Abstract
The synthesis and the biological evaluation of pyrano[3,2-e]indoles and their reaction intermediates are described. The compounds prepared were evaluated for their inhibition of NO production, antioxidant activity and also for their ability to inhibit in vitro the growth of four human tumor cell lines: large lung carcinoma (COR-L23), alveolar basal epithelial carcinoma (A549), amelanotic melanoma (C32) and melanoma (A375). The two reaction intermediates, 5a and 5b, showed the highest inhibition of NO production in murine monocytic macrophage (IC50 = 1.1 μM and IC50 = 2.3 μM respectively). Compound 5a was the most active against melanotic melanoma (IC50 = 11.8 μM) while the other compounds exhibited weak cytotoxicity with IC50 values >50 μM on all cell lines.
Introduction
Pyranoindoles constitute an important class of heterocyclic systems due to their remarkable biological activities. They are reported as μ-opiate receptor ligands [Citation1], restricted phenolic analogs of the neurotransmitter serotonin [Citation2]. They are inhibitors of hepatitis C polymerase [Citation3], COX-2 [Citation4], PLA2 and lipoxygenase [Citation5]. They were reported also to possess anti- inflammatory [Citation6] and antimicrobial activities [Citation7]. Nevertheless, literature survey revealed that the pyrano[3,2-e]indole system has received little attention. The few reports on the derivatives of the latter system indicated that they act as CNS agents [Citation8], inhibitors of poly-ADP- ribosylation reaction [Citation9] and also restricted phenolic analogs of the neurotransmitter serotonin [Citation2,Citation10].
Several anti-inflammatory and neuroprotective drugs have been reported to have an antioxidant and/or radical scavenging mechanism as part of their activity [Citation11–13]. The mechanism of inflammation injury is attributed, in part, to the release of reactive oxygen species (ROS) from activated neutrophiles and macrophages. The over production of these species lead to tissue injuries by damaging macromolecules and lipid peroxidation of the membranes [Citation14–15]. In addition, ROS propagate inflammation by stimulating release of mediators such as NO and cytokines such as interleukin-1, tumor necrosis factor-α, and interferon-γ, which stimulate recruitment of additional neutrophiles and macrophages. Thus free radicals are important mediators that provoke inflammatory processes and, consequently, their neutralization by antioxidants and radical scavengers can attenuate inflammation [Citation16–17].
Most clinically important medicines belong to steroidal or non-steroidal anti-inflammatory chemical therapeutics for treatment of inflammation-related diseases [Citation18]. Although these drugs have a potent activity, yet a long-term administration is required for treatment of chronic diseases. Furthermore, these drugs have various severe side effects. Therefore, new naturally occurring agents with weak side-effects are desirable. Many coumarine- derivatives and analogs are able to scavenge reactive oxygen species (ROS)-free radicals, such as hydroxyl- and superoxide radicals, and also hypochlorous acid. Thus they can influence processes involving free radical-injury [Citation19–20]. They have also been found to inhibit lipid peroxidation [Citation21] and to possess vasorelaxant, anticoagulant, anti-inflammatory [Citation22], anti-tumoral and anti-allergic [Citation23] activities. Thus, on the basis of these evidences we wish to report herein a easy synthesis of pyrano[3,2-e]indoles 6a-b and their precursors 4a-b and 5a-b using 5-hydroxyindole as a starting material.
Material & methods
Chemistry
Commercial reagents were purchased from Aldrich, Acros Organics and Alfa Aesar and used without additional purification. Melting points were determined on a Kofler melting point apparatus. IR spectra were taken with a Perkin Elmer BX FT-IR. Mass spectra were taken on a JEOL JMS GCMate spectrometer at ionising potential of 70 eV (EI) or were performed using a spectrometer LC-MS Waters alliance 2695 (ESI+). 1H-NMR (400 MHz) was recorded on a JEOL Lambda 400 Spectrometer. Chemical shifts are expressed in parts per million downfield from tetramethylsilane as an internal standard. Thin layer chromatography (TLC) was performed on silica gel 60F-264 (Merck).
The von Pechmann reaction is a common method used to produce coumarins and derivatives [24]. Classically, the process consists of the condensation of phenols with β-ketoesters in the presence of an acidic catalyst (aluminium chloride, phosphorus pentoxide, trifluoroacetic acid [Citation25]). Recently Indium(III) chloride [Citation26] has been used as an efficient catalyst giving coumarins in good yields.
