Abstract
Galinsosides A (1) and B (2), new flavanone glucosides together with two known flavanones, 7,3′,4′-trihydroxyflavanone (3) and 3,5,7,3′,4′-pentahydroxyflavanone (4) have been isolated from an ethyl acetate- soluble fraction of Galinsoga parviflora. Their structures were assigned on the basis of spectral studies. Compound 1 showed significant antioxidant and urease inhibitory activity while compound 2 was moderately active. On the other hand, 2 showed inhibitory potential against α-glucosidase.
Introduction
The genus Galinsoga belongs to the family Asteraceae which consists of 3 species, occurring mainly in New Zealand, United Kingdom, Brazil, South Asia and America. In Pakistan it is very common in Baluchistan, Dir, Hunza, Swat, Gilgat, Muree and Kashmir [Citation1]. Galinsoga parviflora is one of the important species of the genus Galinsoga. The flowers of this plant are pink, 15-25 mm in diameter, with pink or red-tipped ray florets and yellow disk florets. The fruit is sparsely, hairy achences (seeds), 1-1.4 mm long, with a single row of long haired pappus. The leaves are small, narrow, elliptical upper leaves. Lower leaves are often three-lobed. Leaves explored pleasant-smelling when crushed. Stems are long, thin, ribbed, sparsely-hairy to smooth, rooting, sprawling, up to 70cm long. The plant can grow in semi-shade or moist soil. The plant is useful in treating nettle by rubbing the roots. The juice of the whole plant is applied to treat wounds. It helps to coagulate the blood of fresh cuts and wounds. Literature survey revealed that an aliphatic ketone [Citation2] has been reported from this species. A methanolic extract of this plant showed strong toxicity in brine shrimp lethality test [Citation3]. Further pharmacological screening revealed strong antioxidant and inhibitory activity against urease and α-glucosidase enzymes which was most pronounced in the ethylacetate fraction. This promoted us to carry out bioassay guided isolation studied of the bioactive compounds from this fraction. As a result, we now report the isolation and structural elucidation of galinsosides A (1) and B (2), the new flavanone glucosides, along with the known flavanones 7,3′,4′-trihydroxyflavanone (3) [Citation4] and 3,5,7,3′,4′-pentahydroxyflavanone (4) [Citation5], respectively, reported for the first time from this species.
The compound 1 showed significant antioxidant and urease inhibitory activity while the compound 2 was moderately active. On the other hand, the latter showed inhibitory potential against α-glucosidase.
Materials and methods
Plant material
The whole plant material of Ganlinsoga parviflora Cav. was collected from District Ziarat (Balochistan) in April, 2006 and identified by Dr. Suriya Khatoon, Plant Taxonomist, Department of Botany, University of Karachi, where a voucher specimen (GP-0062-06) has been deposited.
General experimental procedures
Optical rotations were measured on a JASCO DIP-360 polarimeter. IR spectra were recorded on a 460 Shimadzu spectrometer. EI-MS and HR-FAB-MS were recorded on JMS-HX-110 and JMS-DA 5000 mass spectrometers. The 1H-,13C-NMR, HMQC, and HMBC spectra were recorded on Bruker spectrometers operating at 400 MHz for 1H- and 100 MHz for 13C-NMR, respectively. The chemical shift values are reported in ppm (δ) units and the coupling constants (J) are in Hz. Aluminum sheets precoated with silica gel 60 F254 (20 × 20 cm, 0.2 mm thick; E-Merck) were used for TLC and silica gel (230-400 mesh) was used for column chromatography. Visualization of the TLC plates was carried out under UV at 254 and 366 nm and by spraying with ceric sulfate reagent solution (with heating). The GC was performed on a Shimadzu gas chromatograph (GC-9A) (3% OV-1 silanized chromosorb W, column temperature 180°C, injection port and detector temperature 275-300°C, flow rate 35 mL/min, flame-ionization detector).
Extraction and isolation
The shade dried whole plant material (15 kg) was extracted with MeOH (3 × 60 L) at room temperature. The combined methanolic extract was evaporated under reduced pressure to obtain a thick gummy mass (700 g). It was suspended in water and successively extracted with n-hexane, ethylacetate and n-butanol. The EtOAc soluble fraction (150 g) was subjected to column chromatography eluting with CHCl3, CHCl3-MeOH and MeOH in increasing order of polarity to obtain five fractions (A-E). Fraction B obtained from CHCl3-MeOH (9.5:0.5) was a mixture of two components, which were separated by column chromatography using solvent system CHCl3-MeOH (9.7:0.3) to afford compounds 3 (12 mg) and 4 (15 mg) from the top and the tail fractions, respectively. The fraction C obtained from CHCl3-MeOH (9:1) was further purified by column chromatography eluting with CHCl3-MeOH (9.3:0.7) to afford 1 (10 mg). The fraction D obtained from CHCl3-MeOH (8:2) was rechromatographed and eluted with CHCl3-MeOH (8.5:1.5) to afford 2 (18 mg).
