme is known to date. To rationalize the biological results in terms of structure-activity relationships, we developed a 3D-QSAR predictive approach based on a library of thirty-one molecules (0.3 < IC50 (μM) < 80) acting as antileishmanial agents to get more information on structural features which are essential for the activity [Citation12]. A 3D-QSAR CoMSIA model was built combining steric, electrostatic, hydrophobic and hydrogen bond acceptor fields (q2 = 0.594, r2 = 0.897, r2pred = 0.649).
On the basis of the different 3D contour maps, we were able to elucidate an optimum chemical scaffold () that could have an interesting level of activity. Selection of the most appropriate substituents will lead us to verify our conclusions. This inhibitor should have (i) an imidazole ring, (ii) a sterically hindered, electronegative and hydrophilic group in position 2 of the indole nucleus, (iii) various electronegative groups in position 5 and (iv) a hydrophobic and H-bond acceptor substituent in para- position of the benzyl moiety.
Figure 1. Chemical structure of a potential inhibitor following the conclusions of the model.
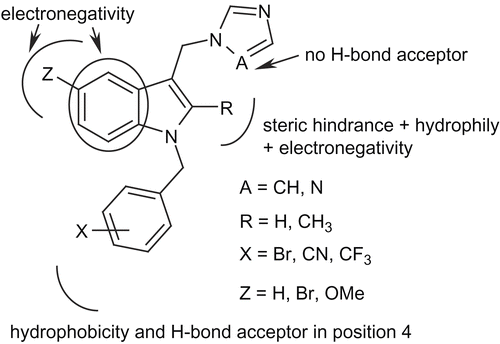
Starting from the conclusions of our original 3D-QSAR model, we describe in this paper the synthesis and evaluation of a series of compounds. Interpretation of the biological results brought us new information on the most appropriate structural characteristics for activity and then a new series of molecules were prepared. Synthetic work and biological results of this series are also discussed.
Materials and methods
Chemistry
Instrumentation. Melting points were determined on an Electrothermal IA 9000 melting point apparatus in open capillary tubes and are not corrected. IR spectra were obtained in KBr pellets with a Perkin-Elmer Paragon 1000 PC spectrometer. 1H and 13C-NMR spectra were recorded on a Bruker AC 250 or AVANCE 400 spectrometer in d6-DMSO as solvent. Chemical shifts are expressed as δ values (ppm) relative to Me4Si as internal standard. Coupling constants J are in Hz. Electrospray ionization (ESI) mass spectra were recorded on a ESQUIRE-LC Ion Trap System. All reactions were monitored by thin-layer chromatography (TLC) using 0.2 mm-thick silica gel plates 60F-254 (5735 Merck). Column chromatography was carried out using silica gel 60 (70–230 Mesh, ASTM, Merck). Chemicals and solvents used were commercially available. Compounds 5a, 5e and 5j were prepared according to reported methods [8, Citation9 and Citation13].
General procedure for the preparation of compounds 2a-e
The method used for the synthesis of 5-methoxy-2-methyl-1H-indole-3-carbaldehyde (2e) is described. To a 100 mL three-necked round flask was introduced anhydrous N,N-dimethylformamide (4.2 mL, 34.35 mmol) at 0 °C under argon followed by slow addition of phosphorus oxychloride (1.3 mL, 13.62 mmol). Solution was mixed at 0 °C for 40 min. A solution of 5-methoxy-2-methyl-1H-indole (1e) (2 g, 12.41 mmol) in 2.5 mL of DMF was slowly added maintaining the temperature below 10 °C. The solution was stirred for 40 min at 0 °C and then 35 °C for additional 40 min. Then pilled ice was added to the flask and a solution of sodium hydroxide (5.5 g, 137.25 mmol dissolved in 14.6 mL of water) was introduced using a dropping funnel. Solution was vigorously stirred during the addition, then heated to 100 °C for 30 min and left to reach room temperature. The brown precipitate was filtered off and washed with large volumes of water. The powder was dried in a ventilated oven at 60 °C for one night. Compound 2e was obtained without further purification.
5-Methoxy-2-methyl-1H-indole-3-carbaldehyde.(2e). Yield: 96%, yellow powder, mp = 190–191 °C; 1H NMR δ (250 MHz, DMSO) 2.68 (s, 3H, CH3), 3.81 (s, 3H, OCH3), 6.83 (dd, 1H, 3J = 10 Hz, 4J = 2.5 Hz, H6), 7.32 (d, 1H, 3J = 10 Hz, H7), 7.60 (d, 1H, 4J = 2.5 Hz, H4), 10.05 (s, 1H, CHO), 11.89 (s, 1H, NH). IR (KBr) cm−1 1480, 1561 (ν C=C); 1632 (ν C=O); 2992 (ν C-Halkane); 3025 (ν C-Harom); 3165 (ν N-H).
General procedure for the preparation of compounds 3a-y
The method used for the synthesis of 4-[(3-formyl-5-methoxy-2-methyl-1H-indol-1-yl)methyl]benzonitrile (3s) is described. To a solution of 5-methoxy-2-methyl-1H-indole-3-carbaldehyde (2e) (300 mg, 1.59 mmol) in 5 mL of anhydrous acetonitrile at 25 °C was added cesium carbonate (1.03 g, 3.17 mmol). The mixture was heated at reflux for 2 h and then 4-(bromomethyl)benzonitrile (340 mg, 1.74 mmol) was added in one portion. The reaction mixture was stirred at reflux for 1 h. After filtration, the solvent was removed under reduced pressure and the residue was partitioned between water and dichloromethane. Organic layer was dried over Na2SO4 and concentrated in vacuo. Column chromatography of the residue on silica gel using dichloromethane as eluent gave the pure product 3s.
4-[(3-Formyl-5-methoxy-2-methyl-1H-indol-1-yl)methyl]benzonitrile (3s). Yield: 99%, yellow powder, mp = 138–139 °C; 1H NMR δ (250 MHz, DMSO) 2.69 (s, 3H, CH3), 3.82 (s, 3H, OCH3), 5.52 (s, 2H, NCH2), 6.87 (dd, 1H, 3J = 8.8 Hz, 4J = 2.4 Hz, H6), 7.05 (d, 2H, 3J = 8.5 Hz, Hbenzyl), 7.44 (d, 1H, 3J = 8.8 Hz, H7), 7.56 (d, 2H, 3J = 8.5 Hz, Hbenzyl), 7.70 (d, 1H, 4J = 2.4 Hz, H4), 10.13 (s, 1H, CHO). IR (KBr) cm−1 1479, 1519 (ν C=C); 1639 (ν C=O); 2223 (ν CN); 2926 (ν C-Halkane); 3060 (ν C-Harom).
General procedure for the preparation of compounds 4a-y and 8b, 8c, 8e
The method used for the synthesis of 4-(3-hydroxymethyl-5-methoxy-2-methyl-1H-indol-1-ylmethyl)benzonitrile (4s) is described. To a solution of 4-[(3-formyl-5-methoxy-2-methyl-1H-indol-1-yl)methyl]benzonitrile (3s) (480 mg, 1.47 mmol) in 10 mL of pure methanol was added portionwise sodium borohydride (530 mg, 4.71 mmol). Reaction was exothermic and effervescent. Mixture was stirred at room temperature for 1 h under argon and water was added with care to the reaction. Reaction mixture was extracted with dichloromethane. Organic layer was dried over Na2SO4 and concentrated in vacuo. Pure product 4s was obtained without further purification.
