
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Compounds containg catechol or bisphosphonate were tested as inhibitors of the zinc metalloproteases, thermolysin (TLN), pseudolysin (PLN) and aureolysin (ALN) which are bacterial virulence factors, and the human matrix metalloproteases MMP-9 and −14. Inhibition of virulence is a putative strategy in the development of antibacterial drugs, but the inhibitors should not interfere with human enzymes. Docking indicated that the inhibitors bound MMP-9 and MMP-14 with the phenyl, biphenyl, chlorophenyl, nitrophenyl or methoxyphenyl ringsystem in the S1′-subpocket, while these ringsystems entered the S2′- or S1 -subpockets or a region involving amino acids in the S1′- and S2′-subpockets of the bacterial enzymes. An arginine conserved among the bacterial enzymes seemed to hinder entrance deeply into the S1′-subpocket. Only the bisphosphonate containing compound RC2 bound stronger to PLN and TLN than to MMP-9 and MMP-14. Docking indicated that the reason was that the conserved arginine (R203 in TLN and R198 in PLN) interacts with phosphate groups of RC2.
Graphical Abstract
Introduction
It is estimated that there are more than 60000 different proteasesCitation1,Citation2. In vertebrates they are involved in regulation of physiologic processes such as cell growth, angiogenesis, blood pressure, coagulation, cell signalling, reproduction, wound repair, hemostasis and homeostasisCitation2–7. Proteases are either secreted from cells or localised inside cells. They are divided into classes and clans depending on the active site residues taking part in the catalytic reaction (Merops database)Citation8–10. The major classes found in all organisms are aspartate-, threonine-, cysteine-, serine- and metallo-proteases, but in addition the classes glutamate-, aspargine- and mixed-proteases have been detected in microorganisms (Merops database)Citation10.
Dysregulation of one or several proteases in humans is often associated with diseaseCitation3,Citation4,Citation11–14 and several proteases are potential targets for therapeutic interventionsCitation15–18. In humans there are around 570 different proteases and approximately 190 of these are metalloproteasesCitation19. Of around 280 cell-secreted human proteases, approximately 120 are metalloproteasesCitation2,Citation19,Citation20. Matrixins or matrix metalloproteases (MMPs) is a family of secreted and membrane associated calcium dependent metalloproteases which contains a catalytic and a structural zinc ionCitation11. MMPs belong to the M10 family of proteases. In humans there are 23 different MMPs, and MMP-9 and -14 are two of the membersCitation11. One or several members of the MMP family are overexpressed and functionally involved in pathological conditions such as chronic venous disease, fibrotic disorders, inflammation, liver diseases, lung diseases, neurological diseases, osteoarthritis, viral infection, cardiovascular diseases and in various cancer formsCitation21. Several investigators both in academia and industry have developed MMP inhibitors interacting with the active site. However, in clinical tests the vast majority of MMP inhibitors have failedCitation22 The most likely reason is that the MMPs are of major importance in many physiological processes such as cell apoptosis, embryogenesis, immune response, morphogenesis, tissue remodelling, tooth enamel formation, reproduction, menstruation, wound healing, angiogenesis and axonal growthCitation13,Citation21,Citation23. MMPs are tightly regulated and expressed in all human tissues and organsCitation13,Citation22,Citation23, and therefore an uncontrolled activity regulation of one or several MMPs by an inhibitor should be avoided. In microorganisms, proteases are involved in processes such as generation of nutrition, growth, survival and invasion into host organismsCitation24–30. Bacterial infectious diseases claim millions of casualties each year, and the spreading of antibiotic multi-resistance among central human pathogenic bacteria is recognised as a major global health concern and a pressing societal challenge. Development of new antibiotics with novel modes of action and innovative strategies to efficiently fight bacterial infections are urgently needed. Inhibition of bacterial virulence rather than directly targeting bacterial growth and viability has gained increasing interests in anti-infective drug discoveryCitation31,Citation32. Such compounds may impose less evolutional pressure for resistance development than classical antibiotics, and have limited impact on the host commensal flora. Several proteases are bacterial virulent factors and therapeutically interesting as putative antibacterial drug targetsCitation26–29,Citation33. However, compounds targeting bacterial virulence have so far not been approved as drugsCitation34,Citation35.
MMP-9 (gelatinase B) is secreted, while MMP-14 belongs to the membrane type metalloproteases (MT-MMPs), and is also called membrane type 1 metalloprotease (MT1-MMP). The MT-MMPs contain either a transmembrane domain or a GPI-membrane anchor, with the catalytic site located outside the cell in the extracellular environmentCitation11. The MMPs are constituted of different structural domains, and both MMP-9 and MMP-14 contain an N-terminal prodomain, followed by a catalytic domain, a hinge region and a C-terminal hemopexin like (HPX) domain. In MMP-14, the HPX domain is followed by the transmembrane domain, while MMP-9 contains three fibronectin-II like (FnII) repeats in the catalytic domainCitation11. The MMPs belong to the clan metzincins and the catalytic zinc ion is bound to the protein through three histidines of the segment (HEXXHXXGXXH/D + M)Citation8,Citation9. The fourth zinc ligand in the inactive proform is the cysteine in the PRCGV motif of the pro-domainCitation36,Citation37. The fourth zinc ligand in the activated MMPs is a water molecule, that also binds to the side chain of the glutamate that follows the first histidine in the zinc binding segmentCitation8,Citation9. MMP-9 can be activated in the extracellular environment by naturally occurring proteases such as trypsin, kallikrein, MMP-2 and MMP-3, but also by mercurial and organomercurial compounds such as HgCl2 and APMA (p-aminophenylmercuric acetate) and bacterial metalloproteases such as thermolysin (TLN) and pseudolysin (PLN)Citation37. MMP-14, like the other MT-MMPs, are activated inside cells by the serine protease furinCitation11. MMP-14 is the most studied enzyme among membrane-linked MMPs, while MMP-9 (gelatinase B) is the most studied among secreted MMPsCitation14,Citation38. Binding of inhibitors to the active site of MMP-14 and MMP-9 have been extensively studied both by kinetic and X-ray crystallographyCitation39–47. The active sites of the MMPs are similar but not identical. Their S1'-subpocket determines the substrate cleavage site, and they all prefer hydrophobic amino acids in this pocketCitation48,Citation49.
TLN from Bacillus thermoproteolyticus is the model enzyme of the M4 family of proteases, which is also termed the thermolysin familyCitation50. These enzymes have a zinc ion in the catalytic site, which has tetrahedral coordination. Two histidines of a HEXXH motif and a glutamic acid located 18–72 residues C-terminal to the HEXXH motif are the three ligands that anchor the zinc ion to the enzyme, while the fourth ligand is a water molecule as in the MMPs, which also binds the side chain of the glutamate following the first histidine in the zinc binding segmentCitation8,Citation9,Citation50. Inhibitors containing a metal binding group replace the catalytic water molecule on the zinc ion when they bind the catalytic siteCitation51. TLN, PLN from Pseudomonas aeruginosa (LasB or elastase of P. aeruginosa) and aureolysin (ALN) from Staphylococcus aureus belong to the subclan MA(E) of the M4 family, also known as the “Glu-zincins”Citation8,Citation9,Citation50. These three proteases have several similarities despite a modest sequence identity (28% between TLN and PLN)Citation52,Citation53. The three dimensional (3D) structures of PLN and TLN have been extensively studied, also in complex with inhibitors, and reveal large similarities in the overall structure. The main structural differences are that PLN consists of a slightly more open substrate binding cleft than TLN, and that PLN has one structural calcium while TLN has threeCitation53–55. For ALN only the 3 D-structure of the free enzyme is knownCitation56. Although PLN is not as well characterised as TLN, it appears that the slight difference in substrate specificity between the two enzymes is mainly due to the size of the S1′-subpocket and a more open substrate binding cleft in PLN than in TLN. PLN has a broader substrate specificity than most other M4 family members including TLN, although all these enzymes prefer a hydrophobic amino acid at the P1’ position. Furthermore, for substrate degradation four subsites of PLN require to be occupiedCitation50,Citation53,Citation55.