Compound (1):
Light yellow solid; (10 mg). [α]D25 -97.2o (c 0.31, MeOH); CD curve [θ]333 + 6658 (max), [θ]298 -3128 (min) [θ]249 + 3609 (max), [θ]242 + 2123 (min), UV (MeOH) λmax (log ϵ) 329 (2.48), 286 (2.73) nm; IR (KBr) λmax: 3452,1670, 1588, and 1225 cm−1; EI-MS m/z (rel. int.) 286 (18), 150 (75), 136 (23), 120 (10); HR-FAB-MS m/z 449.1458 [M+H]+ (calcd for C22H25O10, 449.1451). 1H-NMR (400 MHz CD3OD) δ: 2.76 (2H, dd, J = 17.0,12.4 Hz, H-3), 3.52 (1H, t, J = 9.2 Hz, H-4′), 3.62 (2H, dd, J = 11.0,4.4 Hz, H-6′′), 3.82 (1H, t, J = 9.2, Hz, H-3′′), 3.92 (1H, s, OMe-7), 3.96 (1H, dd, J = 9.2,3.7 H-2′′), 5.01 (1H, d, J = 7.5, H-1′′), 5.18 (1H, dd, J = 12.4,3.0 Hz, H-2), 6.38 (1H, dd, J = 8.5,2.1 Hz, H-6), 6.49 (1H, d, J = 2.1 Hz, H-8), 6.59 (1H, dd, J = 8.2, 2.1 Hz, H-4′), 6.92 (1H, d, J = 2.1 Hz, H-6′), 6.97 (1H, d, J = 8.2 Hz, H-3′). 13C-NMR (100 MHz, CD3OD): 78.2 (C-2), 42.4 (C-3), 196.9 (C-4), 134.2 (C-5), 109.6 (C-6), 162.8 (C-7), 99.6 (C-8), 165.2 (C-9), 115.7 (C-10), 131.3 (C-1′), 148.2 (C-2′), 118.1 (C-3′), 116.4 (C-4′), 153.7 (C-5′), 116.2 (C-6′), 101.4 (C-1′′), 74.6 (C-2′′), 78.1 (C-3′′), 71.2 (C-4′′), 78.4 (C-5′′), 62.1 (C-6′′).
Compound (2):
Light yellow solid; (18 mg). [α]D25 -98.2o (c 0.31, MeOH); IR (KBr) νmax: 3451, 1670, 1585, and 1114 cm−1; EI-MS m/z (rel. int.) 302 (15), 166 (81), 152 (18), 136 (68), 120 (8); HR-FAB-MS m/z 465.1389 [M+H]+ (calcd for C22H25O11, 465.1397). 1H-NMR (400 MHz CD3OD) δ: 2.44 (2H, dd, J = 17.0,3.1 Hz, H-3), 3.54 (1H, t, J = 9.5 Hz, H-4′′), 3.65 (2H, dd, J = 11.2,4.5 Hz, H-6′′), 3.67 (1H, m, H-5′′), 3.75 (2H, dd, J = 11.2,5.1 Hz H-6′′), 3.85 (1H, t, J = 9.5 Hz, H-3′′), 3.92 (1H, s, OMe-7), 3.97(1H, dd, J = 9.5,3.7 Hz, H-2′′), 5.02 (1H, d, J = 7.5 Hz, H-1′′), 5.20 (1H, dd, J = 12.6,3.1 Hz, H-2), 6.41 (1H, dd, J = 8.1,2.0 Hz, H-6′), 6.56 (1H, d,J = 2.0 Hz, H-8), 6.60 (1H, d, J = 8.1 Hz, H-5′), 6.69 (1H, d, J = 2.0 Hz, H-2′), 6.72 (1H, d, J = 2.0 Hz, H-6). 13C NMR, (100 MHz, CD3OD): 78.2 (C-2), 41.3 (C-3), 196.5 (C-4), 160.4 (C-5), 98.5 (C-6), 164.9 (C-7), 98.9 (C-8), 165.7 (C-9), 110.9 (C-10), 125.8 (C-1′), 148.2 (C-2′), 146.0 (C-3′), 149.5 (C-4′), 115.7 (C-5′), 116.9 (C-6′), 56.7, (OMe), 101.6 (C-1′′), 74.8 (C-2′′), 78.3 (C-3′′), 71.1 (C-4′′), 78.3 (C-5′′), 62.4 (C-6′′).