4-(3-Hydroxymethyl-5-methoxy-2-methyl-1H-indol-1-ylmethyl)benzonitrile (4s). Yield: 98%, brown powder, mp = 113–114 °C; 1H NMR δ (250 MHz, DMSO) 2.32 (s, 3H, CH3), 3.80 (s, 3H, OCH3), 4.63–4.64 (m, 3H, CH2OH), 5.51 (s, 2H, NCH2), 6.71 (dd, 1H, 3J = 8.8 Hz, 4J = 2.7 Hz, H6), 7.11–7.14 (m, 3H, H4, Hbenzyl), 7.26 (d, 1H, 3J = 8.8 Hz, H7), 7.80 (d, 2H, 3J = 8,2 Hz, Hbenzyl). IR (KBr) cm−1 1486, 1585 (ν C=C); 2216 (ν CN); 2927 (ν C-Halkane); 3035 (ν C-Harom); 3421 (ν O-H).
Procedure for the preparation of compound 7c
A solution of 4-propylbenzaldehyde diethylacetal (6c) (3 g, 13.49 mmol) in 13.5 mL of glacial acetic acid and 6.5 mL of water was refluxed for 17 h. Then it was cooled to 0 °C and 30 mL of a cold 6 M sodium hydroxide solution were slowly added. Reaction mixture was extracted with diethyl ether and organic layer was dried over Na2SO4 and concentrated in vacuo. Column chromatography on silica gel using dichloromethane as eluent afforded the pure product 7c.
4-Propylbenzaldehyde (7c). Yield: 97%, non-colored oil; 1H NMR δ (250 MHz, DMSO) 0.93 (t, 3H, 3J = 4.9 Hz, CH3), 1.58–1.70 (m, 2H, CH2CH3), 2.68 (t, 2H, 3J = 7.0 Hz, CH2CH2), 7.46 (d, 2H, 3J = 7.9 Hz, Hbenzyl), 7.86 (d, 2H, 3J = 7.9 Hz, Hbenzyl), 9.99 (s, 1H, CHO). IR (NaCl) cm−1 1456, 1512 (ν C=C); 1701 (ν C=O); 2927 (ν C-Halkane); 3035 (ν C-Harom).
Procedure for the preparation of compound 7e
To a solution of 4-butylbenzaldehyde diethylacetal (6e) (3 g, 12.69 mmol) in 24 mL ethanol were added 6 mL of water and 1.8 mL (25.38 mmol) of DMSO. The solution was refluxed for 1.5 h. A large amount of solvent was removed under reduced pressure. Residue was taken up in water and dichloromethane and product was extracted with dichloromethane. Organic layer was dried over Na2SO4 and concentrated in vacuo. Pure product 7e was isolated without purification.
4-Butylbenzaldehyde (7e). Yield: 100%, non-colored oil; 1H NMR δ (250 MHz, DMSO) 0.93 (t, 3H, 3J = 4.9 Hz, CH3), 1.33–1.38 (m, 2H, CH2CH3), 1.58–1.64 (m, 2H, CH2CH2), 2.68–2.73 (m, 2H, CH2CH2), 7.46 (d, 2H, 3J = 8.2 Hz, Hbenzyl), 7.86 (d, 2H, 3J = 8.2 Hz, Hbenzyl), 9.99 (s, 1H, CHO). IR (NaCl) cm−1 1461, 1574 (ν C=C); 1695 (ν C=O); 2931 (ν C-Halkane); 3040 (ν C-Harom).
General procedure for the preparation of compounds 9a-e
The method used for the synthesis of 4-methylbenzyl bromide (9a) is described. To a solution of 4-methylphenylmethanol (8a) (3 g, 24.5 mmol) in 15 mL anhydrous diethylether under argon was added dropwise phosphorus tribromide (1.28 mL, 13.5 mmol). Solution was stirred at room temperature for 2.5 h under argon. Water was added and reaction mixture was extracted with diethylether. Organic layers were dried over Na2SO4 and concentrated in vacuo. Pure product 9a was isolated without purification.
4-Methylbenzyl bromide (9a). Yield: 100%, non-colored needles, mp = 35–36 °C; 1H NMR δ (250 MHz, DMSO) 2.33 (3H, s, CH3), 4.71 (2H, s, CH2Br), 7.19 (2H, d, 3J = 7.9 Hz, Hbenzyl), 7.37 (2H, d, 3J = 7.9 Hz, Hbenzyl). IR (KBr) cm−1 810 (ν C-Br); 1435, 1507 (ν C=C); 2927 (ν C-Halkane).
General procedure for the preparation of compounds 5b-y
The method used for the synthesis of 1-(4-cyanobenzyl)-3-(1H-imidazol-1-ylmethyl)-5-methoxy-2-methyl-1H-indole (5s) is described. To a solution of 4-(3-hydroxymethyl-5-methoxy-2-methyl-1H-indol-1-ylmethyl)benzonitrile (4s) (153 mg, 0.50 mmol) in 8 mL of acetonitrile under argon was added 1,1’-carbonyldiimidazole (122 mg, 0.75 mmol). The mixture was stirred for 4 h at room temperature and solvent was removed under reduced pressure. Residue was diluted with water and dichloromethane and extracted. Organic layer was dried over Na2SO4 and concentrated in vacuo. Column chromatography using a gradient from pure dichloromethane to a mixture dichloromethane/ethanol: 19/1 afforded the product 5s. Finally, product is triturated in diisopropylic ether to get crystals.
1-(4-Cyanobenzyl)-3-(1H-imidazol-1-ylmethyl)-1H-indole (5b). Yield: 45%, white powder, mp = 125–126 °C; 1H NMR δ (250 MHz, DMSO) 5.38 (2H, s, CH2-Im), 5.57 (2H, s, NCH2), 6.88 (1H, s, Himid), 7.07 (1H, ddd, 3J = 7.6 Hz, 3J = 8.2 Hz, 4J = 2.4 Hz, H5), 7.13 (1H, ddd, 3J = 3J = 8.2 Hz, 4J = 2.4 Hz, H6), 7.20 (1H, s, Himid), 7.35 (2H, d, 3J = 8,5 Hz, Hbenzyl), 7.44 (1H, d, 3J = 8.2 Hz, H7), 7.60 (1H, d, 3J = 7.6 Hz, H4), 7.65 (1H, s, H2), 7.79–7.84 (3H, m, Himid, Hbenzyl). 13C NMR δ (250 MHz, DMSO) 41.6 (CH2-Im), 49.0 (NCH2), 110.6 (C4), 110.8 (C7), 111.5 (C3), 119.1 (C6), 119.2 (C5), 119.8 (CN), 120.0 (C2), 122.4 (Cimid), 127.2 (C9), 128.2 (2C, CHbenzyl), 128,7 (Cbenzyl), 128.9 (Cimid), 133.0 (2C, CHbenzyl), 136.6 (C8), 137.6 (Cimid), 144.3 (Cbenzyl). IR (KBr) cm−1 1466, 1503 (ν C=C and ν C=N); 2228 (ν C≡N). MS: 245.1 (MH+-67), 313.1 (MH+).
3-(1H-Imidazol-1-ylmethyl)1-(4-trifluoromethylbenzyl)-1H-indole (5c). Yield: 61%, white powder, mp = 103–104 °C; 1H NMR δ (250 MHz, DMSO) 5.38 (2H, s, CH2-Im), 5.57 (2H, s, NCH2), 6.88 (1H, s, Himid), 7.07 (1H, ddd, 3J = 7.6 Hz, 3J = 8.2 Hz, 4J = 2.4 Hz, H5), 7.13 (1H, ddd, 3J = 3J = 8.2 Hz, 4J = 2.4 Hz, H6), 7.21 (1H, s, Himid), 7.40 (2H, d, 3J = 8.6 Hz, Hbenzyl), 7.46 (1H, d, 3J = 8.2 Hz, H7), 7.60 (1H, d, 3J = 7.6 Hz, H4), 7.66 (1H, s, H2), 7.72 (2H, d, 3J = 8.6 Hz, Hbenzyl), 7.81 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 41.6 (CH2-Im), 48.9 (NCH2), 110.8 (2C, C4,7), 111.4 (C3), 119.2 (C5), 119.8 (C6), 119.9 (C2), 122.4 (Cimid), 125.9 (CF3), 127.2 (C9), 128.1 (2C, CHbenzyl), 128.4 (1C, d, 2J = 32 Hz, Cbenzyl), 128.7 (Cimid), 128.9 (2C, CHbenzyl), 136.6 (C8), 137.5 (Cimid), 143.5 (Cbenzyl). IR (KBr) cm−1 1155 (ν CF3); 1467, 1503 (ν C=C and ν C=N); 3102 (ν C-Harom). MS: 288.1 (MH+-67), 356.0 (MH+).