Accordingly, we attempted the von Pechmann reaction on 5-hydroxyindole or 5-hydroxy-1-methylindole using Indium(III) chloride as a catalyst for the preparation of compounds 6a-b (). Thus, when 5-hydroxy-1- methylindole 3b was allowed to react with an equimolar amount of ethyl acetoacetate under von Pechmann conditions using Indium(III) chloride as a catalyst, the reaction product was found to be the derivative 4a and not the expected pyranoindole 6b (). When the reaction was repeated using an excess of ethyl acetoacetate, also no pyranoindole was formed and the product was identified as the indole derivative 4b.
Figure 1. Structures of 6a and 6b.
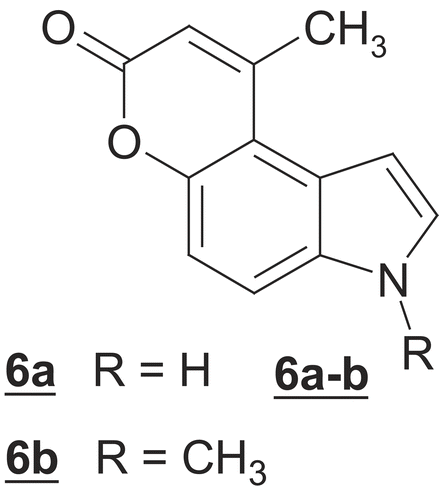
Scheme 1. Reagents: (i) ethyl acetoacetate (1 mol for 4a, excess for 4b); Indium(III)chloride; reflux 2h;(ii) but-2-ynoic acid; DCC; DMAP; CH2Cl2 /DMF (10:1); r.t. 2h; (iii) PtCl4; 1,4-dioxane/1,2-dichloroethane (1:1); reflux 4h.
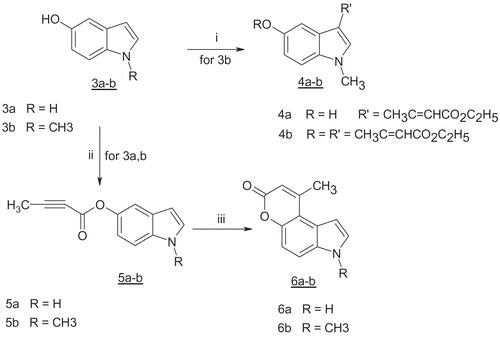
These failures prompted us to try an another synthetic strategy previously described in the literature [Citation27] by esterification of the 5-hydroxyindoles 3a-b with but-2-ynoic acid to give the esters 5a-b. When these esters were heated under reflux in dioxane-dichloroethane in the presence of PtCl4 as a catalyst, an intramolecular cyclization occurred giving 9-methyl-3H-pyrano[3,2-e]indol-7-one (6a), (40%) and 3,9-dimethyl-3H-pyrano[3,2-e]indol-7-one (6b) (45%) ().
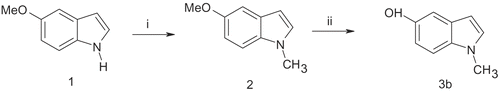
On the other hand, 5-hydroxy-1-methylindole 3b could be prepared from 5-methoxyindole 1 by treatment with NaH to obtain the N-anion followed by reaction with methyl iodide to give 5-methoxy-1-methylindole 2. Selective demethylation of the methoxyl group of compound 2 by heating under reflux in anhydrous pyridine hydrochloride gave the 5-hydroxy-1-methylindole 3b ().
Scheme 2. Reagents: (i) NaH, CH3I, DMF, 25°C; (ii) pyiridine hydrochloride; 3h reflux.
Procedure for the preparation of 5-methoxy-1-methylindole 2
To a stirred cold solution (0˚C) of 5-methoxyindole (1) (2.0 g, 13.60 mmol) in dry DMF (60 mL), was added NaH 60% oil dispersion (0.54 g, 20.40 mmol). After 15 min stirring at this temperature, iodomethane (2.54 mL, 40.80 mmol) was added and the mixture was further stirred at room temperature for 1h. Water (200 mL) was then added to the reaction mixture and the solid product obtained was filtered, washed with water and dried. It is obtained as pure white solid, yield 92%, mp 114°C. IR (KBr): 2922, 1621, 1496, 1242, 1151, 1025, 802, 725 cm−1. 1H-NMR (d6-DMSO) δ 7.30 (d, 1H, Ar, J = 8.8 Hz); 7.23 (d, 1H, Ar, J = 2.9 Hz);); 7.04-7.01 (m, 1H, Ar); 6.76 (dd, 1H, Ar, J = 1.9, 8.8 Hz); 6.30 (d, 1H, Ar, J = 2.9 Hz); 3.72 (s, 6H, NCH3 and OCH3). MS (ESI+): 162 (M+ +1).