Acid hydrolysis of compounds 1 and 2
A solution of compound (1 & 2) (each 8 mg) in MeOH (5 mL) containing 1N HCl (4 mL) was refluxed for 4 h, concentrated under reduced pressure, and diluted with H2O (8 mL). It was extracted with EtOAc. The aqueous phase in each case was concentrated to obtain the glycone while the organic phase provided the corresponding aglycones. In both the cases the glycone could be identified as D-glucose by the sign of its optical rotation ([α]D20+ 52°) from 1 and ([α]D20 + 51.3°) from 2, respectively. It was also confirmed based on the retention time of its TMS ether (α-anomer 4.1 min, β-anomer 7.8 min) with a standard. Removal of ethylacetate from 1 gave an inseparable mixture of products which could not be further worked up due to paucity of material. The aglycon from 2 crystallized from CHCl3 as yellow needles m.p. 222 °C. Its color reactions as well as physical and spectral data showed complete agreement to those reported in the literature for (2S) 3′,4′,5-trihydroxy-7-methoxyflavonone [Citation6].
Antioxidant assay
The free radical scavenging activity was measured by 1,1-diphenyl-2-picryl-hydrazil (DPPH) [Citation7]. The solution of DPPH of 0.3 mM was prepared in ethanol. Five microlitres of each sample of different concentration (62.5 μg - 500 μg) was mixed with 95 μL of DPPH solution in ethanol. The mixture was dispersed in 96 well plates and incubated at 37°C for 30 min. The absorbance at 515 nm was measured by micro titre plate reader (Spectramax plus 384 Molecular Device, USA) and percent radical scavenging activity was determined in comparison with the methanol treated control. BHA is used as standard
DPPH scavenging effect (%) = Ac – As/Ac × 100
Where, Ac = Absorbance of control (DMSO treated), As = Absorbance of sample.
Enzyme inhibition assays
Urease (Jack bean) solution (25 μL) was mixed with 5 μL of diluted compound (ranging from 25-500 μg) and the mixture was incubated at 30°C. Aliquots were taken after 15 min and immediately transferred to assay mixtures containing urea (100 mM) in buffer (40 μL) and again incubated for 30 min in 96 well plates. Urease activity was determined by measuring ammonia production using the indophenol method as described by Viquaruddine [Citation8]. The 50 μL each of phenol reagent (1% w/v phenol and 0.005% w/v sodium nitroprusside) and 70 μL of alkali reagent (0.5% w/v NaOH and 0.1 % active chloride NaOCl) were added to each well. The increasing absorbance was measured after 50 min at wavelength 630 nm using microtitre plate reader (Spectramax plus 384 Molecular Device, USA). All reactions were performed in triplicates in a final volume of 200 μL. All the assays were performed at pH 8.2 (0.01 M K2HPO4. 3H2O, 1mM EDTA and 0.01 M LiCl2). Thiourea was used as standard and percentage inhibitions were calculated from formula, 100–(OD test well/OD control) × 100 [Citation8].
α-Glucosidase inhibition assay is based on the breakdown of substrate to produce a colored product, followed by measuring the absorbance over a period of time [Citation9]. α-Glucosidase (Sigma, type III, from yeast) was dissolved in buffer A (0.1 mol/L potassium phosphate, 3.2 mmol/L-MgCl2, pH 6.8) (0.1 units/mL) and p-nitrophenyl- α-D-glucopyranoside dissolved in buffer A at 6 mmol/L was used as substrate. 102 μL Buffer B (0.5mol/L potassium phosphate, 16 mmol/L-MgCl2, pH 6.8), 120 μL sample solution (0.6 mg/mL in dimethylsulfoxide), 282 μL water and 200 μL substrate were mixed. This mixture was incubated in water-bath at 37˚C for 5 min and then 200 μL enzyme solution was added and mixed. The enzyme reaction was carried out at 37˚C for 30 min and then 1.2 ml 0.4 mol/L glycine buffer (pH = 10.4) was added to terminate the reaction. Enzymatic activity was quantified by measuring the absorbance at 410 nm. 1-Deoxynojiromycin hydrochloride was used as positive control [Citation9].