5-Bromo-1-(4-bromobenzyl)-3-(1H-imidazol-1-ylmethyl)-1H-indole (5d). Yield: 78%, orange powder, mp = 142–143 °C; 1H NMR δ (250 MHz, DMSO) 5.35 (2H, s, CH2-Im), 5.44 (2H, s, N-CH2), 6.89 (1H, s, Himid), 7.17 (2H, d, 3J = 8.4 Hz, Hbenzyl), 7.20 (1H, s, Himid), 7.28 (1H, dd, 3J = 8.8 Hz, 4J = 2.0 Hz, H6), 7.46 (1H, d, 3J = 8.8 Hz, H7), 7.54 (2H, d, 3J = 8.4 Hz, Hbenzyl), 7.69 (1H, s, H2), 7.80 (1H, d, 4J = 2.0 Hz, H4), 7.81 (1H, s, Himid). 13C NMR δ (400 MHz, DMSO) 40.9 (CH2-Im), 48.7 (N-CH2), 110.9 (C3), 112.7 (2C, C5, Cindole), 119.5 (Cimid), 120.8 (Cbenzyl), 121.2 (C4), 124.5 (Cindole), 128.5 (Cimid), 128.6 (C9), 129.4 (2C, CHbenzyl), 129.8 (Cbenzyl), 130.1 (C2), 131.7 (2C, CHbenzyl), 134.9 (C8), 137.3 (Cimid). IR (KBr) cm−1 798 (ν C-Br); 1466, 1562 (ν C=C and ν C=N); 2917 (ν C-Halkane); 3082 (ν C-Harom). MS: 379.2 (MH+-67).
5-Bromo-1-(4-trifluoromethylbenzyl)-3-(1H-imidazol-1ylmethyl)-1H-indole (5f). Yield: 31%, white powder, mp = 162–163 °C; 1H NMR δ (400 MHz, DMSO) 5.37 (2H, s, CH2-Im), 5.58 (2H, s, NCH2), 6.90 (1H, s, Himid), 7.22 (1H, s, Himid), 7.28 (1H, dd, 3J = 8.8 Hz, 4J = 2.0 Hz, H6), 7.38 (2H, d, 3J = 8.0 Hz, Hbenzyl), 7.46 (1H, d, 3J = 8.8 Hz, H7), 7.72 (2H, d, 3J = 8.0 Hz, Hbenzyl), 7.73 (1H, s, H2), 7.82 (1H, d, 3J = 2.0 Hz, H4), 7.85 (1H, s, Himid). 13C NMR δ (400 MHz, DMSO) 41.6 (CH2-Im), 49.7 (NCH2), 106.9 (C4), 111.8 (C7), 113.6 (C3), 120.5 (C5), 122.1 (Cimid), 123.0 (CF3), 125.6 (C2), 126.7 (2C, CHbenzyl), 128.7 (2C, CHbenzyl), 129.1 (C9), 129.6 (Cbenzyl), 131.2 (Cimid), 135.1 (C6), 135.9 (C8), 138.2 (Cimid), 143.6 (Cbenzyl). IR (KBr) cm−1 744 (ν C-Br); 1159 (ν CF3); 1467, 1503 (ν C=C and ν C=N); 2855 (ν C-Halkane); 3123 (ν C-Har.). MS: 367.1 (MH+-67), 434.1 (MH+).
1-(4-Bromobenzyl)-3-(1H-imidazol-1-ylmethyl)-5-methoxy-1H-indole (5g). Yield: 57%, light-colored powder, mp = 145–146 °C; 1H NMR δ (250 MHz, DMSO) 3.76 (3H, s, OCH3), 5.33 (2H, s, CH2-Im), 5.38 (2H, s, N-CH2), 6.80 (1H, dd, 3J = 8.8 Hz, 4J = 2.4 Hz, H6), 6.88 (1H, s, Himid), 7.10 (1H, d, 3J = 2.4 Hz, H4), 7.15 (2H, d, 3J = 8.2 Hz, Hbenzyl), 7.21 (1H, s, Himid), 7.33 (1H, d, 3J = 8.8 Hz, H7), 7.54 (2H, d, 3J = 8.2 Hz, Hbenzyl), 7.57 (1H, s, H2), 7.85 (1H, s, Himid). 13C NMR δ (400 MHz, DMSO) 42.2 (CH2-Im), 49.6 (N-CH2), 56.5 (OCH3), 101.8 (C4), 111.5 (C3), 112.2 (Cindole), 112.7 (Cindole), 120.4 (Cimid), 121.5 (Cbenzyl), 128.3 (C9), 129.3 (Cimid), 129.9 (Cbenzyl), 130.3 (2C, CHbenzyl), 132.3 (C2), 132.5 (2C, CHbenzyl), 138.1 (C8), 138.7 (Cimid), 154.8 (C5). IR (KBr) cm−1 792 (ν C-Br); 1490 (ν C=N); 1577 (ν C=C); 2941 (ν C-Halkane); 3091 (ν C-Harom). MS: 330.1 (MH+-67).
4-{[5-Methoxy-3-(1H-imidazol-1-ylmethyl)-1H-indol-1-yl]methyl}benzonitrile (5h). Yield: 46%, brown powder, mp = 88–89 °C; 1H NMR δ (400 MHz, DMSO) 3.77 (3H, s, OCH3), 5.34 (2H, s, CH2Im), 5.52 (2H, s, NCH2), 6.80 (1H, dd, 3J = 8.8 Hz, 4J = 2.0 Hz, H6), 6.89 (1H, s, Himid), 7.11 (1H, d, 4J = 2.0 Hz, H4), 7.22 (1H, s, Himid), 7.21–7.32 (3H, m, H7, Hbenzyl), 7.59 (1H, s, Himid), 7.79–7.83 (3H, m, H2, Hbenzyl). 13C NMR δ (400 MHz, DMSO) 41.3 (CH2-Im), 48.9 (NCH2), 55.6 (OCH3), 100.9 (C4), 110.3 (C3), 110.8 (C5), 111.3 (C7), 111.9 (C6), 118.8 (CN), 119.6 (C2), 127.4 (C9), 127.8 (2C, CHbenzyl), 128.4 (Cimid), 129.2 (Cimid), 131.4 (Cbenzyl), 132.7 (2C, CHbenzyl), 137.3 (Cimid), 144.2 (C8), 154.0 (Cbenzyl). IR (KBr) cm−1 1487, 1579 (ν C=C and ν C=N); 2226 (ν C≡N); 2937 (ν C-Halkane). MS: 275.1 (MH+-67), 343.1 (MH+).