Procedure for the preparation of 5-hydroxy-1-methylindole 3b
A mixture of 2 and anhydrous pyridine hydrochloride (1:16) was heated to reflux for 3h. The reaction mixture was left to cool to room temperature, then ice water was added. The product was extracted with Et2O. The organic layers were washed with a solution of HCl (2N), dried (MgSO4) and concentrated to yield 3b as yellow solid, yield 60%, mp = 156°C, IR (KBr): 3177, 2924, 1621, 1489, 1234, 1145, 949, 795, 719 cm−1. 1H-NMR (d6-DMSO) δ 8.67 (s, 1H, OH); 7.19 (s, 1H, Ar); 7.17-7.15 (m, 1H, Ar); 6.81 (d, 1H, Ar, J = 3.0 Hz); 6.62 (dd, 1H, Ar, J = 1.9, 8.8 Hz); 6.18 (d, 1H, Ar, J = 2.9 Hz); 3.04 (s, 3H, NCH3). MS (ESI+): 148 (M+ +1).
General procedure for the preparation of Ethyl 3-(indol-3-yl)but-2-enoates 4a-b
To a mixture of 5-hydroxy-1-methyl-1H-indole (3b) and ethyl acetoacetate (equimolar ratio for 4a, and excess for 4b), indium(III) chloride (10 mol%) was added under nitrogen. The reaction mixture was heated under reflux for 2h, and then it was left to cool to room temperature. Ice water was added and then the reaction mixture was extracted by ethyl acetate. The organic layers were collected and washed with brine, dried over MgSO4 and evaporated under reduced pressure. The solid residue was washed with Et2O, to give the pure compounds 4a-b.
Ethyl 3-(5-hydroxy-1-methyl-1H-indol-3-yl)but-2-enoate 4a: pink solid, yield 58 %, mp = 200°C. IR (KBr): 3400, 2931, 1705, 1488, 1373, 1329, 1201, 1083, 1016, 851, 788 cm−1. 1H-NMR (d6-DMSO) δ 8.32 (s, 1H, Ar); 7.08-7.06 (m, 2H, Ar, OH); 6.52-6.49 (m, 2H, Ar); 6.45 (s, 1H, C=CH); 3.78-3.74 (q, 2H, CH2); 3.67 (s, 3H, NCH3); 1.83 (s, 3H, C-CH3); 0.82-0.78 (t, 3H, CH3). MS (EI) m/z: 259 (M+ , 1).
Ethyl 3-[5-(2-ethoxycarbonyl-1-methylvinyloxy)-1-methyl-1H-indol-3-yl]but-2-enoate 4b: pink solid, yield 65%, mp = 180°C; IR (KBr): 3412, 2984, 1705, 1622, 1473, 1373, 1168, 1088, 1027, 805 cm−1. 1H-NMR (d6-DMSO) δ 8.94 (s, 1H, Ar); 7.32 (d, 1H, Ar, J7,6 = 8.8 Hz); 6.87 (s, 1H, Ar); 7.32 (d, 1H, Ar, J6,7 = 8.8 Hz); 6.04-6.01 (m, 2H, C=CH); 4.11-4.09 (q, 2H, CH2); 3.90-3.88 (q, 2H, CH2); 3.76 (s, 3H, NCH3); 1.42 (s, 6H, C-CH3); 1.24-1.20 (t, 3H, CH3); 0.95-0.92 (t, 3H, CH3). MS (EI) m/z: 371 (M+ , 14).
General procedure for the preparation of indol-5-yl-but-2-ynoates 5a-b
To a solution of 5-hydroxyindole (3a) or 5-hydroxy-1-methyl-1H-indole (3b) in a mixture of CH2Cl2 and DMF (10:1), but-2-ynoic acid (1.1 eq), DCC (1 eq) and DMAP (cat) were added. The reaction mixture was stirred at room temperature for 2h, and then concentrated under reduced pressure. The solid residue was suspended in ethyl acetate, filtered and the filtrate was washed with brine. The collected organic layers were dried over anhydrous MgSO4 and evaporated under reduced pressure. The solid residue was purified by silica gel chromatography using ethyl acetate:petroleum ether, 2:8 as an eluent to give the pure compounds 5a or 5b respectively.