PLN, TLN and ALN are secreted bacterial virulence factors, and inhibitors may be new antibacterial drugs, either alone or used as adjuvant to traditional antibacterial treatment. PLN, TLN and ALN have structural resemblance with human MMPs. In order to have a therapeutic value, compounds targeting these virulence factors should not interfere strongly with the function of human MMPs, due to the importance of MMPs in physiological processes. Identifying structural determinants for strong binding to the bacterial M4 proteases and human MMPs would therefore be of pivotal importance for the development of new antibacterial drugs. In this study, we have studied several catechol containing and bisphosphonate containing compounds for their inhibition of TLN, PLN, ALN, MMP-9 and MMP-14 in order to identify new M4 inhibitors and investigate structural determinants that might be important for selective binding using inhibition kinetic and molecular modelling.
Results and discussion
Km values for the fluorescence quenched substrates
At conditions used in the present work (1% DMSO in all assays), the Km values of the substrate McaPLGL(Dpa)AR-NH2 with APMA-activated recombinant MMP-9 (rMMP-9(A)), trypsin-activated MMP-9 from THP-1 cells (MMP-9(T)) and MMP-14 were 4 ± 1, 6 ± 2 and 4.9 ± 0.4 µM, while the Km values of the substrate McaRPPGFSAFK(Dnp)-OH with ALN, PLN and TLN were 76 ± 7, 24 ± 8 and 6 ± 1 µM, respectively. The estimated Km values for ALN and PLN must be regarded as uncertain since the highest substrate concentration used was 10 µM due to quenching. The obtained Km values are very similar to those previously obtained for theses enzymes without DMSOCitation57 or with a DMSO concentration of 5%Citation58. Previously it was reported that TLN is inactivated in most organic cosolvents, but can tolerate up to 10% DMSO to enhance substrate solubilityCitation59. For TLN, the Km value was also determined with a DMSO concentration of 2%. This resulted in a Km value of 7 ± 1 μM which is not significantly different from the value obtained in the presence of 1% DMSO.
Quenching experiments with catechol containing compounds and bisphosphonates
All compounds were first tested for possible quenching of the formed fluorescence product. The experiments were performed with varying concentrations of the putative inhibitors (0 − 100 μM) against varying fluorescence product (McaPL-OH) concentration as previously described for PAC-1 and Isatin derivativesCitation58, and as described in the experimental section. These experiments revealed that in contrast to the PAC-1 and Isatin derivativesCitation58, neither the catechol derivatives nor the bisphosphonates used in the present work quenched the fluorescence product. However, some of the compounds showed fluorescence at the emission and excitation wavelength used, but that did not affect the inhibitory assays as enzymatic reactions were followed continuously.
Inhibitory effects of catechol containing compounds
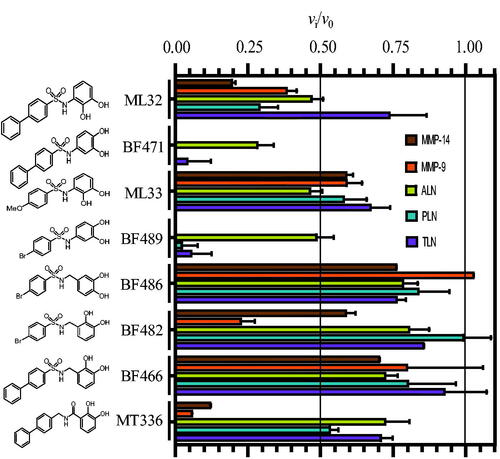
The inhibitorial power of 100 μM of the eight catechol containing compounds was tested against the two human metalloproteases MMP-9(T) and MMP-14 and the three bacterial metalloproteases ALN, PLN and TLN (. In order to detect putative slow and slow-tight binding, the catechol derivatives were first incubated along with the enzyme for 15 min at 37 °C. In controls without inhibitor present, enzymes and buffer were preincubated under identical conditions. Thereafter, the enzyme reaction was started by adding the relevant chromogenic substrate and the rate was followed continuously for 30 min. Except for BF486, which did not affect MMP-9, and BF482 which did not affect PLN, all cathecol containg compounds showed inhibition of the five proteases (. Both BF471 and BF489 showed more than 50% inhibition of all five proteases, while ML32 reduced the activity more than 50% for four of the proteases (not for TLN). The activity was reduced with more than 50%, by MT336 for the two MMPs, by BF482 for MMP-9(T) and by ML33 for ALN (. In all other inhibitory studies with catechol containing compounds, the activity was reduced between 0 and 45%.
Figure 1. Inhibitory effect of 100 μM of the catechol containing compounds on the activity of the human metalloproteases, MMP-9 and MMP-14, and the bacterial metalloproteases TLN, PLN and ALN. The inhibition experiments were performed by using a fixed concentration of 4 μM of both the MMP-9 and MMP-14 substrate McaPLGL(Dpa)AR-NH2 and the ALN, PLN and TLN substrate McaRPPGFSAFK(Dnp)-OH. The vi/v0 (mean ± sd) were based on 4–6 experiments.
X-ray crystallography studies showed that BF471 binds to the active site of MMP-8Citation60, and hence we could expect that all tested catechol derivatives should bind the active site. To assure that this is correct, the inhibitors BF471 and BF489 were tested against varying McaRPPGFSAFK(Dnp)-OH using TLN as described in the Experimental section. The results showed that the two catechol derivatives competed with the substrate (data not shown) and the Ki values obtained were 57 ± 6 μM for BF471 and 73 ± 11 μM for BF489. It should be noted that the reaction was started by adding the enzyme to the substrate-inhibitor mixtures. In the case of slow binding, the full potential of these inhibitors would not be realised by this inhibitory assay. When the activity was reduced by more than 60%, experiments were performed with varying concentrations of the catechol derivatives. IC50 values were obtained from dose response plots, and Ki values were determined from the IC50 values based on substrate competitive inhibition. By the use of Ki values, we can compare the binding strength of the compounds for the different enzymes and not only comparing the compounds ability to bind one enzyme. The obtained Ki values of the catechol contaning compounds for the five tested proteases are given in . There are several possible explanations for the deviation in determined Ki value of BF471 and BF489 for TLN from the dose response plots and the double inverse plots. One possible explanation is that the catechol compounds are slow binders, and hence less inhibitory activity is observed without preincubation of enzyme and inhibitor.
Table 1. Ki values of the catechol containing compounds for TLN, PLN, ALN, MMP-14, and MMP-9(T).
and show that the position of the OH-groups in the catechol moiety affects the binding. Moving the OH-group from position 2 to position 4 (ML32 vs. BF471 and ML33 vs. BF489) largely strengthen the binding for four of the enzymes, while the effect was less for ALN. Another striking effect occurs with the addition of a methylene group between the catechol moiety and the sulphonamide group, which resulted in weaker binding (BF489 vs. BF486 and ML32 vs. BF466). Changing the sulphonyl group to a methylene group and the methylene between the catechol moiety and the sulphonylamide moiety to a carbonyl (BF466 vs. MT336) resulted in stronger binding for the two MMPs, but with only limited activity changes for the bacterial enzymes. Overall, it appeared that the structural differences between the catechol derivatives had little effect on the ALN activity, while the activity of the other four enzymes varied correspondingly. None of the catechol derivatives showed stronger binding to the bacterial than the human enzymes. The largest differences in binding strengths between the human MMPs and the bacterial proteases were seen for MT336.