3-(1H-Imidazol-1-ylmethyl)-5-methoxy-1-(4-trifluoromethylbenzyl)-1H-indole (5i). Yield: 52%, light-colored powder, mp = 84–85 °C; 1H NMR δ (400 MHz, DMSO) 3.76 (3H, s, OCH3), 5.34 (2H, s, CH2Im), 5.52 (2H, s, NCH2), 6.79 (1H, dd, 3J = 8.8 Hz, 4J = 2.0 Hz, H6), 6.88 (1H, s, Himid), 7.10 (1H, d, 4J = 2.0 Hz, H4), 7.22 (1H, s, Himid), 7.32–7.38 (3H, m, H7, Hbenzyl), 7.60 (1H, s, Himid), 7.71 (2H, d, 3J = 8.0 Hz, Hbenzyl), 7.81 (1H, s, H2). 13C NMR δ (400 MHz, DMSO) 41.3 (CH2-Im), 48.8 (NCH2), 55.6 (OCH3), 100.9 (C4), 110.7 (2C, C3, C5), 111.3 (C7), 111.9 (C6), 119.5 (C2), 125.7 (2C, CHbenzyl), 127.4 (C9), 127.7 (2C, CHbenzyl), 128.4 (Cimid), 129.2 (Cimid), 131.5 (Cbenzyl), 137.3 (Cimid), 143.2 (C8), 154.0 (Cbenzyl). IR (KBr) cm−1 1162 (ν CF3); 1491, 1579 (ν C=C and ν C=N). MS: 318.1 (MH+-67), 386.1 (MH+).
1-(4-Cyanobenzyl)-3-(1H-imidazol-1-ylmethyl)-2-methyl-1H-indole (5k). Yield: 54%, yellow oil; 1H NMR δ (250 MHz, DMSO) 2.46 (3H, s, CH3), 5.38 (2H, s, CH2-Im), 5.59 (2H, s, NCH2), 6.87 (1H, s, Himid), 7.06–7.17 (5H, m, H5, H6, Hbenzyl, Himid), 7.40 (1H, d, 3J = 8.0 Hz, H7), 7.60 (1H, dd, 3J = 7.5 Hz, 4J = 0.2 Hz, H4), 7.77–7.79 (3H, m, Hbenzyl, Himid). 13C NMR δ (400 MHz, DMSO) 10.0 (CH3), 40.4 (CH2-Im), 45.7 (NCH2), 107.7 (C3), 109.8 (C4), 110.1 (C9), 117.9 (Cimid), 118.8 (CN), 119.3 (C5), 119.8 (C7), 121.5 (C6), 127.0 (Cbenzyl), 127.2 (2C, CHbenzyl), 128.4 (Cimid), 132.8 (2C, CHbenzyl), 135.7 (C2), 136.2 (C8), 137.1 (Cimid), 144.2 (Cimid). IR (NaCl) cm−1 1467, 1502 (ν C=C and ν C=N); 2228 (ν C≡N); 2927 (ν C-Halkane). MS: 259.1 (MH+-67).
3-(1H-Imidazol-1-ylmethyl)-2-methyl-1-(4-trifluoromethylbenzyl)-1H-indole (5l). Yield: 50%, yellow powder, mp = 134–135 °C; 1H NMR δ (400 MHz, DMSO) 2.48 (3H, s, CH3), 5.38 (2H, s, CH2-Im), 5.59 (2H, s, N-CH2), 6.87 (1H, s, Himid), 7.10 (1H, ddd, 3J = 7.2 Hz, 3J = 6.8 Hz, 4J = 1.6 Hz, H5), 7.11 (1H, ddd, 3J = 7.6 Hz, 3J = 6.8 Hz, 4J = 1.2 Hz, H6), 7.14 (1H, s, Himid), 7.20 (2H, d, 3J = 8.0 Hz, Hbenzyl), 7.41 (1H, dd, 3J = 7.6 Hz, 4J = 1.6 Hz, H7), 7.60 (1H, dd, 3J = 7.2 Hz, 4J = 1.2 Hz, H4), 7.71 (2H, d, 3J = 8.0 Hz, Hbenzyl), 7.77 (1H, s, Himid). 13C NMR δ (400 MHz, DMSO) 10.0 (CH3), 40.7 (CH2-Im), 45.6 (N-CH2), 107.6 (C3), 117.9 (C4), 119.3 (C5), 119.8 (Cimid), 121.4 (C6), 122.2 (CF3), 125.8 (2C, q, 3J = 4,0 Hz, CHbenzyl), 127.0 (2C, CHbenzyl), 127.7 (C9), 128.2 (Cbenzyl), 128.4 (Cimid), 135.7 (C8), 136.2 (C2), 137.1 (Cimid), 143.3 (Cbenzyl). IR (KBr) cm−1 1159 (ν CF3); 1467, 1508 (ν C=C and ν C=N); 2922 (ν C-Halkane); 3014 (ν C-Harom). MS: 303.4 (MH+-67).
3-(1H-Imidazol-1-ylmethyl)-1-(4-methylbenzyl)-2-methyl-1H-indole (5m). Yield: 45%, light-yellow powder, mp = 73–74 °C; 1H NMR δ (250 MHz, DMSO) 2.27 (3H, s, ArCH3), 2.47 (3H, s, indoleCH3), 5.36 (2H, s, CH2-Im), 5.41 (2H, s, NCH2), 6.86 (1H, s, Himid), 6.91–6.94 (2H, m, H5, H6), 7.01–7.14 (5H, m, Hbenzyl, Himid), 7.42 (1H, d, 3J = 7.0 Hz, H7), 7.59 (1H, d, 3J = 6.7 Hz, H4), 7.74 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 10.4 (indoleCH3), 21.1 (ArCH3), 41.0 (CH2-Im), 46.1 (NCH2), 107.5 (C3), 110.2 (C4), 118.0 (C7), 119.6 (C9), 119,7 (Cimid), 119.8 (C5), 126.6 (2C, CHbenzyl), 127.2 (Cbenzyl), 128.7 (Cimid), 129.6 (2C, CHbenzyl), 135.6 (C6), 136.1 (C2), 136.5 (C8), 136.7 (Cimid), 137.3 (Cbenzyl). IR (KBr) cm−1 1463, 1508 (ν C=C and ν C=N); 2942 (ν C-Halkane); 3087 (ν C-Harom). MS: 248.1 (MH+–67), 338.2 (MH++23).
1-(4-Ethylbenzyl)-3-(1H-imidazol-1-ylmethyl)-2-methyl-1H-indole (5n). Yield: 59%, light-yellow powder, mp = 99–100 °C; 1H NMR δ (250 MHz, DMSO) 1.16 (3H, t, 3J = 7.3 Hz, CH2CH3), 2.49 (3H, s, indoleCH3), 2.58 (2H, t, 3J = 7.3 Hz, CH2CH3), 5.37 (2H, s, CH2-Im), 5.42 (2H, s, NCH2), 6.86 (1H, s, Himid), 6.94 (2H, d, 3J = 7.9 Hz, Hbenzyl), 7.04–7.17 (5H, m, H5, H6, Hbenzyl, Himid), 7.40–7.43 (1H, m, H7), 7.57–7.60 (1H, m, H4), 7.75 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 10.4 (indoleCH3), 16.0 (CH2CH3), 28.2 (CH2CH3), 40.7 (CH2-Im), 46.1 (NCH2), 107.5 (C3), 110.2 (C4), 118.0 (C7), 119.6 (C9), 119.8 (C5), 121.5 (Cimid), 126.6 (2C, CHbenzyl), 127.2 (Cbenzyl), 128.5 (2C, CHbenzyl), 128.7 (Cimid), 135.9 (C6), 136.1 (C2), 136.5 (C8), 137.3 (Cbenzyl), 143.1 (Cimid). IR (KBr) cm−1 1266 (ν C-N); 1415, 1467, 1508 (ν C=C and ν C=N); 2958 (ν C-Halkane); 3091 (ν C-Harom). MS: 262.1 (MH+–67), 352.2 (MH++23).