Indol-5-yl but-2-ynoate 5a: white solid, yield 62%, mp = 108°C. IR (KBr): 3410, 2237, 1705, 1480, 1261, 1121, 884 cm−1. 1H-NMR (CDCl3) δ 8.25 (bs, 1H, NH); 7.40 (s, 1H, Ar); 7.20-7.23 (m, 1H, Ar); 7.03-7.01 (m, 1H, Ar); 6.95 (dd, 1H, Ar, J = 1.9, 8.8 Hz); 6.60-6.50 (m, 1H, Ar); 2.10 (s, 3H, C-CH3). GC-MS m/z: 199 [M+].
1-Methyl-1H-indol-5-yl but-2-ynoate 5b: white solid, yield 64%, mp = 90°C; IR (KBr): 3402, 2233, 1711, 1625, 1487, 1246, 1217, 1123, 1033, 882 cm−1. 1H-NMR (CDCl3) δ 7.28 (s, 1H, Ar); 7.23-7.19 (t, 1H, Ar); 7.01 (d, 1H, Ar, J = 3.00 Hz); 6.98 (dd, 1H, Ar, J = 2.0, 8.8 Hz); 6.46 (d, 1H, Ar, J = 3.0 Hz); 3.70 (s, 3H, NCH3); 2.06 (s, 3H, C-CH3). MS (ESI+): 214 (M+ +1). GC-MS m/z: 213 [M+].
General procedure for the preparation of pyrano[3,2-e]indoles 6a-b
To a solution of compound 5a or 5b (1 eq) in 1,4-dioxane and 1,2-dichloroethane (1:1) PtCl4 (5 mol%) was added. The reaction mixture was heated under reflux for 4h then concentrated under reduced pressure. The residue was dissolved in ethyl acetate, and washed with brine. The organic layer was then dried (MgSO4) and concentrated under reduced pressure to give a solid residue. The solid product was purified by silica gel chromatography using ethyl acetate:petroleum ether, (1:1) as an eluent to give the pure compounds 6a-b.
9-Methyl-3H-pyrano[3,2-e]indol-7-one 6a: white solid, yield 45%, mp = 170°C. IR (KBr): 3400, 2925, 1700, 1660, 1610, 1580, 1410, 1248, 1050, 641 cm−1. 1H-NMR (d6-DMSO) δ 8.40 (bs, 1H, NH); 7.82 (d, 1H, Ar, J = 8.8 Hz); 7.70 (d, 1H, Ar, J = 3.0 Hz); 7.35 (d, 1H, Ar, J = 8.8 Hz); 6.94 (d, 1H, Ar, J = 3.0 Hz); 6.50 (s, 1H, Ar); 2.72 (s, 3H, CH3). GC-MS m/z: 199 [M+].
3,9-Dimethyl-3H-pyrano[3,2-e]indol-7-one 6b: white solid, yield 40%, mp = 154°C. IR (KBr): 3329, 2927, 1698, 1678, 1627, 1580, 1413, 1244, 1050, 641 cm−1. 1H-NMR (d6-DMSO) δ 7.77 (d, 1H, Ar, J = 8.8 Hz); 7.58 (d, 1H, Ar, J = 2.9 Hz); 7.20 (d, 1H, Ar, J = 8.8 Hz); 6.86 (d, 1H, Ar, J = 2.9 Hz); 6.39 (s, 1H, Ar); 3.87 (s, 3H, NCH3); 2.68 (s, 3H, CH3). MS (ESI+): 214 (M+ +1).
1,1-Diphenyl-2-picrylhydrazyl (DPPH) assay
This experimental procedure was adapted from Wang et al. [Citation28] modified as reported in Conforti et al. [Citation29]. The absorbance was measured using a Perkin Elmer Lambda 40 UV/VIS spectrophotometer at 517 nm against blank, which was without DPPH. All tests were run in triplicate and averaged. Ascorbic acid was used as a positive control. Decreasing of DPPH solution absorbance indicates an increase of DPPH radical scavenging activity. This activity is given as % DPPH radical-scavenging that is calculated in the equation:
Cell line and cell culture
The murine monocytic macrophage cell line RAW 264.7 (ECACC No.: 91062702) was grown in plastic culture flask in Dulbecco’s Modified Eagle’s Medium (DMEM) with L-glutamine supplemented with 10% foetal bovine serum (FBS) and 1% antibiotic/antimitotic solution (penicillin/streptomycin) under 5% CO2 at 37°C. After 4-5 days cells were removed from culture flask by scraping and centrifugated for 10 min under 1500 rpm. The medium was then removed and the cells were resuspended with fresh DMEM. Four cancer cell lines, large lung cell carcinoma COR-L23 (ECACC No.: 92031919), amelanotic melanoma C32 (ATCC No.: CRL-1585), alveolar basal epithelial carcinoma A549 (EACC No.: 86012804) and melanoma were used in this experiment A375 (EACC No.: 88113005). The COR-L23, C32 cells were cultured in RPMI 1640 medium, while A549 and A375 cells were cultured in D-MEM medium. Both medium were supplemented with 10% foetal bovine serum, 1% L-glutamine, 1% penicillin/streptomycin.