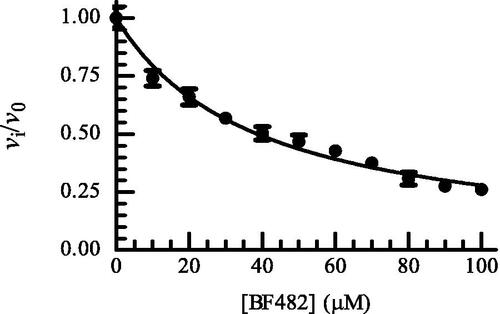
Except for ML33, all catechol derivatives have previously been tested for binding to MMP-2, MMP-8 and MMP-9Citation60. For six of them, IC50 values for the enzymes between 2 and 12 µM were reported. The exception was MT336, which had IC50 values between 4 and 56 µM. The Ki values for MMP-9 in the present study were higher than the IC50 values in the previous study, the exception was MT336 for which we obtained a lower value. It is not easy to point out a single factor in the experiments contributing to deviations between the studies. Both studies used a pH of 7.5 but the buffer compositions were slightly different. In the present study, we used MMP-9 purified from THP-1 cells and activated by trypsin, and hence the enzyme has its C-terminal hemopexin domain intactCitation57. Tauro et al.Citation60 used a commercial active MMP-9 produced in E-coli that only contained the catalytic domain and the fibronectin-like (FnII) module. We do not believe that the use of the two different variants of MMP-9 should affect binding of the catechol derivatives, as we previously have shown that small MMP inhibitors, like galardin and an azasugar-based hydroxamate compound had, the same strength of binding to different N- and C-terminal truncated variants of recombinant MMP-9 and trypsin activated MMP-9 from THP-1 cellsCitation57. A factor that may contribute to differences is that after preincubation of MMP-9 with catechol derivatives and adding of the substrate for starting the reaction, we followed the reaction continuously for 30 min, while Tauro et al.Citation60 used an endpoint assay allowing the reaction to proceed for 2 to 4 h before the fluorescence was measured. We did some tests to determine if some of these differences could affect the binding results. In the test experiments, we used rMMP-9(A) which differs from the MMP-9(T) by a slightly different N-terminal and a largely truncated C-terminal HPX domainCitation57. One hundred µM of BF466 was tested where the 0.1 M Hepes buffer pH 7.5 was exchanged to a 0.1 M Tris-HCl buffer pH 7.5. The rMMP-9(A) was preincubated with BF466 for 0, 15 and 30 min at 37 °C and the reaction was started by the addition of the substrate McaPLGL(Dpa)AR-NH2 (4 µM in the assay) following the reaction continuously for 3 h. The controls without BF466 were treated identically. The obtained vi/v0 values for the three time points were 0.74 ± 0.04 (N = 4), 0.68 ± 0.02 (N = 4) and 0.73 ± 0.05 (N = 4). These results fit well with obtained data for MMP-9(T) in Hepes buffer () suggesting that neither the buffer nor the origin of the MMP-9 affected the binding of this inhibitor. Further, the compound could not be regarded as a slow binder of MMP-9. We also tested if the use of an end point assay could affect the results. Here rMMP-9(A) was preincubated with different concentrations of BF482 for 30 min in 0.1 M Hepes pH 7.5 and the reaction was started by the addition of McaPLGL(Dpa)AR-NH2 (4 µM in the assay). The reaction was allowed to proceed for 4 h, and the reaction was stopped by addition of EDTA (end concentration 10 mM) and the relative fluorescence intensity determined. This resulted in an IC50 value of 40 ± 1 µM and a Ki value of 20.1 ± 0.7 µM (), while the vi/v0 value for 100 µM inhibitor was similar to that in . This suggests that the use of either initial rate assays or end point assays is not a reason for the obtained differences between this study and that of Tauro et al.Citation60.
Figure 2. Dose response plot of BF482 for MMP-9(A). The enzyme with and without inhibitor was pre-incubated for 30 min at room temperature and the reaction was started by adding McaPLGL(Dpa)AR-NH2 (4 μM in assay). The reaction was allowed to proceed for 4 h at 37 °C and stopped by the addition of EDTA (10 mM end concentration). The relative fluorescence was determined with the Clariostar plate reader as described in the Experimental Section, where vi and v0 are the reaction rates in the presence and absence of BF482, respectively. Each point on the curve shows the mean ± sd (N = 5 for all points except for two points where N = 4). The regression coefficient r2 is 0.97, with a determined IC50 value of 38.8 ± 0.9 μM and a Ki value of 19.4 ± 0.4 μM.
Docking
Docking of the catechol containing compounds into the X-ray structure of MMP-9 (PDB ID: 5cuh) and MMP-14 (PDB ID: 1bqq) showed that the biphenyl, bromophenyl and methoxyphenyl group of the compounds enter the S1' -subpocket. The interaction modes of BF471 in MMP-9 and MMP-14 were very similar to that observed in the X-ray structure of BF471 with MMP-8Citation60. In contrast, docking catechol containing compounds into TLN (PDB ID: 5dpe) and PLN (PDB ID: 1u4g) showed docking poses both with the diphenyl, bromophenyl and methoxyphenyl groups entering the S1- or the S1′/S2′ -subpockets. The cathecol containing compounds bind quite weak to ALN, and docking indicated binding modes quite different from the other proteases (. It seems like the side chain of R203 in TLN, corresponding to R200 in ALN and R197 in PLN, is the main reason for the binding pose differences between the human and bacterial proteases. For most of the compounds, the arginine side chain orientation hinders the biphenyl, bromophenyl and methoxyphenyl groups of the compounds to enter deeply into the S1'-subpocket (. This arginine is known as functionally important for TLN-like proteases, and is suggested to interact with a backbone carbonyl group of the substratesCitation61 and is located at the border between the S1′- and S2′-subpockets. shows the most important amino acids in the different protease subpockets for binding the compounds in the present study.
Figure 3. BF471 docked into PLN, TLN, ALN and MMP-9, and ML32 docked into ALN and MMP-9. Zinc is shown in dark grey. The side chain of zinc coordinating amino acids are shown in blue. The side chains of some amino acids of importance for ligand binding are displayed with the following colour coding of atoms: oxygen: red; nitrogen: blue, carbon: yellow, hydrogen: grey. Colour coding of ligand atoms: oxygen: red; sulphur: green; nitrogen: blue; hydrogen; grey; carbon (BF471): pink; carbon (ML32): orange.
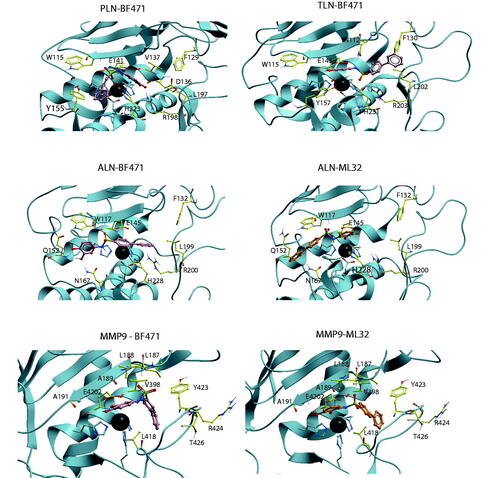
Table 2. Functionally important amino and amino acids in the S1-, S1′- and S2′-subpockets of TLN, PLN, ALN, MMP-9, and MMP-14 contributing to the binding of catechol or bisphosphonate containing compounds tested in the present study.