3-(1H-Imidazol-1-ylmethyl)-2-methyl-1-(4-propylbenzyl)-1H-indole (5o). Yield: 48%, brown oil; 1H NMR δ (250 MHz, DMSO) 0.88 (3H, t, 3J = 4.9 Hz, CH2CH2CH3), 1.51–1.60 (2H, m, CH2CH2CH3), 2.48 (3H, s, indoleCH3), 2.54 (2H, t, 3J = 7.0 Hz, CH2CH2CH3), 5.36 (2H, s, CH2-Im), 5.42 (2H, s, NCH2), 6.86 (1H, s, Himid), 6.94 (2H, d, 3J = 7.9 Hz, Hbenzyl), 7.02–7.10 (3H, m, H5, H6, Himid), 7.13 (2H, d, 3J = 7.9 Hz, Hbenzyl), 7.36–7.39 (1H, m, H7), 7.55–7.60 (1H, m, H4), 7.74 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 10.4 (indoleCH3), 14.1 (CH2CH2CH3), 24.5 (CH2CH2CH3), 37.3 (CH2CH2CH3), 40.0 (CH2-Im), 46.1 (NCH2), 107.5 (C3), 110.2 (C4), 118.0 (C7), 119.6 (C9), 119.8 (C5), 121.5 (Cimid), 126.6 (2C, CHbenzyl), 127.2 (Cbenzyl), 128.7 (Cimid), 129.0 (2C, CHbenzyl), 135.9 (C6), 136.1 (C2), 136.5 (C8), 137.3 (Cbenzyl), 141.5 (Cimid). IR (NaCl) cm−1 1266 (ν C-N); 1415, 1461, 1502 (ν C=C and ν C=N); 2937 (ν C-Halkane); 3087 (ν C-Harom). MS: 276.2 (MH+–67).
3-(1H-Imidazol-1-ylmethyl)-1-(4-isopropylbenzyl)-2- methyl-1H-indole (5p). Yield: 11%, orange powder, mp = 53–54 °C; 1H NMR δ (250 MHz, DMSO) 1.18 (6H, d, 3J = 6.7 Hz, 2 CH3 iPr), 2.49 (3H, s, indoleCH3), 2.86 (1H, m, 3J = 6.7 Hz, CH iPr), 5.37 (2H, s, CH2-Im), 5.42 (2H, s, NCH2), 6.86 (1H, s, Himid), 6.95 (2H, d, 3J = 8.2 Hz, Hbenzyl), 7.03–7.10 (2H, m, H5, H6), 7.13 (1H, s, Himid), 7.19 (2H, d, 3J = 8.2 Hz, Hbenzyl), 7.41 (1H, d, 3J = 7.6 Hz, H7), 7.58 (1H, d, 3J = 6.7 Hz, H4), 7.75 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 10.4 (indoleCH3), 24.3 (2C, 2 CH3 iPr), 35.5 (CH iPr), 40.9 (CH2-Im), 46.1 (NCH2), 107.4 (C3), 110.2 (C4), 118.0 (C7), 119.7 (C9), 119.9 (Cimid), 121.5 (C5), 126.6 (2C, CHbenzyl), 127.0 (2C, CHbenzyl), 127.2 (Cbenzyl), 128.5 (Cimid), 136.0 (C6), 136.1 (C2), 136.5 (C8), 137.3 (Cimid), 147.7 (Cbenzyl). IR (KBr) cm−1 1471, 1508 (ν C=C and ν C=N); 2947 (ν C-Halkane); 3094 (ν C-Harom). MS: 276.2 (MH+–67).
1-(4-Butylbenzyl)-3-(1H-imidazol-1-ylmethyl)-2-methyl-1H-indole (5q). Yield: 49%, brown oil; 1H NMR δ (250 MHz, DMSO) 0.89 (3H, t, 3J = 7.0 Hz, CH2CH2CH2CH3), 1.22–1.33 (2H, m, CH2CH2CH2CH3), 1.36–1.58 (2H, m, CH2CH2CH2CH3), 2.48 (3H, s, indoleCH3), 2.50 (2H, t, 3J = 7.0 Hz, CH2CH2CH2CH3), 5.36 (2H, s, CH2-Im), 5.41 (2H, s, NCH2), 6.86 (1H, s, Himid), 6.94 (2H, d, 3J = 8.2 Hz, Hbenzyl), 7.01–7.15 (5H, m, H5, H6, Hbenzyl, Himid), 7.39–7.43 (1H, m, H7), 7.56–7.60 (1H, m, H4), 7.75 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 10.4 (indoleCH3), 14.2 (CH2CH2CH2CH3), 22.2 (CH2CH2CH2CH3), 35.5 (CH2CH2CH2CH3), 34.9 (CH2CH2CH2CH3), 40.7 (CH2-Im), 46.1 (NCH2), 107.5 (C3), 110.2 (C4), 118.0 (C7), 119.6 (C9), 119.8 (C5), 121.5 (Cimid), 126.6 (2C, CHbenzyl), 127.2 (Cbenzyl), 128.7 (Cimid), 129.0 (2C, CHbenzyl), 135.8 (C6), 136.1 (C2), 136.5 (C8), 137.3 (Cbenzyl), 141.7 (Cimid). IR (NaCl) cm−1 1272 (ν C-N); 1415, 1467, 1503 (ν C=C and ν C=N); 2916 (ν C-Halkane); 3094 (ν C-Harom). MS: 290.2 (MH+–67).
1-(4-Bromobenzyl)-3-(1H-imidazol-1-ylmethyl)-5-methoxy-2-methyl-1H-indole (5r). Yield: 39%, dark-yellow powder, mp = 80–81 °C; 1H NMR δ (250 MHz, DMSO) 2.43 (3H, s, CH3), 3.77 (3H, s, OCH3), 5.34 (2H, s, CH2-Im), 5.40 (2H, s, NCH2), 6.74 (1H, dd, 3J = 8.8 Hz, 4J = 2.4 Hz, H6), 6.86 (1H, s, Himid), 6.94 (2H, d, 3J = 8.6 Hz, Hbenzyl), 7.11 (1H, d, 4J = 2.4 Hz, H4), 7.14 (1H, s, Himid), 7.30 (1H, d, 3J = 8.8 Hz, H7), 7.52 (2H, d, 3J = 8.6 Hz, Hbenzyl), 7.76 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 11.0 (CH3), 41.6 (CH2-Im), 46.4 (NCH2), 56.5 (OCH3), 101.3 (C4), 108.1 (C3), 111.4 (Cindole), 111.5 (Cindole), 120.2 (Cimid), 121.2 (Cbenzyl), 128.4 (C9), 129.2 (Cimid), 129.4 (2C, CHbenzyl), 132.2 (C2), 132.6 (2C, CHbenzyl), 137.1 (C8), 137.9 (Cimid), 138.8 (C5), 155.0 (Cbenzyl). IR (KBr) cm−1 789 (ν C-Br); 1483, 1623 (ν C=C and ν C=N); 2927 (ν C-Halkane); 3091 (ν C-Harom). MS: 344.0 (MH+-67).
1-(4-Cyanobenzyl)-3-(1H-imidazol-1-ylmethyl)-5-methoxy-2-methyl-1H-indole (5s). Yield: 46%, yellow powder, mp = 124–125 °C; 1H NMR δ (250 MHz, DMSO) 2.42 (3H, s, CH3), 3.77 (3H, s, OCH3), 5.35 (2H, s, CH2-Im), 5.54 (2H, s, NCH2), 6.73 (1H, dd, 3J = 8.0 Hz, 4J = 0.2 Hz, H6), 6.88 (1H, s, Himid), 7.11–7.16 (4H, m, H4, Hbenzyl, Himid), 7.28 (1H, d, 3J = 8.0 Hz, H7), 7.80 (3H, d, Hbenzyl, Himid). 13C NMR δ (250 MHz, DMSO) 10.4 (CH3), 46.1 (CH2-Im), 55.4 (NCH2), 55.9 (OCH3), 100.8 (C4), 107.7 (C3), 110.4 (C7), 110.8 (C5), 111.0 (C6), 119.1 (CN), 119.6 (Cimid), 127.5 (2C, CHbenzyl), 127.8 (Cbenzyl), 128.6 (C9), 131.6 (Cimid), 133.1 (2C, CHbenzyl), 136.5 (C2), 137.3 (C8), 144.7 (Cimid), 154.5 (Cbenzyl). IR (KBr) cm−1 1486, 1503 (ν C=C and ν C=N), 2232 (ν C≡N), 2925 (ν C-Halkane); 3091 (ν C-Harom). MS: 289.4 (MH+-67).