Cells counts and viability were performed using a standard trypan blue cell counting technique. The cell concentration was adjusted to 1 × 106 cells/mL for RAW 264.7 and 2 × 104 for COR-L23, C32, A549 and A375 in the same medium. 100 μL of the above concentration were cultured in 96-well plate for one days to become nearly confluent. Concentrations ranging from 5–200 μg/mL of the samples were prepared from the stock solutions by serial dilution in DMEM to give a volume of 100 μL in each well of a microtiter plate (96-well). Then cells were cultured with vehicle, compounds for 48 h for human tumor cells and for 24 h in the presence of 1 μg/mL LPS for RAW 264.7.
Inhibition of nitric oxide (NO) production in LPS-stimulated RAW 264.7 cells
The presence of nitrite, a stable oxidized product of NO, was determined in cell culture media by Griess reagent (1% sulfanamide and 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride in 2.5% H3PO4 ) [Citation30]. 100 μL of cell culture supernatant was removed and combined with 100 μL of Griess reagent in a 96-well plate followed by spectrophotometric measurement at 550 nm using a microplate reader (GDV DV 990 B/V, Roma, Italy). Nitrite concentration in the supernatants was determined by comparison with a sodium nitrite standard curve.
Assay for cytotoxic activity
Cytotoxicity was determined using the 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma) assay reported by Tubaro et al. [Citation31] with some modification. All the cell lines used were capable of attachment to form a homogeneous monolayer on plastic substratum of the culture wells, which is ideal for the 3-(4,5- dimethylthiazol- 2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
The assay for each concentration of samples was performed in triplicates and the culture plates were kept at 37°C with 5% (v/v) CO2 for one days. After incubation, 100 μL of medium was removed from each well. Subsequently, 100 μL of 0.5% w/v MTT (Sigma, Italy), dissolved in phosphate buffered saline, was added to each well and allowed to incubate for a further 4 h. After 4 h of incubation, 100 μL of DMSO was added to each well to dissolve the formazan crystals. Absorbance values at 550 nm were measured with a microplate reader (GDV DV 990 B/V, Roma, Italy). Cytotoxicity was expressed as IC50 which is the concentration to reduce the absorbance of treated cells by 50% with reference to the control (untreated cells).
Statistical analysis
Data were expressed as means ± SD. Statistical analysis was performed by using Student’s t test. Differences were considered significant at P ≤ 0.05. The inhibitory concentration 50% (IC50) was calculated from the Prism dose–response curve (Prism Graphpad, Prism version 4.0 for Windows, GraphPad Software, San Diego, CA, USA) obtained by plotting the percentage of inhibition versus the concentrations.
Results and discussion
All compounds prepared, 4a-b, 5a-b and 6a-b, were screened for their antioxidant activity, inhibition of NO production and also for their in vitro growth inhibitory activity of human tumor cell lines (C32: amelanotic melanoma; A375: melanoma; COR-L23: large lung carcinoma; A549: alveolar basal epithelial carcinoma). To evaluate the inhibition of NO production activity of the compounds prepared, the murine monocytic macrophage cell line RAW 264.7 was employed as a model for inhibition of inflammation mediators such as NO. Indomethacin was used as a reference drug. Once activated by inflammatory stimulation, macrophages produce a large number of cytotoxic molecules. It is well characterized that treatment of RAW 264.7 macrophages with LPS (1 μg/mL) for 24 h induces NO production (26.4 μM from the basal level of 0.6 μM) [Citation32], as assessed by measuring the accumulation of nitrite, a stable metabolite of NO, in the media based on Griess reaction. The beneficial effect of coumarin derivatives on the inhibition of production of the inflammatory mediators in macrophages can be mediated through oxidative degradation of products of phagocytes, such as O2- and HOCl. As shown in , incubation of RAW 264.7 cells with prepared compounds induced a significant inhibitory effect on the LPS-induced nitrite production. At 50 μM compound 5a is as efficient as 5b with 99 (±2.2) and 97 (±2.1) % inhibition of LPS-induced NO production in RAW 264.7 cells () whereas the compound 5a is more potent than 5b with an IC50 value of 1.1 and 2.3 μM (). These results are important if compared to the reference drug indomethacin which showed an IC50 of 147.5 μM. 4b and 6a showed activity similar to reference drug indomethacin with IC50 values of 38.9 and 43.7 μM ( and ). 4a showed the lower activity with an IC50 value of 83.5 μM while 6b didn’t show any activity. All compounds did not show any cytotoxicity on RAW 264.7 cells at tested concentrations (data not shown).