The highest scored pose of BF471 with TLN was with the diphenyl moiety into the S2′-subpocket, with the sulphonamide group interacting between the side chain of R203, the side chain of N112 in the S2'-subpocket and H231 (. Both hydroxyl groups of the BF471 catechol ring interacted with the Zn atom, while the hydroxyl group in position 3 also created a hydrogen bond with E143. However, the best pose of BF489 in TLN had the bromophenyl entirely into the S1′-subpocket, while a hydroxyl of the catechol ring interacted with the backbone of W115 (S1-subpocket) and the amide with E143, thereby obtaining a binding pose quite similar to that observed for BF471 and the other catechol containing compounds in MMP-9 and MMP-14 (.
The highest scored pose of BF471 in PLN was with the diphenyl moiety into the S1-subpocket interacting close to Y155, while the catechol moiety entered into the region between the S1′- and S2′-subpocket with the hydroxyl groups interacting with R198 (. The NH-group formed a hydrogen bond with E141. Moving the hydroxyl group from position 2 (ML32) to position 4 (BF471) of the catechol moiety resulted in increased binding affinity towards all enzymes. In PLN the change allowed interactions between both hydroxyl groups of BF471 and R198, while only one hydroxyl group of ML32 interacted with R198 which may explain the higher PLN affinity of BF471 than of ML32.
The compounds showed weak affinity for ALN (), and only BF471 reduced the enzymatic activity with more than 60% at a concentration of 100 µM and a Ki value of 49 μM was determined. BF471 obtained a docking pose in ALN quite different from those in the other enzymes (. The catechol hydroxyl groups formed hydrogen bonds with Q152 and N167 in the S1-subpocket, while the NH group formed hydrogen bonds with the backbone of W117 and with the biphenyl ring-system located above the Zn2+and R200. The possibility of two hydrogen bonds (with Q152 and N167) is perturbed for ML32 with the hydroxyl group in position 2 (. Instead the hydroxyl groups of ML32 were occupied with Zn2+, while diphenyl ring system was highly exposed to solvent without clear interactions with amino acids in ALN. Based on the docking it was not easy to explain the increased binding affinity of BF471 for ALN compared with ML32. Both compounds docked quite similar into the enzyme. However, the meta-hydroxyl group (position 3) of ML32 was located further from Y157 in the S1-subpocket without the possibility of a hydrogen bond, and in addition the sulphonamide group in ML32 was located further from H231 than the corresponding group in BF471.
Docking indicated that ML32 and ML33 bound similarly to MMP-9 and -14. The hydroxyl group in position 2 of both compounds formed hydrogen bonds with the two oxygen atoms of the side chain carboxyl group of E402 (MMP-9 numbering). The hydroxyl group in position 2 seems to replace the zinc-bound water in the free enzyme, as it also interacted with the catalytic zinc. The two oxygen atoms at the sulphonylamide group formed hydrogen bonds with the main chain NH groups of L187 and A189, while the amide hydrogen of the compounds formed hydrogen bonds with the main chain carbonyl of P421. Thus the main difference in binding strength between ML32 and ML33 for MMP-9 and -14 is mainly attributed to the difference in interaction with the S1'-subpocket by the diphenyl and the methoxyphenyl group.
The position of the OH– groups in the catechol in relation to the position of the amide nitrogen bound to the catechol seemed important for inhibitory capacity of the compounds. Moving the hydroxyl in position 2 of ML32 and ML33 into position 4 (BF471 and BF489) seemed to strengthen binding to MMP-9 and MMP-14 (, . BF471 docked into MMP-9 with the hydroxyl group in position 3 forming a hydrogen bond with the side chain of E402, while the hydroxyl group in postion 4 interacted with the backbone CO of A191. Both hydroxyl groups of BF471 and BF489 also interacted with Zn2+, while only the hydroxyl group in position 2 of ML32 and ML33 interacted with Zn2+. Introduction of a methyl group between sulphonylamide moiety and the catechol ring of BF489 giving compound BF486 resulted in decreased inhibition of all enzymes (. Docking indicated that the bromophenyl group of BF486 was deeper into the S1'-subpocket of MMP-9 and -14 than that of BF489, and one of the oxygen at the sulphonylamide group formed a hydrogen bond with the main chain nitrogen of Y423 and not with A189 and L188 as seen for BF489. Furthermore, one side chain carbonyl oxygen of E402 formed a hydrogen bond with the NH group of the sulphonylamide moiety of BF486, while the other E402 carbonyl oxygen interacted with the hydroxyl group in positon 3 of the catechol. In addition, the position 3 hydroxyl group interacted with the backbone CO of A191. These overall changes result in weaker binding of BF486 than of BF489.
MT336 differs from BF466 in that the sulphonyl group of BF466 is replaced by a methylene group, and the methyl group between the sulphonylamide moiety and the catechol ring by a carbonyl group. Hence MT336 contains an amide bond, which improved the binding for MMP-9 and MMP-14 compared to BF466, but not for the bacterial enzymes ( and ). Docking into MMP-9 and MMP-14 showed that the carbonyl of MT336 was much closer to the catalytic zinc atom than any of the oxygen atoms of the sulphonylamide moiety of BF466, and in addition, the hydroxyl group in position 2 of the catechol was also closer to Zn2+.
Inhibitory effects of bisphosphonates
shows the inhibition of 100 μM of the seven bisphosphonates. Replacing the catechol moiety of ML32 and BF471 with a bisphosphonate giving MT242, and the catechol moiety of ML33 giving LS4, had either no effect or reduced the binding ( and ). Adding a methoxy group to position 4 of the phenyl group (RC14 to LS4) had almost no effect of the binding, except for PLN where the binding was strengthened. The addition of a strong electron withdrawing group (NO2) at the position 4 of the phenyl group, giving RC2 resulted in a much stronger binding to PLN and TLN than to the two MMPs ( and ). However, for ALN the binding strength was almost similar to that without the NO2 group (RC14) or with a methoxy group at the phenyl ring (LS4) (. The binding of RC2 to ALN was even weaker than for the two human MMPs (. Removal of the sulphonyl group from the bisphosphonate of RC14 giving GD16 had limited effect on the binding of the five proteases (. Addition of a chloride ion at position 4 of the phenyl group of GD16 giving ML45 resulted in enhanced binding to all proteases except for TLN ( and ). Addition of an additional phenyl ring to position 4 of the phenyl group of GD16, giving MT363, gave much stronger binding for four of the proteases. However, the binding was reduced for ALN ( and ). Most bisphosphonates bound weaker to ALN than to the other proteases.
Figure 4. Inhibitory effect of 100 μM bisphosphonate containing compounds on the activity of the human metalloproteases, MMP-9 and MMP-14, and the bacterial metalloproteases TLN, PLN, and ALN. The inhibition experiments were performed by using a fixed concentration of 4 μM of both the MMP-9 and MMP-14 substrate McaPLGL(Dpa)AR-NH2 and the ALN, PLN and TLN substrate McaRPPGFSAFK(Dnp)-OH. The vi/v0 (mean ± sd) were based on 4–6 experiments.