3-(1H-Imidazol-1-ylmethyl)-5-methoxy-2-methyl-1-(4-trifluoromethylbenzyl)-1H-indole (5t). Yield: 63%, yellow powder, mp = 135–136 °C; 1H NMR δ (250 MHz, DMSO) 2.44 (3H, s, CH3), 3.78 (3H, s, OCH3), 5.35 (2H, s, CH2-Im), 5.54 (2H, s, N-CH2), 6.74 (1H, dd, 3J = 8.8 Hz, 4J = 2.4 Hz, H6), 6.87 (1H, s, Himid), 7.12 (1H, d, 4J = 2.4 Hz, H4), 7.15 (1H, s, Himid), 7.18 (2H, d, 3J = 8.0 Hz, Hbenzyl), 7.30 (1H, d, 3J = 8.8 Hz, H7), 7.70 (2H, d, 3J = 8.0 Hz, Hbenzyl), 7.77 (1H, s, Himid). 13C NMR δ (400 MHz, DMSO) 10.1 (CH3), 40.4 (CH2-Im), 45.7 (N-CH2), 55.6 (OCH3), 100.5 (C4), 107.4 (C3), 110.5 (Cindole), 110.7 (Cindole), 119.3 (Cimid), 125.7 (2C, q, 3J = 4.0 Hz, CHbenzyl), 126.5 (CF3), 126.9 (2C, CHbenzyl), 127.5 (C9), 127.9 (1C, q, 2J = 32.0 Hz, Cbenzyl), 128.4 (Cimid), 131.3 (C2), 136.2 (C8), 137.1 (Cimid), 143.4 (Cbenzyl), 154.1 (C5). IR (KBr) cm−1 1160 (ν CF3); 1485, 1623 (ν C=C and ν C=N); 2939 (ν C-Halkane); 3049 (ν C-Harom). MS: 333.2 (MH+-67).
3-(1H-Imidazol-1-ylmethyl)-5-methoxy-2-methyl-1-(4-methylbenzyl)-1H-indole (5u). Yield: 57%, white powder, mp = 116–117 °C; 1H NMR δ (250 MHz, DMSO) 2.12 (3H, s, ArCH3), 2.26 (3H, s, indoleCH3), 3.77 (3H, s, OCH3), 5.33 (2H, s, CH2-Im), 5.36 (2H, s, NCH2), 6.73 (1H, dd, 3J = 8.9 Hz, 4J = 2.8 Hz, H6), 6.86–6.91 (3H, m, Hbenzyl, Himid), 7.10–7.13 (4H, m, H4, Hbenzyl, Himid), 7.30 (1H, d, 3J = 8.9 Hz, H7), 7.74 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 10.5 (indoleCH3), 21.0 (ArCH3), 41.0 (CH2-Im), 46.2 (NCH2), 55.9 (OCH3), 100.6 (C4), 107.3 (C3), 110.8 (C7), 110.9 (C9), 119.6 (Cimid), 126.6 (2C, CHbenzyl), 127.7 (C4), 128.7 (Cimid), 129.6 (2C, CHbenzyl), 131.6 (Cbenzyl), 135.7 (C6), 136.5 (C2), 136.7 (C8), 137.3 (Cimid), 154.3 (C5). IR (KBr) cm−1 1218 (ν C-N); 1435, 1408, 1582 (ν C=C and ν C=N); 2916 (ν C-Halkane); 3107 (ν C-Harom). MS: 278.2 (MH+–67), 368.2 (MH++23).
1-(4-Ethylbenzyl)-3-(1H-imidazol-1-ylmethyl)-5-methoxy-2-methyl-1H-indole (5v). Yield: 47%, white powder, mp = 101–102 °C; 1H NMR δ (250 MHz, DMSO) 1.15 (3H, t, 3J = 7.6 Hz, CH2CH3), 2.45 (3H, s, indoleCH3), 2.61 (2H, q, 3J = 7.6 Hz, CH2CH3), 3.77 (3H, s, OCH3), 5.34 (2H, s, CH2Im), 5.37 (2H, s, NCH2), 6.73 (1H, dd, 3J = 8.9 Hz, 4J = 2.5 Hz, H6), 6.86–6.93 (3H, m, Hbenzyl, Himid), 7.10–7.16 (4H, m, H4, Hbenzyl, Himid), 7.30 (1H, d, 3J = 8.9 Hz, H7), 7.76 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 10.2 (indoleCH3), 15.7 (CH2CH3), 27.9 (CH2CH3), 40.9 (CH2-Im), 45.9 (NCH2), 55.6 (OCH3), 100.3 (C4), 107.0 (C3), 110.5 (C7), 110.6 (C9), 119.3 (Cimid), 126.3 (2C, CHbenzyl), 127.4 (Cimid), 128.1 (2C, CHbenzyl), 128.4 (Cbenzyl), 131.3 (Cbenzyl), 135.7 (C6), 136.2 (C2), 137.0 (C8), 142.7 (Cimid), 153,9 (C5). IR (KBr) cm−1 1218 (ν C-N); 1440, 1479, 1582 (ν C=C and ν C=N); 2959 (ν C-Halkane); 3111 (ν C-Harom). MS: 291.8 (MH+–67), 381.7 (MH++23).
3-(1H-Imidazol-1-ylmethyl)-5-methoxy-2-methyl-1-(4-propylbenzyl)-1H-indole (5w). Yield: 49%, white powder, mp = 93–94 °C; 1H NMR δ (250 MHz, DMSO) 1.23 (3H, t, 3J = 7.3 Hz, CH2CH2CH3), 1.86–1.94 (2H, m, CH2CH2CH3), 2.78 (3H, s, indoleCH3), 2.80 (2H, t, 3J = 7.0 Hz, CH2CH2CH3), 4.11 (3H, s, OCH3), 5.64 (2H, s, CH2-Im), 5.71 (2H, s, NCH2), 7.07 (1H, dd, 3J = 8.6 Hz, 4J = 2.4 Hz, H6), 7.21–7.27 (3H, m, Hbenzyl, Himid), 7.45–7.49 (4H, m, H4, Hbenzyl, Himid), 7.65 (1H, d, 3J = 8.5 Hz, H7), 8.10 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 10.5 (indoleCH3), 14.1 (CH2CH2CH3), 24.5 (CH2CH2CH3), 37.3 (CH2CH2CH3), 41.0 (CH2-Im), 46.2 (NCH2), 55.9 (OCH3), 100.6 (C4), 107.3 (C3), 110.8 (C7), 110.9 (C9), 119.6 (Cimid), 126.5 (2C, CHbenzyl), 127.7 (Cbenzyl), 128.7 (Cimid), 129.0 (2C, CHbenzyl), 131.7 (Cbenzyl), 136.0 (C6), 136.5 (C2), 137.3 (C8), 141.4 (Cimid), 154.3 (C5). IR (KBr) cm−1 1227 (ν C-N); 1420, 1484, 1586 (ν C=C and ν C=N); 2929 (ν C-Halkane); 3103 (ν C-Harom). MS: 306.2 (MH+–67).