Figure 2. Percentage inhibition of NO production in LPS-induced RAW 264.7 macrophages for compounds 5a and 5b.
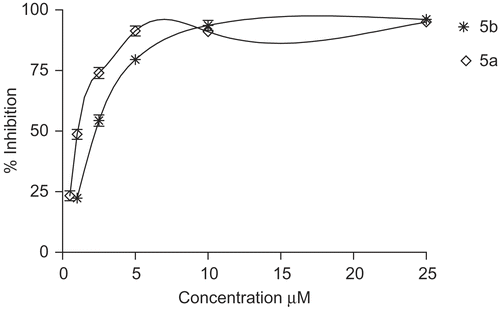
Figure 3. Percentage inhibition of NO production in LPS induced RAW 264.7 macrophages for compounds 6a, 4a and 4b.
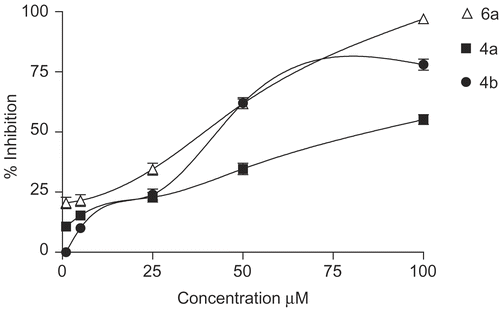
Table I. IC50 values of inhibition of NO production in LPS-induced RAW 264.7 macrophages for compounds.
Many non-steroidal anti-inflammatory drugs have been reported to act either as inhibitors of free radical production or as radical scavengers [Citation33]. Several compounds with antioxidant properties offer protection in rheumatoid arthritis and inflammation and find uses for the treatment of these pathologies [Citation34]. Thus, we tested the new derivatives with regard to their antioxidant ability and in comparison with well-known antioxidant agents, for example, ascorbic acid. The interaction of the examined compounds with the stable free radical DPPH (1,1-diphenyl-2-picrylhydrazyl) [Citation35] was studied at 1000 to 50 μM after 30 min (). Compounds 4a-b and 5a interact with DPPH in a concentration dependent manner (), whereas compounds 6a-b and 5b do not show any interaction under tested concentrations. Slight differences are observed between the compounds 4b and 5a with the concentration whereas compound 4a presents reducing ability at 100 μM.
Figure 4. Percentage inhibition on DPPH radical of compounds 4a, 4b and 5a.
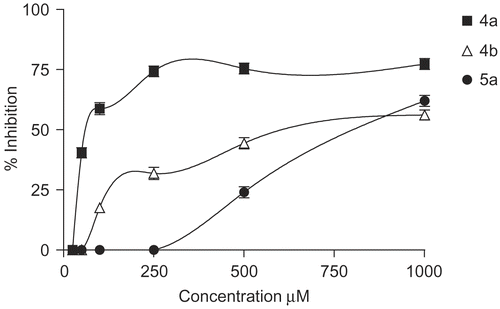
Table II. Radical scavenging activity of analyzed compounds.
Furthermore the pyranoindoles and their synthetic precursors were evaluated for their in vitro cytotoxic properties on four human cancer cell lines. For each cell lines, there was a linear relationship between cell number and absorbance, measured at 550 nm in both control and drug-treated wells. After 48 hours of treatment, the cytotoxicity of the compounds under test was determined. The cytotoxic effects of indoles derivatives on the growth of human tumor cell lines are given in . The 4a, 6b and 6a compounds exhibited weak cytotoxicity with IC50 values >50 μM on all cell lines. The most antiproliferative compound against C32 cell line was 5a followed by 5b with IC50 values of 11.8 and 21.8 μM, respectively. 4b showed the same activity on COR-L23 and A375 cell lines with IC50 values of 39.3 and 40.4 μM, respectively ().