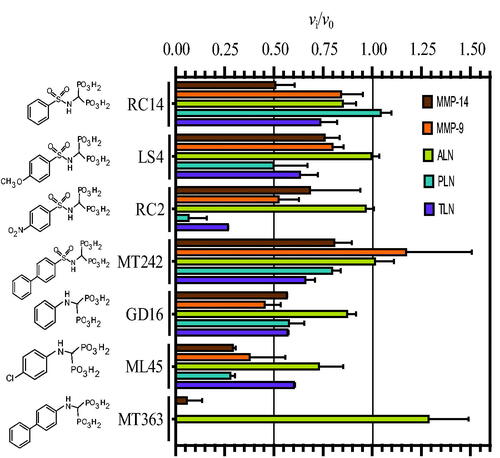
Table 3. The obtained Ki values of the various bisphosphonate containing compounds for TLN, PLN, ALN, MMP-14 and MMP-9.
The seven bisphosphonate compounds were also previously tested against various MMPs including MMP-9 and MMP-14Citation62,Citation63. As for the catechol derivatives, the results for some of the compounds varied slightly from the present results for MMP-9 and MMP-14. A difference between the assays was that in the present study we used a preincubation time of 15 min at 37 °C, while Rubino et al. 2011Citation62 and Tauro et al. 2013Citation63 used 30 min at 25 °C. Another important difference was that in the present work we followed the reaction continuously for 30 min after preincubation, while Rubino et al. 2011Citation62 and Tauro et al. 2013Citation63 used an end point assay where the reaction was stopped by adding 3% of acetic acid after 2–4 h of incubation, followed by fluorescence measurements. These differences in assay conditions are not likely explanations for the small differences in the obtained binding values as previously discussed for the catechol derivatives.
Docking
Docking indicated that like the catechol containg compounds, the bisphosphonates bind MMP-9 and MMP-14 with the phenyl, biphenyl, chlorophenyl, nitrophenyl or methoxyphenyl ringsystem into the S1′-subpocket (. The phosphate and sulphonamide groups were located in the region of Zn2+, E402, A189, L187, L188, and V398 (MMP-9 numbering). However, docking into TLN and PLN was not conclusive, and docking poses with these ringsystems into the S1- or S1′/S2′-subpockets of TLN and PLN were observed for most of the compounds. Notable features from the binding studies were that RC2 inhibits TLN and PLN stronger than ALN and the two human MMPs, while MT363 is a decent inhibitor of all enzymes, except ALN (, ).
Figure 5. MT363 and RC2 docked into PLN, TLN MMP-9 (MT363) and MMP-14 (RC2). TLN, PLN and MMP-9. Zinc is shown in dark grey. The side chain of zinc coordinating amino acids are shown in blue. The side chains of some amino acids of importance for ligand binding are displayed with the following colour coding of atoms: oxygen: red; nitrogen: blue; carbon: yellow; hydrogen: grey. Colour coding of ligand atoms: oxygen: red; sulphur: green; nitrogen: blue; phosphate: orange; hydrogen: grey; carbon – MT363: purple; carbon – ML32: orange.
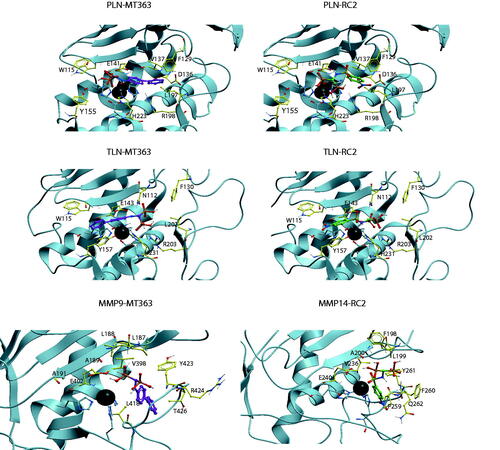
Docking of RC2 into PLN showed that one of the phosphate groups interacted with Zn2+, H223 and E141, while the other phosphate group interacted with R198 (two hydrogen bonds) and H223. The NH-group interacted with the backbone of A113. The NO2-group was located within the S1′-subpocket close to the side chain R198, the backbone NH-group of G187 and the side chain of the zinc-coordinating H140 (. However, docking poses in PLN with the nitrophenyl group of RC2 in the S1-subpocket were also observed. In TLN, poses quite similar to that described in PLN were observed, but the highest scored was with the nitrophenyl group into the S1-subpocket interacting with Y157 and with the nitro group exposed to solvent. The sulphonamide group interacted with N112 and H231. One of the phosphate groups interacted with Zn2+, and the side chains of R203 and E143, while the other interacted with the side chain of R203 and N112. In MMP-9 and -14, RC2 bound with the the nitrophenyl group into the S1′-subpocket (. In MMP-14, the phosphate groups were involved in a network of interactions with Zn2+ and E240, backbone NH and CO of A200, and the backbone NH groups of L199 and Y261, in addition to being exposed to solvent. The phosphate groups of RC2 had fewer strong interactions with the enzyme in the MMPs than in PLN and TLN, which may contribute to stronger interactions of RC2 with TLN and PLN than with the MMP-9 and -14.
Except for ALN, MT363 binds quite strong to all enzymes. In PLN the biphenyl group of MT363 was located within the S2'-subpocket, while one the phosphate groups interacted with Zn2+, E141 and the backbone NH of W115, while the other interacted with N112, F114, in addition to being quite solvent exposed. However, docking poses with the biphenyl group into the S1-subpocket were also obtained. In TLN, MT363 docked best with the biphenyl group into the S1-subpocket obtaining stacking interactions with Y157, while also poses with the biphenyl into the S2′-subpocket were obtained. In the best docking pose the phosphate groups interacted with Zn2+, R203 and H231, while the other pointed into the S2′-subpocket and interacted with N112 (. In the MMPs, the biphenyl moiety was in the S1′-subpocket, while the bisphosphonate group interacted with Zn2+, the side chain of E402 (MMP-9 numbering), V398, the backbone of A189, in addition to being exposed to the solvent.
Conclusion
Several of the tested MMP-inhibitors were identified as strong TLN and PLN inhibitors, while only BF471 inhbited ALN activity with more than 60%. Both tested catechol containing compounds and bisphosphonates bound MMP-9 and −14 with the the phenyl, biphenyl, chlorophenyl, bromophenyl, nitrophenyl or methoxyphenyl ringsystem into the structurally flexible S1′-subpocket. In TLN, PLN and ALN, the contribution of a postively charged arginine (TLN; R203, PLN; R198, ALN; R200) at the entrance of the S1′-subpocket hinders that these functional groups fully enter the S1′-subpocket of the bacterial enzymes. Instead these groups occupy the S1- or the S2′-subpocket, or are located at the entrance of the S1′-subpocket. However, interactions with the arginine seem to be an important factor for strong binding to the bacterial proteases. RC2- bound stronger to TLN and PLN than to the MMPs. RC2 may be used as a scaffold to identify new compounds that bind much stronger to the bacterial virulence factors TLN and PLN than to the human MMPs, and hence have a therapeutic potential as virulence factor inhibitors and represent a new strategy in the fight against drug resistant bacterial infections.