Percentage inhibition= Ac – As/Ac × 100
Where Ac = Absorbance of control and As = Absorbance of sample
Compound 1 showed significant antioxidant and urease inhibitory activity while compound 2 was moderately active. On the other hand the latter showed inhibitory potential against α-glucosidase enzyme.
Results and discussion
The ethylacetate soluble fraction of the methanolic extract of the whole plant of Galinsoga parviflora was subjected to a series of column chromatographic techniques to obtain compounds 1-4 and their structures established by UV, IR, MS and NMR spectroscopy.
Galinsoside A (1) () was isolated as light yellow amorphous solid. It gave violet colourations with FeCl3 The molecular formula C22H24O10 was established by HR-FAB-MS showing [M+H]+ peak at m/z 449.1458 (calcd for C22H25O10, 449.1451) having eleven degree of unsaturation. The IR spectrum revealed the presence of hydroxyl groups (3452 cm−1), a methoxyl group (1225 cm−1) and a conjugated carbonyl (1670 cm−1) in the molecule. It showed UV absorption maxima at 285 and 329 nm, characteristic of a flavanone-O-glycoside [Citation10,Citation11]. The UV spectrum did not show any bathochromic shift on addition of NaOAc and AlCl3/HCl revealing the absence of free hydroxyl groups at C-7, C-5 and C-3, respectively. The EIMS gave a distinct peak at m/z 286 and further two fragment ions were observed at m/z 150 and 136 due to retro-Diels-Alder fragmentation, confirming the presence of one methoxyl group in ring A and two hydroxyl groups in the ring B of the flavanone skeleton. The presence of flavanone structure was further indicated by an AMX system with resonances at δ 5.18 (1H, dd, J = 12.4, 3.0 Hz, H-2), 2.76 (1H, dd, J = 17.0, 12.4 Hz, H-3ax) and 2.42 (1H, dd, J = 17.0, 3.0 Hz, H-3eq) [Citation12-Citation14]. In the aromatic region of the 1H-NMR spectrum of 1 further signals which appeared at δ 6.97 (1H, d, J = 8.2 Hz, H-3′), 6.92 (1H, d, J = 2.1 Hz, H-6′) and 6.59 (1H, dd, J = 8.2, 2.1 Hz, H-4′) were assigned to the protons of the trisubstituted ring B and the signals appearing at δ 7.51 (1H, d, J = 8.5 Hz, H-5), 6.49 (1H, d, J = 2.1 Hz, H-8) and 6.38 (1H, dd, J = 8.5, 2.1 Hz, H-6) were assigned to the protons of the monosubstituted ring A. The methoxyl protons appeared at δ 3.92 (3H, s, OMe-7). The signals for the sugar moiety appeared at δ 5.01 (1H, d, J = 7.5 Hz, H-1′′), 3.96 (1H, dd, J = 9.2, 3.7 Hz, H-2′′), 3.82 (1H, t, J = 9.2 Hz, H-3′′, 3.52 (1H, t, J = 9.2 Hz, H-4′′), 3.65 (1H, m, H-5′′) and methylene protons at δ 3.62 (1H, dd, J = 11.0, 4.4 Hz) and 3.74 (1H, dd, J = 11.0, 5.1 Hz). The 13C-NMR (BB, DEPT) spectra of 1 corroborated the presence of one methyl, two methylene, twelve methine and seven quaternary carbons. The signals at δ 78.2, 42.4 and 196.9 were typical of C-2, C-3 and C-4 carbons of the flavanone skeleton [Citation13,Citation14]. The attachment of the sugar moiety was deduced at C-2′ due to the upfield shift of C-2’ and downfield shifts of C-1’ and C-3’ compared to literature values [Citation15,Citation16]. The sugar was identified as D-glucose by comparing its 13C-NMR signals with the reference data [Citation17,Citation18]. Acid hydrolysis of 1 provided glycone which could be separated and identified as D-glucose through sign of its optical rotation and retention time of its TMS ether in gas chromatography (GC). The larger coupling constant of the anomeric proton allowed us to assign the β configuration to the glucose moiety. The position of the methoxyl group was ascertained through HMBC as its protons at δ 3.92 showed 3J correlation with C-7 (δ 162.8). It was further authenticated by NOE experiment in which NOEs’ were observed between the methoxyl protons at δ 3.90 with neighboring proton at C-6 (δ 6.38) and C-8 (δ 6.49) respectively. The position of the O-β-D-glucopyranoside moiety was assigned to C-2′ as the anomeric proton at δ 5.01 showed 3J correlation with C-2′ (δ 148.2). The absolute configuration at C-2 was assigned ‘S’ on the basis of CD spectrum which showed similar Cotton effects as reported in literature for related flavonoids [Citation19-Citation21]. Thus the structure of galinsoside A (1) was assigned as (2S)-5′-hydroxy-7-methoxyflavanone 2’-O-β-D-glucopyranoside.