Table III. Cytotoxic activities against human tumor cell lines of compounds.
Conclusion
In conclusion the antiradical activity of the tested compounds can explain, at least in part, the inhibition of NO production and antiproliferative activities. Compound 5a is the most active in vitro. It was found to possess inhibitory activity against the production of NO and against the growth of human amelanotic melanoma cells (C32). Furthers investigations are in progress.
Declaration of interest: The authors report no conflicts of interest.
References
- Hinze C, Sundermann B, Schick H, Henkel B. Preparation of spirocyclic pyrano[4,3-b] indoles and related compounds as μ-opiate receptor ligands WO 2005063769 A1 20050714, CA 143:133272 AN 2005:612304.
- Macor JE, Ryan K, Newman ME. The synthesis of pyrano[3,2-e]indoles and pryano[2,3-f] indoles as rotationally restricted phenolic analogs of the neurotransmitter serotonin. Tetrahedron 1992;48(6):1039–1052.
- Gopalsamy A, Collette MS, Ellingboe JW, Laporte MG, Mansour TS, Burns CJ, Park K, Condon SM. Preparation of pyranoindole derivatives to treat infection with hepatitis C virus. WO 2003099275 A1 20031204 CA 140:16714 AN 2003:950835.
- Kreft AF, Caufield CE, Failli AA, Caggiano TJ, Greenfield AA, Kubrak DM. Preparation of pyranoindole inhibitors of COX-2. US 5776967 A 19980707 CA 129:122569 AN 1998:457268.
- Musser JH, Kreft AF, Failli AA, Demerson CA, Shah US, Nelson JA. Substi-tuted indole-, indene-, pyranoindole- and tetrahydrocarbazolealkanoic acid derivatives as inhibitors of PLA2 and lipoxygenase. US 5420289 A1 9950530 CA 124:8801 AN 1995:661173.
- Nakkady SS, Fathy MM, Hishmat OH, Mahmond SS, Ebeid MY., New indole, aminoindole and pyranoindole derivatives with anti-inflammatory activity, Boll Chim Farmaceu 2000;39(2):59–66.
- Gadaginamath GS, Kavali RR. Synthesis and antimicrobial activity of novel 4H-pyrano[2,3-f]indole derivatives, Ind J Chem Section B 1999;38B(2):178–182.
- Fu JM. Preparation of substituted 2,3,7,8,9,10,11,12-octahydro azepino[4,5-b] pyrano[3,2-e]indoles as CNS agents. WO 2002004456 A1 20020117, CA 136:118395 AN 2002:51468.
- Zaalishvili T, Kharadze D, Khostaria T, Arabuli N, Kolkhidashvili K, Kutalia K, Omiadze T, Kirmelashvili L, Edilashvili L, Zaalishvili G, Tsetskhladze Z. Design of indole derivatives and studying their inhibitory ability of poly-ADP-ribosylation reaction. Bull Georg Acad Sci 2005;172(2): 314–316.
- Macor JE, Blank DH, Post RJ. The synthesis of conformationally/rotationally restricted analogs of the neurotransmitter serotonin. Tetrahedron Lett 1994;35(1):45–48.
- Perry EK, Pickering AT, Wang WW, Houghton PJ, Perry NS. Medicinal plants and Alzheimer’s disease: from ethnobotany to phytotherapy. J Pharm Pharmacol 1999;51:527–534.
- Lin CC, Huang PC. Antioxidant and hepatoprotective effects of Acathopanax senticosus. Phytother Res 2002;14:489–494.
- Repetto MG, Llesuy SF. Antioxidant properties of natural compounds used in popular medicine for gastric ulcers. Braz J Med Biol Res 2002;35:523–534.
- Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 1995;41:1819–1828.
- Winrow VR, Winyard PG, Morris CJ, Blake DR. Free radicals in inflammation: second messengers and mediators of tissue destruction. Br Med Bull 1993;49:506–522.
- Delaporte RH, Sánchez GM, Cuellar AC, Giuliani A, Palazzo de Mello JC. Anti-inflammatory activity and lipid peroxidation inhibition of iridoid lamiide isolated from Bouchea fluminensis (Vell.) Mold. (Verbenaceae). J Ethnopharmacol 2002;82:127–130.