Experimental section
Materials
TRIS, DMSO, Na2HPO4 and sodium acetate were from Merck (Darmstadt, Germany). EDTA was from Fluka (Buchs, Switzerland). Acrylamide, Commassie Brilliant Blue G-250 and Triton X-100 were from BDH (Poole, UK). RPMI 1640, streptomycin, penicillin, phorbol 12-myristate 13-acetate (PMA), Hepes, Brij-35, Silver nitrate, alkaline phosphatase-conjugated antibodies and gelatine were purchased from Sigma (St Louis, MO, USA). Gelatine-Sepharose, Q-Sepharose and Sephadex G-50 (fine) were from GE-Healthcare (Uppsala, Sweden). DC Protein Assay and unlabelled molecular weight standards were from BioRad (Richmond, CA, USA). Magic Marker molecular weight standards were from Invitrogen (Carlsbad, CA, USA). Western Blotting Luminol reagent and HRP-conjugated donkey anti-goat secondary antibody were from Sancta Cruz (Santa Cruz, CA, USA). HRP-conjugated goat anti-rabbit secondary antibody was from Southern Biotech (Birmingham, AL, USA). Foetal bovine serum was from Biochrom AG (Berlin, Germany). Human MT1-MMP/MMP-14 (catalytic domain), TLN and PLN were from Calbiochem (San Diego, CA, USA) and Aureolysin was from BioCentrum Ltd (Kraków, Poland). McaPLGL(Dpa)AR-NH2 (ES001) and McaRPPGFSAFK(Dnp)-OH (ES005) were from R&D Systems (Minneapolis, MN, USA).
Synthesis of compounds
Synthesis of compounds tested in the present study was reported previouslyCitation60,Citation62,Citation63.
Biosynthesis of proMMP-9
The human leukemic monocyte cell-line THP-1 was a kind gift from Dr. K. Nilsson, Department of Pathology, University of Uppsala, Sweden. The cells were cultured in RPMI 1640 medium with 10% foetal bovine serum, 50 μg/ml of streptomycin, and 100 units/ml of penicillin. To isolate secreted cell-synthesized proMMP-9, the cells were washed 3 times in serum-free medium and then cultured for 72 h in serum-free RPMI 1640 medium with 0.1 μM PMA as described previouslyCitation64,Citation65. Conditioned medium was harvested, loose cells were pelleted by centrifugation at 1200 rpm (200 g) for 10 min. ProMMP-9 was thereafter isolated and detected as described below.
Purification and activation of proMMP-9 from the THP-1 cells
The proMMP-9 in conditioned medium from the THP-1 cells was partly purified as described previouslyCitation57,Citation65,Citation66. SDS-electrophoresis under reducing conditions, followed by either silver or Coomassie Blue staining, showed two bands, a major band at 92 kDa and a minor band at 28 kDa. Western blotting revealed that the 92 kDa band was proMMP-9, and the 28 kDa band was TIMP-1. The amount of proMMP-9 was estimated spectrophotometrically at 280 nm using ε280nm=114,360 M−1 cm−1 Citation67, ignoring the contribution of TIMP-1.
The purified proMMP-9 was activated by trypsin, by mixing approximately 300 µg of proMMP-9 with trypsin (31 μg/ml) for 10 min at 37 °C in 0.1 M Hepes pH 7.5, 0.005% Brij35% and 10 mM CaCl2. The activation and processing of MMP-9 were terminated by adding a 50 times excess of SBTI (2.7 mg/ml) in relation to trypsin and after 10 min incubation at room temperature, the mixture was transferred and kept on ice during the kinetic and inhibition kinetic measurements. After activation, the activity was determined with 10 µM of McaPLGL(Dpa)AR-NH2 in 0.1 M Hepes pH 7.5, 0.005% Brij35% and 10 mM CaCl2 in a total assay volume of 100 µL, at 37 °C. Initial rates were measured at an excitation wavelength of 320 nm and an emission wavelength of 405 nm with a slit width of 10 nm using a Perkin Elmer LS 50 Luminescence spectrometer and the FL WinLab Software Package (Perkin Elmer). The amount of active MMP-9 was determined by active site titration using galardin as described previouslyCitation57.
Expression, purification and activation of recombinant human proMMP-9 in Sf9 insect cells
The expression and purification of recombinant human full-length proMMP-9 (rpMMP-9) from Sf9 insect cells were performed as described previouslyCitation57. The amount of proMMP-9 was estimated spectrophotometrically at 280 nm using ε280nm=114,360 M−1 cm−1 Citation57. Activation of the recombinant proMMP-9 was performed with APMA (auto-activation) as described previouslyCitation57. The amount of active MMP-9 was determined by active site titration using galardin also described previouslyCitation57.
Determination of Km values
Km values were determined for McaPLGL(Dpa)AR-NH2 with APMA-activated recombinant MMP-9 (rMMP-9(A)), trypsin-activated MMP-9 from THP-1 cells (MMP-9(T)) and MMP-14, and for McaRPPGFSAFK(Dnp)-OH with ALN, PLN and TLN. Substrate concentrations used were 1–10 µM in a total volume of 100 µL of 0.1 M Hepes pH 7.5 containing 10 mM CaCl2, 0.005% Brij-35 and 1.0% DMSO. Substrate concentrations above 10 µM resulted in quenching as reported previouslyCitation58. Initial rate experiments were performed as described above for the determination of enzyme activity of MMP-9 during activation and the same excitation and emission wavelengths were used for both substrates.
Determination of IC50 and Ki values
The various inhibitors were dissolved in 100% DMSO giving an inhibitor concentration of 10 mM. All the inhibitory and control experiments contained a total and fixed concentration of 1.0% DMSO. The inhibitory constant IC50 of the various compounds were performed with inhibitor concentrations ranging from 10−10 to 10−4 M in the assay, with a fixed substrate concentration of 4.0 µM in a total volume of 100 µL 0.1 M Hepes pH 7.5, 10 mM CaCl2, 0.005% Brij-35 and 1.0% DMSO, except for ALN where the substrate concentration was 5.0 µM. The fixed enzyme concentration were as follows; 0.28 nM MMP-9(T), 0.05 nM MMP-9(A), 1.0 nM MMP-14, 1.4 nM ALN, 0.5 nM PLN and 0.21 nM TLN. Enzymes with and without inhibitors were pre-incubated for 15 min at 37 °C, the initial rate assays were started by adding the substrate and the reaction was followed for 30 min. Assays were performed using a Spectra Max Gemini EM micro-plate reader (Molecular Devices) or a Clario Star micro plate reader (CLARIOstar® BMG LABTECH). Assays were performed at 37 °C, using an excitation wavelength of 320 nm and an emission wavelength of 405 nm with a slit width of 10 nm. The IC50 values were calculated either in Sigma Plot (Enzyme kinetics 1.3 module) or in Graph Pad Prism 5 using EquationEquations (1)(1)
(1) or (Equation2
(2)
(2) ) depending on the concentration span of the used inhibitor:
(1)
(1)
(2)
(2)
where vi is the enzyme activity in the presence of inhibitor, v0 the activity in the absence of inhibitor, pI=−log [Inhibitor] in M and pIC50 = −log IC50 in M. All experiments were performed in at least triplicate.
For substrate competitive inhibitors, EquationEquations (3)(3)
(3) shows the relation between IC50 and Ki values based on the fixed concentration of substrate used and the enzymes Km value for the substrate:
(3)
(3)
Quenching experiments
Some of the catechol and biphosphate derivatives showed a concentration dependent fluorescence at wavelengths used for the McaPLGL(Dpa)AR-NH2 and McaRPPGFSAFK(Dnp)-OH substrates. To determine to which extent these derivatives could quench the time dependent enzymatic increase in the fluorescence product of the processed substrate, quenching experiments were performed as described previouslyCitation58. Briefly, the fluorescence (λex=320 nm, λem=405 nm, slit width = 10 nm) of various concentrations of the fluorescent product of the substrate McaPLGL(Dpa)AR-NH2, McaPL-OH (0–100 nM), was determined in absence and presence of various concentrations of the catechol and bisphosphonate derivatives (0–100 µM). Primary and secondary plots were used to determine whether the catechol and bisphosphonate derivatives quenched the McaPL-OH fluorescence.