3-(1H-Imidazol-1-ylmethyl)-1-(4-isopropylbenzyl)-5-methoxy-2-methyl-1H-indole (5x). Yield: 33%, white powder, mp = 96–97 °C; 1H NMR δ (250 MHz, DMSO) 1.18 (6H, d, 3J = 6.7 Hz, CH3 iPr), 2.45 (3H, s, indoleCH3), 2.84–2.88 (1H, m, 3J = 6.7 Hz, CH iPr), 3.77 (3H, s, OCH3), 5.34 (2H, s, CH2-Im), 5.36 (2H, s, NCH2), 6.73 (1H, dd, 3J = 8.9 Hz, 4J = 2.5 Hz, H6), 6.86–6.93 (3H, m, Hbenzyl, Himid), 7.10 (1H, d, 4J = 2.5 Hz, H4), 7.13–7.20 (3H, m, Hbenzyl, Himid), 7.31 (1H, d, 3J = 8.9 Hz, H7), 7.76 (1H, s, Himid). 13C NMR δ (250 MHz, DMSO) 10.2 (indoleCH3), 24.0 (2C, CH3 iPr), 33.2 (CH iPr), 41.6 (CH2-Im), 45.8 (NCH2), 55.6 (OCH3), 100.3 (C4), 106.9 (C3), 110.5 (C7), 110.6 (C9), 119.3 (Cimid), 126.3 (2C, CHbenzyl), 126.7 (2C, CHbenzyl), 127.4 (Cimid), 128.3 (Cbenzyl), 131.3 (Cbenzyl), 135.8 (C6), 136.2 (C2), 137.0 (C8), 147.4 (Cimid), 154.0 (C5). IR (KBr) cm−1 1224 (ν C-N); 1425, 1485, 1582 (ν C=C and ν C=N); 2955 (ν C-Halkane); 3088 (ν C-Harom). MS: 305.7 (MH+–67).
1-(4-Butylbenzyl)-3-(1H-imidazol-1-ylmethyl)-5-methoxy-2-methyl-1H-indole (5y). Yield: 42%, white powder, mp = 92–93 °C; 1H NMR δ (250 MHz, DMSO) 0.89 (3H, t, 3J = 7.3 Hz, CH2CH2CH2CH3), 1.24–1.37 (2H, m, CH2CH2CH2CH3), 1.46–1.58 (2H, m, CH2CH2CH2CH3), 2.44 (3H, s, indoleCH3), 2.63 (2H, t, 3J = 7.6 Hz, CH2CH2CH2CH3), 3.77 (3H, s, OCH3), 5.33 (2H, s, CH2-Im), 5.36 (2H, s, NCH2), 6.72 (1H, dd, 3J = 8.7 Hz, 4J = 2.5 Hz, H6), 6.86–6.92 (3H, m, Hbenzyl, Himid), 7.10–7.14 (4H, m, H4, Hbenzyl, Himid), 7.30 (1H, d, 3J = 8.7 Hz, H7), 7.75 (1H, s, Himid). 13C NMR δ (400 MHz, DMSO) 10.5 (indoleCH3), 14.2 (CH2CH2CH2CH3), 22.2 (CH2CH2CH2CH3), 33.6 (CH2CH2CH2CH3), 34.9 (CH2CH2CH2CH3), 40.9 (CH2-Im), 46.2 (NCH2), 55.9 (OCH3), 100.6 (C4), 107.3 (C3), 110.8 (C7), 110.9 (C9), 119.6 (Cimid), 126.6 (2C, CHbenzyl), 127.7 (Cimid), 128.7 (Cbenzyl), 128.9 (2C, CHbenzyl), 131.7 (Cbenzyl), 135.9 (C6), 136.5 (C2), 137.3 (C8), 141.6 (Cimid), 154.3 (C5). IR (KBr) cm−1 1224 (ν C-N); 1436, 1488, 1584 (ν C=C and ν C=N); 2922 (ν C-Halkane); 3095 (ν C-Harom). MS: 319.8 (MH+–67), 409.8 (MH++23).
Biological evaluation
In vitro determination of biological activity was done on promastigotes from the Leishmania mexicana (MHOM/MX/95/NAN1). They were maintained in Schneider’s insect medium (Sigma Chemical Co., St-Louis, MO, USA), plus 10% heat-inactivated fœtal calf serum (Sigma), penicillin and streptomycin at 26 °C, by passage every 7 days. Promastigotes were inoculated into 96-well plates (Nunc Inc., Napperville, IL, USA). Cultures were exposed for 96 h at 26 °C to test molecules 5a-y with a triplicate culture for each concentration (100, 10 and 1 μM). Antiproliferative effect was determined by a Uptiblue® micromethod based on the conversion of the fluorochrome by mitochondrial dehydrogenases. The fluorescence was measured at 590 nm with an excitation at 550 nm [14].
Results and discussion
Preparation and evaluation of 1-benzyl-3-(imidazol-1-ylmethyl)indoles 5a-l, 5r-t
A general synthetic route has already been published by our Group [Citation8] to prepare this class of compounds. The expected 1-benzyl-3-(imidazol-1-ylmethyl)indoles were obtained in four steps from commercially available indole scaffolds. The synthesis was performed taking into account our experience in this domain. Experimental conditions are described in . The first step of our synthetic route was a Vilsmeier-Haack formylation [Citation15] of the indole nucleus 1. Reaction of phosphorous oxychloride with DMF at low temperature led to the formation of an electrophile. To the reaction medium were slowly added indoles 1a-e bearing adequate substituents in positions 2 and 5. Derivatives 2a-e were prepared as powders in good yields. N-substitution in position 1 of the indole rings was performed using cesium carbonate as a base in acetonitrile under reflux. After two hours, various para-substituted benzyl halides were introduced and reacted with the intermediate for one additional hour. Compounds 3a-l and 3r-t were obtained in moderate to good yields. Reduction of the carbaldehyde moiety from 3a-l and 3r-t with the use of sodium borohydride in methanol afforded the primary alcohols 4a-l and 4r-t in satisfactory yields [Citation16]. Finally, N,N’-carbonyldiimidazole (CDI) was added to a solution of 4a-l and 4r-t in acetonitrile and after two hours of reaction, final compounds 5a-l and 5r-t were isolated in yields ranging from 11 to 78% [Citation17–20].
Figure 2. Synthetic route used for the preparation of 3-(imidazol-1-ylmethyl)indoles 5a-y.
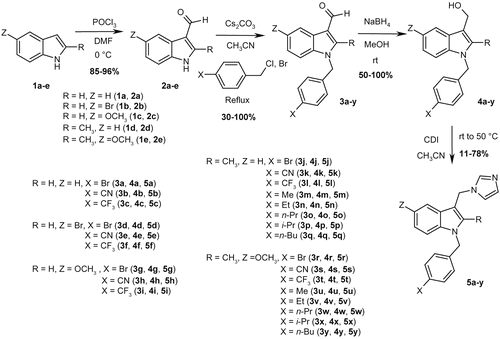
These original molecules were evaluated for their inhibitory activity on L. mexicana promastigotes. Experimental results are reported in and compared to their predictive values from the 3D-QSAR model.
Table 1. Biological results of compounds 5a-l, 5r-t and comparison with predicted values of activity (boxes with * represent molecules used in the previous 3D-QSAR model).
The biological results are relatively heterogeneous (2.3 < IC50 (μM) < 32). Compounds with a CN group in position X acting as a H-bond acceptor (5b, 5h, 5k, 5s) are much less active than expected by the model. Large gaps are observed between predicted and experimental IC50 values. With compound 5b (R = H, Z = H, X = 4-CN, IC50 = 13.1 μM) the difference is 4.4 between both values. Compounds 5h (R = H, Z = OCH3, X = 4-CN, IC50 = 32.0 μM), 5k (R = CH3, Z = H, X = 4-CN, IC50 = 21.8 μM) and 5s (R = CH3, Z = OCH3, X = 4-CN, IC50 = 12.7 μM) are 17- to 50-fold less active. This might be linked to the unique molecule used in the model with such a group or to the fact that the hydrophobic parameter must also be considered in this position. This last hypothesis remains the most probable because substitution from CN to CF3 led to more active compounds (5c, 5f, 5i, 5l and 5t) in the micromolar range, closer to the predicted values. In addition brominated compounds (5d, 5g, 5r) are as active as their CF3 analogues. Moreover, combining several structural parameters, a lipophilic group on para-position of the benzyl moiety (X = Br), a methyl group in R and an electro-donating group in position 5 of the indole ring (Z = OCH3), led to one of the most active compound 5r with an IC50 value of 2.3 μM.