Figure 1. Structures of compounds 1 & 2.
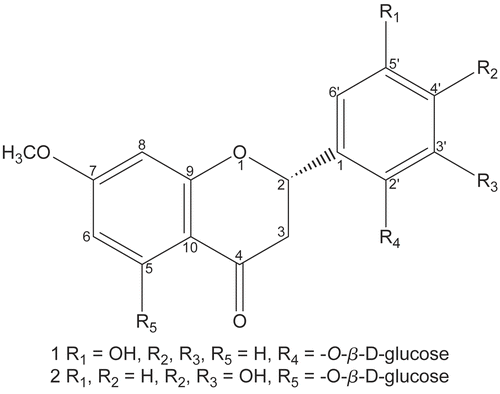
Galinsosides B (2) () was isolated as light yellow amorphous solid which gave positive FeCl3 and Quastel tests, the latter indicating the presence of an ortho dihydroxy moiety. The molecular formula C22H24O11 was established by HR-FAB-MS showing [M+H]+ peak at m/z 465.1389 (calcd for C22H25O11, 465.1397) having eleven degree of unsaturation. The EI-MS spectrum showed two characteristic fragments at m/z 166 and 136 due to the retro-Diels-Alder fragmentation, confirming hydroxyl and methoxyl groups in ring A and two hydroxyl groups in ring B of the flavanone skeleton. The IR spectrum of 2 was similar to 1 but some differences were observed in 1H- and 13C-NMR spectra. In the aromatic region of the 1H-NMR spectrum, the proton signals of the trisubtitued phenyl ring appeared at δ 6.69 (1H, d, J = 2.0 Hz, H-2′), 6.60 (1H, d, J = 8.1 Hz, H-5′) and 6.41 (1H, dd, J = 8.1, 2.0 Hz, H-6′) and two meta coupled aromatic protons at δ 6.72 (2H, d, J = 2.0 Hz, H-6) and 6.56 (2H, d, J = 2.0 Hz, H-8) were assigned to the protons of disubstituted ring A. The methoxyl protons appeared at δ 3.92 (3H, s, OMe-7). The sugar moiety could be identified through acid hydrolysis, which again provided D-glucose. The 13C-NMR (BB, DEPT) spectra of 2 corroborated the presence of one methyl, two methylene, eleven methine and eight quaternary carbons. The signals at δ 78.4, 41.3 and 196.5 were typical of C-2, C-3 and C-4 carbons of the flavanone skeleton. The larger coupling constant of the anomeric proton revealed the β configuration of glucose moiety. The position of the methoxyl group was ascertained through HMBC as its protons at δ 3.92 showed 3J correlation with C-7 (δ 162.8). It was further authenticated by NOE experiment in which NOEs’ were observed between the methoxyl protons at δ 3.90 with neighboring proton at C-6 (δ 6.38) and C-8 (δ 6.49) respectively. The position of the O-β-D-glucopyranoside moiety was assigned to C-5′ as the anomeric proton at δ 6.60 showed 3J correlation with C-5 (δ 160.4). Final evidence to the structure was provided by the aglycone obtained during acid hydrolysis which could be identified as (2S) 3′,4′,5-trihydroxy-7-methoxyflavanone. On the basis of these evidences, the structure of 2 was assigned as (2S)-3′,4′-dihydroxy-7-methoxyflavanone 5-O-β-D-glucopyranoside.
Pharmacological screening of both 1 and 2 revealed interesting results. Compound 1 which has an O-β-D-gloucopyrnnoside moiety at C-2′ showed significant antioxidant and inhibitory activity against the enzyme urease while the compound 2 showed weak to moderate activity. Thus the sugar unit at C-2′ appears to act as a pharmacophoric group. Contrary to that, the latter showed significant activity against the enzyme α-glucosidase ().
Table 1. IC50 (μM) values of compounds 1 and 2 in the antioxidant assay, and also against urease and α-glucosidase.