Docking
The internal Coordinate Mechanics (ICM) program versionCitation68 was used for docking of catechol containing compounds (ML32, BF471, ML33, BF489, BF486, BF482, BF466, MT336) and bisphosphonate containing compounds (RC14, LS4, RC2, MT242, GD16, ML45, MT363) into the target proteases. The X-ray crystal structures of PLN (PDB-code:1u4g), TLN (PDB-code: 5dpe), MMP-9 (PDB-code: 5cuh, ALN (PDB-code: 1bqb) and MMP-14 (PDB-code:1bqq) were collected from the PDB database and used for docking. Crystallographic water molecules were removed along with the co-crystallized small molecule inhibitors. Hydrogen atoms were added and optimised using the ECEPP/3 force field before the structures were refined and minimised. The various inhibitors were built using ICM and minimised before docking. The binding modes of the inhibitors in the X-ray structure complexes PLN (1u4g), TLN (5dpe) and MMP-9 (5cuh) were used to define the binding pocket for docking into these enzymes, using grid maps that included all amino acids within 5 Å of the cocrystallized inhibitors. However, X-ray crystal structures with small molecule inbitors were not available for ALN and MMP-14. For ALN, the X-ray structure without inhibitor (1bqb) was superimposed with the PLN complex (1u4g) and the inhibitor in the PLN complex was used to create docking grids including all amino acids within 5 Å of the PLN inhibitor. For MMP-14, the X-ray crystal structure of MMP-8 with the inhibitor BF471 (PDB id: 5h8x) was superimposed with the MMP-14 structure in complex with TIMP-2 (1bqq) and binding mode of BF471 in MMP-8 was used to create docking grids within 5 Å of BF471. After ceating grid maps, semi-flexible docking was performed where the enzymes were kept rigid while the ligands were structurally flexible. Each docking was run in three parallels. Ligand conformer sampling in vacuo and Monte Carlo global energy optimisation were used to generate docking posesCitation69, while the poses were scored using the Virtual Ligand Scoring (VLS) module of the ICM program. The VLS scoring function uses steric, entropic, hydrogen bonding, hydrophobic and electrostatic terms to calculate the score and also includes a correction term proportional to the number of atoms in the ligand to avoid bias towards larger ligandsCitation70.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Artenstein AW, Opal SM. Proprotein convertases in health and disease. N Engl J Med 2011;365:2507–18.
- Winberg JO, Matrix Proteinases: biological significance in health and disease. In: Karamanos NK, ed. Extracellular matrix: pathobiology and signaling. Berlin: de Gruyter; 2012: 235–238.
- De Groef L, Van Hove I, Dekeyster E, et al. MMPs in the neuroretina and optic nerve: modulators of glaucoma pathogenesis and repair? Invest Ophthalmol Vis Sci 2014;55:1953–64.
- Quiros PM, Langer T, Lopez-Otin C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol 2015;16:345–59.
- Ricard-Blum S, Vallet SD. Proteases decode the extracellular matrix cryptome. Biochimie 2016;122:300–13.
- Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta 2010;1803:39–54.
- Wolberg AS, Mast AE. Tissue factor and factor VIIa-hemostasis and beyond. Thromb Res 2012;129:S1–S4.
- Cerda-Costa N, Gomis-Ruth FX. Architecture and function of metallopeptidase catalytic domains. Protein Sci 2014;23:123–44.
- Gomis-Ruth FX, Botelho TO, Bode W. A standard orientation for metallopeptidases. Biochim Biophys Acta 2012;1824:157–63.
- Rawlings ND, Barrett AJ, Thomas PD, et al. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 2018;46:D624–D632.
- Hadler-Olsen E, Fadnes B, Sylte I, et al. Regulation of matrix metalloproteinase activity in health and disease. Febs J 2011;278:28–45.
- Hadler-Olsen E, Winberg JO, Uhlin-Hansen L. Matrix metalloproteinases in cancer: their value as diagnostic and prognostic markers and therapeutic targets. Tumour Biol 2013;34:2041–51.
- Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010;141:52–67.
- Sbardella D, Fasciglione GF, Gioia M, et al. Human matrix metalloproteinases: an ubiquitarian class of enzymes involved in several pathological processes. Mol Aspects Med 2012;33:119–208.
- Geurts N, Opdenakker G, Van den Steen PE. Van den Steen PE. Matrix metalloproteinases as therapeutic targets in protozoan parasitic infections. Pharmacol Ther 2012;133:257–79.
- Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. Febs J 2011;278:16–27.
- Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov 2014;13:904–27.
- Yadav L, Puri N, Rastogi V, et al. Matrix metalloproteinases and cancer – roles in threat and therapy. Asian Pac J Cancer Prev 2014;15:1085–91.
- Overall CM, Blobel CP. In search of partners: linking extracellular proteases to substrates. Nat Rev Mol Cell Biol 2007;8:245–57.
- Theocharis AD, Gialeli C, Hascall V, Karmanos N, Extracellular matrix: a functional scaffold. In Karamanos NK, ed. Extracellular matrix: pathobiology and signaling. Berlin: de Gruyter; 2012: 3–19.
- Cui N, Hu M, Khalil RA. Biochemical and biological attributes of matrix metalloproteinases. Prog Mol Biol Transl Sci 2017;147:1–73.
- Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer 2002;2:657–72.
- Kessenbrock K, Wang CY, Werb Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol 2015; 44-46:184–90.
- Ballok AE, O'Toole GA. Pouring salt on a wound: Pseudomonas aeruginosa virulence factors alter Na + and Cl- flux in the lung. J Bacteriol 2013;195:4013–9.
- Dubin G. Extracellular proteases of Staphylococcus spp. Biol Chem 2002;383:1075–86.
- Jensen LM, Walker EJ, Jans DA, Ghildyal R. Proteases of human rhinovirus: role in infection. Methods Mol Biol 2015;1221:129–41.
- Maeda H. Role of microbial proteases in pathogenesis. Microbiol Immunol 1996;40:685–99.
- Matsumoto K. Role of bacterial proteases in pseudomonal and serratial keratitis. Biol Chem 2004;385:1007–16.
- Shinoda S, Miyoshi S. Proteases produced by vibrios. Biocontrol Sci 2011;16:1–11.
- Silva-Almeida M, Pereira BAS, Ribeiro-Guimarães ML, Alves CR. Proteinases as virulence factors in Leishmania spp. infection in mammals. Parasit Vectors 2012;5:160.
- Dickey SW, Cheung GYC, Otto M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat Rev Drug Discov 2017;16:457–71.
- Munguia J, Nizet V. Pharmacological targeting of the host-pathogen interaction: alternatives to classical antibiotics to combat drug-resistant superbugs. Trends Pharmacol Sci 2017;38:473–88.
- Brotz-Oesterhelt H, Sass P. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int J Med Microbiol 2014;304:23–30.
- Hauser AR, Mecsas J, Moir DT. Beyond antibiotics: new therapeutic approaches for bacterial infections. Clin Infect Dis. 2016;63:89–95.