Unfortunately, among the newly prepared molecules, none with a submicromolar IC50 value could be isolated. This might be linked to the fact that in our 3D-QSAR model, we only considered one molecule in this range of activity [Citation12]. Therefore, in order to confirm the importance of the lipophilic parameter in position X and to propose original substituents, we used the Craig Diagram [Citation21] which correlates the Hammett electronic substituent constant (σ) with the Hansch hydrophobicity constant (π) for substituents located on para-position of aromatic rings ().
Figure 3. Craig Diagram (σ: Hammett electronic substituent constant and π: Hansch hydrophobicity substituent constant).
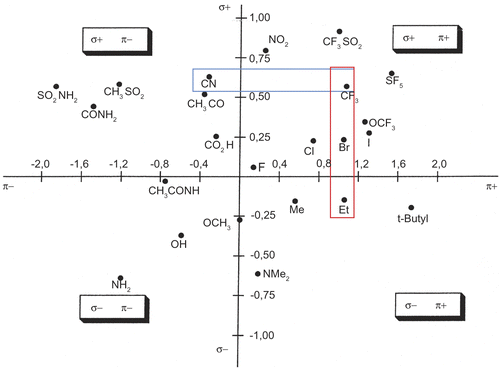
According to this diagram, it is possible to reverse the initial electronic character of the most active compound 5r (X = Br) while preserving a necessary positive π value. This suggested us to design a new series of compounds with various lipophilic and electro-donating alkyl substituents (from methyl to n-butyl in position X). These molecules bear the same scaffold as previously described for compound 5r (R = CH3 and Z = OCH3) and will be compared to their non-substituted analogues (R = CH3 and Z = H).
Preparation and evaluation of 1-(4-alkylbenzyl)-3-(imidazol-1-ylmethyl)indoles 5m-q, 5u-y
We decided to apply the successful route exposed in . However preparation of the different benzyl bromides 9a-e were firstly carried out. This could be performed in two or three steps from the corresponding benzaldehyde acetals or benzaldehydes using conventional methods. Acetals were hydrolysed to aldehydes 7c, 7e [Citation22–Citation23] in excellent yields, superior to 97%. Then those were reduced by sodium borohydride in methanol at room temperature affording the corresponding benzyl alcohols 8a-e in quantitative yields. Finally, reaction of the benzyl alcohols 8a-e with phosphorus tribromide in diethyl ether [Citation24] led to the benzyl bromides 9a-e (). In most cases, no purification was needed and benzyl bromides were isolated with yields up to 96%.
Figure 4. Preparation of benzyl bromides 9a-e from benzaldehyde acetals.
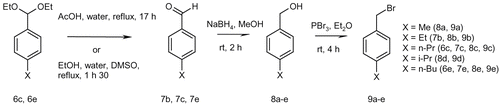
The same synthetic route was then applied to the preparation of the desired compounds bearing electro-donating alkyl side chains on para- position of the benzyl moiety as described on . N-benzylation occurred with 82–100% yield, reduction was quantitative and finally CDI coupling reaction provided us with the ten compounds 5m-q and 5u-y in 11 to 59% yield. Biological results of the newly synthesized molecules were measured using the same protocol as above. Once again, no molecule with a submicromolar IC50 value was observed (). Activities are homogeneous in a micromolar range (2.5 < IC50 (μM) < 5.4). Whatever Z substituents, variation of the alkyl side chain from C1 to C4 in position X conferred no beneficial effect. Steric hindrance might not have any positive or negative effect on the level of activity.
Table 2. Biological results of compounds 5m-q and 5u-y with alkyl side chains on para-position of the benzyl group.
Comparison with derivatives bearing positive σ value substituents such as Br or CF3 (compounds 5j, 5l or 5r, 5t, ) led to similar activities suggesting that electronic parameter does not seem to play a major role in this series. On the other hand the hydrophobic parameter in para-position of the benzyl moiety may be essential in the research of active compounds. Additional investigations should be undertaken around R and Z positions to optimize our series.
Conclusion
Twenty-two compounds were synthesized from a 3D-QSAR model, eighteen of them showed an IC50 value lower than 10 μM. With the help of the Craig diagram, the para- substituent of the benzyl moiety was optimized in terms of hydrophobicity. We confirmed that a lipophilic group was essential for a better in vitro biological activity. Starting from that point, we should synthesize other derivatives focusing our research in the discovery of the most appropriate groups in positions 2 and 5 of the indole nucleus. This might be done in close relation with the conclusions of our model. We also aim to evaluate the activity of the final compounds of this study on Leishmania-infected macrophages to understand their biological behavior such as membrane.
Acknowledgement
Declaration of interest: The authors report no conflicts of interest.
References
- For more details on leishmaniasis see the following website location: http://www.who.int/tdr/publications/publications/pr17.htm.
- Davies CR, Kaye P, Croft SL, Sundar S. Br Med J 2003; 326:377–382.
- Blum J, Desjeux P, Schwartz E, Beck B, Hatz C. J Antimicrob Chemother 2004; 53:158–166.
- Mishra J, Saxena A, Singh S. Curr Med Chem 2007; 14:1153–1169.
- Yardley V, Croft SL. Int J Antimicrob Agents 2000; 13:243–248.
- Mittal MK, Rai S, Ashutosh R, Gupta S, Sundar S, Goyal N. Am J Trop Med Hyg 2007; 76:681–688.
- Basselin M, Denise H, Coombs GH, Barrett MP. Antimicrob Agents Chemother 2002; 46:3731–3738.
- Marchand P, Le Borgne M, Na YM, Pagniez F, Abdala H, Le Baut G, Le Pape P. J Enz Inhib Med Chem 2002; 17:353–358.
- Na YM, Lebouvier N, Le M, Pagniez F, Alvarez N, Le Pape P, Le Baut G. J Enz Inhib Med Chem 2004; 19:451–457.
- Pagniez F, Abdala H, Marchand P, Le Borgne M, Le Baut G, Robert-Piessard S, Le Pape P. J Enz Inhib Med Chem 2006; 21:277–283.
- Veen M, Lang C. Biochem Soc Trans 2005; 33:1178–1181.
- Giraud F, Logé C, Le Borgne M, Pagniez F, Na YM, Le Pape P. SAR QSAR Environ Res 2006; 17:299–309.
- Le Borgne M, Na YM, Pagniez F, Le Baut G, Le Pape P, Abdala H. (2002) PCT Patent WO 02/24685.
- Le Pape P, Pagniez F, Abdala H. Acta Parasitol 2002; 47:79–81.
- Noland WE, Reich C. J Org Chem 1967; 32:828–832.
- Robinson B. Chem Rev 1969; 69:785–797.
- Jones CD, Winter MA, Hirsch KS, Stamm N, Taylor HM, Holden HE, Davanport JD, Krumkalns EV, Suhr RG. J Med Chem 1990; 33:416–429.
- Ogata M. Ann NY Acad Sci 1988; 544:12–31.
- Staab HA. Angew Chem Int Ed 1962; 1:351–367.
- Totleben MJ, Freeman JP, Szmuszkovicz J. J Org Chem 1997; 62:7319–7323.
- Patrick GL. Chimie pharmaceutique. 2nd ed. France: De Boeck Université; 2002, p. 274.
- Kawasaki M, Goto M, Kawabata S, Kometani T. Tetrahedron: Asymmetry 2001; 12:585–596.
- Kametani T, Kondoh H, Honda T, Ishizone H, Suzuki Y, Mori W. Chem Lett 1989; 18:901–904.
- Shanks D, Amorati R, Fumo MG, Pedulli GF, Valgimigli L, Engman L. J Org Chem 2006; 71:1033–1038.