Acknowledgements
Authors are thankful to the Higher Education Commission of Pakistan providing financial support for spectroscopic analysis of compounds at International Centre for Chemical Sciences, HEJ Research Institute of Chemistry, University of Karachi.
Declaration of interest: The authors report no conflicts of interest.
References
- Ali SI, Qaiser M. “Flora of Pakistan”, Chenopodiaceae Jointly published by Department of Botany, University of Karachi, Karachi, Pakistan & Missouri Botanical Press, Missouri Botanical Garden, St. Louis, Missouri, U.S.A, 2001;204:185.
- Bernard A, Eokes L. Electron impact induced fragmentation of cholesterol and related C-5 unsaturated steroids. J Org Chem 1977;42:725.
- Meyer BN, Ferrigni NR, Putnam JE, Jacobsen LB, Nichols DE, McLaughlin JL. Brine shrimp a convenient general bioassay for active plant constituents. Planta Med 1982;45:31.
- Kitanaka S, Takido M. Demethyltorosaflavones C and D from Cassia nomame. Phytochemistry 1992;31:2927.
- Kiehlmann E, Edmones PM. Li Isomerization of dihyroquercentin. J Nat Prod 1995; 58: 450.
- Tian JK, Sun F,Ching Yi. Chemical constituents from the roots of Ranunculus ternatus. J Asian Nat Prod Res 2006;8:35.
- Scalbert A, Johnson IT, Saltmarsh M. Polyphenols antioxidant and beyond. Am J Clin Nutr 2005;81:215.
- Ahmad VU, Hussain J, Hussain H, Reza AJ, Lodhi MA, Chaudary MI. Frist Natural Urease Inhibitor from Euphorbia decipiens. Chem.Pharm.Bull.2003;51: 719
- Xiancui Li, Rongil Niu, Xiao Fan, Han Lijun, Zhang Lixin. Macroalage as a source of alpha glucosidase inhibitors. Chinese J. Oceanology and Limnology 2005;23(3):354.
- Mabry TJ, Markham KR. “The Flavonoids”. Chapman and Hall, London, 1975; P. 79.
- Mabry TJ, Markham KR, Thomas MB. “Systematic Identification of Flavonids”, Springer New York 1970; P. 271.
- Rao EV, Venkataratnam G, Vilain C. Flavonoids from Tephrosia fulvinervis. Phytochemistry 1985;24:2427.
- Wu TS, Hsu MY, Damu AG, Kuo PC, Su CR, Li CY, Sun HD. Constituents of leaves of Phellodendron chinese Var Glabriusculum. Heterocycles 2003;60:397.
- Ahmad I, Anis I, Malik A, Nawaz SA,Choudhary MI. Cholinesterase inhibitory constituents from Onosma hispida. Chem Pharm Bull 2003;51:412.
- Yoshikawa M, Shimada H, Nishida N, Li Y, Toguchida I, Yamahara J, Matsuda H. Antidiabetic principles of natural medicines 11. aldose reductase and α-glucosidase inhibitors from Brazilian natural medicine, the leaves of Myrcia multiflora Dc.(Myrtaceae) structures of myrciacitrins 1 and 11 myrciacitrins 1 and 11. Chem Pharm Bull 1998;46:113.
- Matsuda H, Nishida N, Yoshikawa M. Antidiabetic principles of natural medicines V aldose reductase inhibitors from Myrcia multiflora Dc. (2) structures of myrciacitrins 111, IV and V. Chem Pharm Bull 2002;50(3):429.
- Perffer PE, Valentine KM, Parrish FW. Deuterium-induced differential isotope shift 13C resonance reassignments of mono- and disaccharides. J Am Chem Soc 1979;101:1265.
- Gorin PAJ, Mazurek M. Further studies on the assignment of signals in 13C magnetic resonance spectra of aldoses and derived methyl glycosides. Can J Chem 1975;53:1221
- Baruah NC, Gthyagarajan RP, Herz W, Govindan V. New flavonoids from Inaula cappa. Phytochemistry 1979;18: 2003
- Iinuma M, Ohyama M, Tanaka T, Mizuno M, Hong SK. Three 29 49 69-trioxygenated flavanones in roots of Echinosophora koreensis. Phytochemistry 1992;31:665.
- Gaffield W. Circular dichrosim optical rotatory diospersion and absolute configuration of flavonones, 3-hydroxyflavanones of aglycone chirality in flavanone glycosides. Tetrahedron 1970;26:4093.