- Geronikaki AA, Gavalas AM. Antioxidants and anti-inflammatory diseases: synthetic and natural antioxidants with anti-inflammatory activity. Comb Chem High Throughput Screen 2006;9:425–442.
- Jackson Roberts L, Morrow JD In Hardman JG, Limbird LE, Goodman Gilman A, Editors Goodman and Gilman’s. The Pharmacological Basis of Therapeutics 10th ed. McGraw-Hill; USA. 2001. pp 687–731.
- Larson RA. The antioxidants of higher plants. Phytochemistry 1988;27:969–978.
- Mora A, Paya M, Rios JL, Alcaraz M. Structure-activity relationships of polymethoxyflavones and other flavonoids as inhibitors of non-enzymic lipid peroxidation. J Biochem Pharmacol 1990;40:793–797.
- Hoult JRS, Paya M. Pharmacological and biochemical actions of simple coumarins: natural products with therapeutic potential. Gen Pharmac 1996;27:713–722.
- Terasaka T, Kinoshita T, Kuno M, Seki N, Tanaka K, Nakanishi I. Structure-Based Design, Synthesis, and Structure-Activity Relationship Studies of Novel Non-nucleoside Adenosine Deaminase Inhibitors. J Med Chem 2004;47:3730–3743.
- Menciu C, Duflos M, Fouchard F, Le Baut G, Emig P, Achterrath U, Szelenyl I, Nickel B, Schimdt J, Kutscher B, Gunther E. New N-(Pyridin-4-yl)-(indol-3-yl)acetamides and Propanamides as Antiallergic Agents. J Med Chem 1999;42:638–648.
- Potdar MK, Mohile SS, Salunkhe MM. Coumarin syntheses via Pechmann condensation in Lewis acidic chloroaluminate ionic liquid. Tetrahedron Lett 2001;42:9285–9287.
- Hoefnagel AJ, Gunnewegh EA, Downing RS, van Bekkum H. Synthesis of 7- Hydroxycoumarins catalysed by Solid Acid Catalysts. J Chem Soc Chem Commun 1995; 225-226.
- Subhas Bose D, Rudradas AP, Hari Babu M. The indium(III) chloride-catalyzed von Pechmann reaction: a simple and effective procedure for the synthesis of 4-substituted coumarins. Tetrahedron Lett 2002; 43:9195–9197.
- Chen G, Yee D, Gubernator N, Sames D. Development of fluorogenic substrates for monoamine oxidases (MAO-A and MAO-B). WO 2006026368.
- Wang M, Li J, Rangarajan M, Shao Y, La Voie EJ, Huang CT. Antioxidative Phenolic Compounds from Sage (Salvia officinalis). J Agric Food Chem 1998;46:4869–4873.
- Conforti F, Statti GA, Menichini F. Chemical and biological variability of hot pepper fruits (Capsicum annuum var. acuminatum L.) in relation to maturity stage. Food Chem 2007;102:1096–1104.
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126:131–138.
- Tubaro A, Florio C, Luxich E, Vertua R, Della Loggia R, Yasumoto T. Suitability of the MTT-based cytotoxicity assay to detect okadaic acid contamination of mussels. Toxicon 1996;34: 965–974.
- Kim YH, Kim J, Park H, Kim HP. Anti-inflammatory activity of the synthetic chalcone derivatives: inhibition of inducible nitric oxide synthase-catalyzed nitric oxide production from lipopolysaccharide-treated RAW 264.7 cells. Biol Pharm Bull 2007;30:1450–1455.
- Ahmad S, Israf DA, Lajis NH, Shaari K, Mohamed H, Wahab AA, Ariffin KT, Hoo WY, Aziz NA, Kadir AA, Sulaiman MR, Somchit MN. Cardamonin, inhibits pro-inflammatory mediators in activated RAW 264.7 cells and whole blood. Eur J Pharmacol 2006;538:188–194.
- Costa D, Gomes A, Reis S, Lima JL, Fernandes E. Hydrogen peroxide scavenging activity by non-steroidal anti-inflammatory drugs. Life Sci 2005;76:2841–2848.
- Pekoe G, Van Dyke K, Peden D, Mengoli H, English D. Antioxidation theory of non-steroidal anti-inflammatory drugs based upon the inhibition of luminol-enhanced chemiluminescence from the myeloperoxidase reaction. Agents Actions 1982;12:371–376.