- Zambelloni R, Marquez R, Roe AJ. Development of antivirulence compounds: a biochemical review. Chem Biol Drug Des 2015;85:43–55.
- Nagase H, Woessner JF. Matrix metalloproteinases. J Biol Chem 1999;274:21491–4.
- Woessner Jr., JF, Nagase H, Matrix metalloproteinases and TIMPs. Oxford: Oxford University Press; 2000.
- Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol 2013;48:222–72.
- Antoni C, Vera L, Devel L, et al. Crystallization of bi-functional ligand protein complexes. J Struct Biol 2013;182:246–54.
- Fernandez-Catalan C, Bode W, Huber R, et al. Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. Embo J 1998;17:5238–48.
- Grossman M, Tworowski M, Dym O, et al. The intrinsic protein flexibility of endogenous protease inhibitor TIMP-1 controls its binding interface and affects its function. Biochemistry 2010;49:6184–92.
- Nuti E, Cantelmo RA, Gallo C, et al. N-O-isopropyl sulfonamido-based hydroxamates as matrix metalloproteinase inhibitors: hit selection and in vivo antiangiogenic activity. J Med Chem 2015;58:7224–40.
- Nuti E, Casalini F, Avramova SI, et al. N-O-isopropyl sulfonamido-based hydroxamates: design, synthesis and biological evaluation of selective matrix metalloproteinase-13 inhibitors as potential therapeutic agents for osteoarthritis. J Med Chem 2009;52:4757–73.
- Nuti E, Casalini F, Avramova SI, et al. Potent arylsulfonamide inhibitors of tumor necrosis factor-alpha converting enzyme able to reduce activated leukocyte cell adhesion molecule shedding in cancer cell models. J Med Chem 2010;53:2622–35.
- Nuti E, Panelli L, Casalini F, et al. Design, synthesis, biological evaluation, and NMR studies of a new series of arylsulfones as selective and potent matrix metalloproteinase-12 inhibitors. J Med Chem 2009;52:6347–61.
- Rowsell S, Hawtin P, Minshull CA, et al. Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J Mol Biol 2002;319:173–81.
- Tochowicz A, Maskos K, Huber R, et al. Crystal structures of MMP-9 complexes with five inhibitors: contribution of the flexible Arg424 side-chain to selectivity. J Mol Biol 2007;371:989–1006.
- Eckhard U, Huesgen PF, Schilling O, et al. Active site specificity profiling of the matrix metalloproteinase family: Proteomic identification of 4300 cleavage sites by nine MMPs explored with structural and synthetic peptide cleavage analyses. Matrix Biol 2016;49:37–60.
- Fabre B, Ramos A, de Pascual-Teresa B. Targeting matrix metalloproteinases: exploring the dynamics of the S1' pocket in the design of selective, small molecule inhibitors. J Med Chem 2014;57:10205–19.
- Adekoya OA, Sylte I. The thermolysin family (M4) of enzymes: therapeutic and biotechnological potential. Chem Biol Drug Des 2009;73:7–16.
- Englert L, Biela A, Zayed M, et al. Displacement of disordered water molecules from hydrophobic pocket creates enthalpic signature: binding of phosphonamidate to the S1′-pocket of thermolysin. Biochim Biophys Acta 2010;1800:1192–202.
- Bever RA, Iglewski BH. Molecular characterization and nucleotide sequence of the Pseudomonas aeruginosa elastase structural gene. J Bacteriol 1988;170:4309–14.
- Galloway DR. Pseudomonas aeruginosa elastase and elastolysis revisited: recent developments. Mol Microbiol 1991;5:2315–21.
- Pauptit RA, Karlsson R, Picot D, et al. Crystal structure of neutral protease from Bacillus cereus refined at 3.0 A resolution and comparison with the homologous but more thermostable enzyme thermolysin. J Mol Biol 1988;199:525–37.
- Thayer MM, Flaherty KM, McKay DB. Three-dimensional structure of the elastase of Pseudomonas aeruginosa at 1.5-A resolution. J Biol Chem 1991;266:2864–71.
- Banbula A, Potempa J, Travis J, et al. Amino-acid sequence and three-dimensional structure of the Staphylococcus aureus metalloproteinase at 1.72 A resolution. Structure 1998;6:1185–93.
- Sylte I, Dawadi R, Malla N, et al. The selectivity of galardin and an azasugar-based hydroxamate compound for human matrix metalloproteases and bacterial metalloproteases. PLOS One 2018;13:e0200237.
- Sjoli S, Nuti E, Camodeca C, et al. Synthesis, experimental evaluation and molecular modelling of hydroxamate derivatives as zinc metalloproteinase inhibitors. Eur J Med Chem 2016;108:141–53.
- Yang JJ, Van Wart HE. Kinetics of hydrolysis of dansyl peptide substrates by thermolysin: analysis of fluorescence changes and determination of steady-state kinetic parameters. Biochemistry 1994;33:6508–15.
- Tauro M, Laghezza A, Loiodice F, et al. Catechol-based matrix metalloproteinase inhibitors with additional antioxidative activity. J Enzyme Inhib Med Chem 2016;31:25–37.
- Holden HM, Tronrud DE, Monzingo AF, et al. Slow- and fast-binding inhibitors of thermolysin display different modes of binding: crystallographic analysis of extended phosphonamidate transition-state analogues. Biochemistry 1987;26:8542–53.
- Rubino MT, Agamennone M, Campestre C, et al. Biphenyl sulfonylamino methyl bisphosphonic acids as inhibitors of matrix metalloproteinases and bone resorption. ChemMedChem 2011;6:1258–68.
- Tauro M, Laghezza A, Loiodice F, et al. Arylamino methylene bisphosphonate derivatives as bone seeking matrix metalloproteinase inhibitors. Bioorg Med Chem 2013;21:6456–65.
- Malla N, Berg E, Moens U, et al. Biosynthesis of promatrix metalloproteinase-9/chondroitin sulphate proteoglycan heteromer involves a Rottlerin-sensitive pathway. PLOS One 2011;6:e20616.
- Malla N, Berg E, Uhlin-Hansen L, Winberg JO. Interaction of pro-matrix metalloproteinase-9/proteoglycan heteromer with gelatin and collagen. J Biol Chem 2008;283:13652–65.
- Dawadi R, Malla N, Hegge B, et al. Molecular interactions stabilizing the promatrix metalloprotease-9. Serglycin Heteromer Int J Mol Sci 2020;21:4205.
- Murphy G, Crabbe T. Gelatinases A and B. Methods Enzymol 1995;248:470–84.
- Abagyan R, Totrov M, Kuznetsov D. ICM – a new method for protein modeling and design – applications to docking and structure prediction from the distorted native conformation. J Comput Chem 1994;15:488–506.
- Abagyan R, Kufareva I. The flexible pocketome engine for structural chemogenomics. Methods Mol Biol 2009;575:249–79.
- Schapira M, Abagyan R, Totrov M. Nuclear hormone receptor targeted virtual screening. J Med Chem 2003;46:3045–59.
- Tranchant I, Vera L, Czarny B, et al. Halogen bonding controls selectivity of FRET substrate probes for MMP-9. Chem Biol 2014;21:408–13.
- Elkins PA, Ho YS, Smith WW, et al. Structure of the C-terminally truncated human ProMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallogr D Biol Crystallogr 2002;58:1182–92.
- Krimmer SG, Cramer J, Betz M, et al. Rational design of thermodynamic and kinetic binding profiles by optimizing surface water networks coating protein-bound ligands. J Med Chem 2016;59:10530–48.