
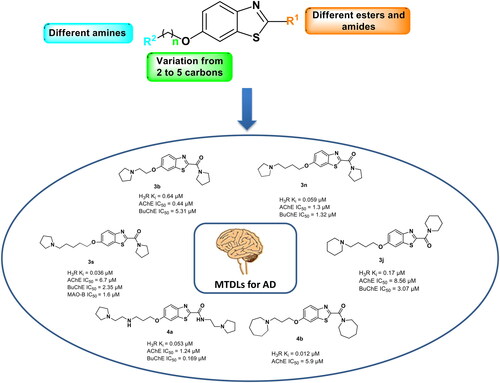
Abstract
Neurodegenerative diseases such as Alzheimer’s disease (AD) are multifactorial with several different pathologic mechanisms. Therefore, it is assumed that multitargeted-directed ligands (MTDLs) which interact with different biological targets relevant to the diseases, might offer an improved therapeutic alternative than using the traditional “one-target, one-molecule” approach. Herein, we describe new benzothiazole-based derivatives as a privileged scaffold for histamine H3 receptor ligands (H3R). The most affine compound, the 3-(azepan-1-yl)propyloxy-linked benzothiazole derivative 4b, displayed a Ki value of 0.012 μM. The multitargeting potential of these H3R ligands towards AChE, BuChE and MAO-B enzymes was evaluated to yield compound 3s (pyrrolidin-1-yl-(6-((5-(pyrrolidin-1-yl)pentyl)oxy)benzo[d]thiazol-2-yl)methanone) as the most promising MTDL with a Ki value of 0.036 μM at H3R and IC50 values of 6.7 µM, 2.35 µM, and 1.6 µM towards AChE, BuChE, and MAO-B, respectively. These findings suggest that compound 3s can be a lead structure for developing new multi-targeting anti-AD agents.
Graphical abstract
Introduction
AD is a progressive irreversible neurodegenerative brain disorder and it is the most common form of dementia.Citation1,Citation2 The histopathological changes observed in AD patients include deficiency in cholinergic neurotransmission, accumulation of amyloid-β (Aβ) peptide in the form of amyloid plaques in the brain, intracellular neurofibrillary tangles and progression of inflammation and damage caused by oxidative stress.Citation3 The drugs approved for treating AD only offer a symptomatic treatment, but fail to stop the neurodegenerative process. Owing to the complex pathology of the disease, a single ligand for a certain target is not sufficient to produce a relevant therapeutic effect.Citation4,Citation5 Therefore, the new pharmacological approach is to design single molecules that modulate multiple targets simultaneously, so-called multitarget-directed ligands (MTDLs). MTDLs are synthesised mainly as potential therapeutics for the treatment of multifactorial diseases such as neurodegenerative diseases.Citation6,Citation7 MTDLs are compounds that affect more than one pathophysiological pathway since they can be rationally designed as structures with pharmacophore moieties that bind to different receptors and enzymes.Citation8 The pathogenesis of most neurodegenerative diseases (especially AD) is linked to different pharmacological targets. Among these targets, neurotransmitters such as ACh, histamine, dopamine and glutamate play major roles in the progressive deterioration of cognitive functions. The combination of H3R antagonist pharmacophore and acetylcholinesterase pharmacophore is one of the most interesting and promising combinations among MTDLs for the treatment of AD.Citation9,Citation10
Histamine H3 receptors (H3Rs) belong to the family of G protein-coupled receptors (GPCR). H3R was first identified on presynaptic histaminergic neurons acting as autoreceptors. Besides its inhibitory activity as an autoreceptor, modulating the release of other neurotransmitters such as noradrenaline, serotonin, dopamine and acetylcholine has been observed by H3 hetero receptor located on non-histaminergic neurons.Citation11 Thus the precognitive use of H3R antagonists/inverse agonists for the treatment of CNS-related disorders, such as depression, schizophrenia, PD, and AD is being investigated.Citation12 Pitolisant, is the first H3R antagonist/inverse agonist which received the approval in Europe and United States for the treatment of narcolepsy with or without cataplexy.Citation13 In the early stage of its discovery, pitolisant showed selectivity towards H3R and induced central histaminergic transmissions in animal models increasing wakefulness and resulted in increased release of dopamine and ACh in the prefrontal cortex of rats. New classes of H3R antagonists with additional pharmacological properties have been described.Citation14 These classes may have synergistic effect leading to the increase of their therapeutic efficacy in the treatment of disorders related to neurotransmitter deficits, such as AD. Accordingly, the development of multitarget compounds including H3 receptor-blocking properties has been of great interest.Citation2,Citation11
Acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) are enzymes responsible for the hydrolysis of acetylcholine and the increase in their activity leads to cognitive disorders.Citation15 Moreover, the inhibition of AChE may not only result in increased levels of ACh but may also affect the Aβ aggregation.Citation16 So far, the anti-Alzheimer’s drugs include acetylcholinesterase inhibitors (AChEI), such as donepezil, rivastigmine and galantamine and the N-methyl-d-aspartate (NMDA) receptor antagonist memantine.Citation3 Unfortunately, these drugs can only slow down the progression of the disease and relief the symptoms but they do not provide effective treatment for AD. In AD, the cognitive decline is caused by neuronal degeneration of brain regions where the cholinergic activity is predominant. The loss of cholinergic transmission can be counteracted by AChE inhibitors, whose efficacy indeed declines during progression of the disease, while at the same time BuChE remains unaltered or increased. Hence, the inhibition of BuChE may provide additional cognitive benefits in the late stages of disease.Citation17 Hence, cholinergic neurotransmission can be enhanced by cholinesterase inhibitors (ChEIs) which counteract such depletion of ACh neurotransmitter.Citation18
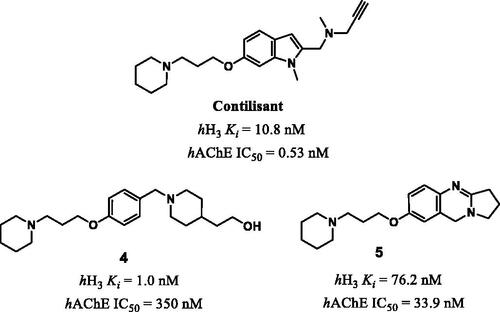
Several studies proposed structures similar to those designed in this work to have dual activity on AChE and H3R such as contilisant, compound 4 and 5 as shown in .Citation11 Therefore, the combination of both H3R inhibition and cholinesterase enzymes inhibition in a single drug might lead to significant synergistic effects on cognitive function and memory. Using these hybrid compounds can lead to huge progress in the treatment of cognitive disorders.Citation9,Citation19
Figure 1. Compounds having dual activities on AChE and H3R.
Herein, novel benzothiazole derivatives as H3R ligands were developed. Benzothiazole is a privileged heterocyclic scaffold used worldwide for a variety of therapeutic applications.Citation20 Several novel benzothiazoles have been developed and exhibited significant antitumour, antimicrobial, antidiabetic, anti-inflammatory, antiviral, antioxidant, antimalarial, antiasthmatic, anthelmintic, and other activities.Citation21–27 New benzothiazole molecules are currently under development and are being evaluated as therapeutic drug candidates for the treatment of neurodegenerative diseases such as Alzheimer’s.Citation28 For instance, riluzole (2-amino-6-trifluoromethoxy) benzothiazole is used to treat amyotrophic lateral sclerosis due to its neuroprotective effect acting as voltage-gated sodium channel blockers, non-competitive inhibition of NMDA receptors and inhibition of glutamate release. Recently, evaluation of riluzole in a clinical trial for AD was reported.Citation29
Inspired by the similarity in the pharmacophoric features of the H3R antagonists and AChE inhibitors, the activity of the synthesised compounds on AChE enzyme to reach multitarget inhibition was tested.Citation14 The dual-acting compounds (3b, 3h, 3j, 3n, 3s, 3t, 4a, and 4b) were also tested against BuChE as well as MAO-B enzyme to extend the multitarget range against AD. MAO-B enzyme inhibition was assessed since some literature stated that MAO-B activity is increased in the brain of patients with neurodegenerative diseases and suggested that MAO-B regulates β-amyloid production via γ-secretase.Citation30,Citation31 Moreover, molecular modelling studies have revealed possible interactions between active compounds and both AChE and BuChE as well as H3R.
Results and discussion
Compounds’ design
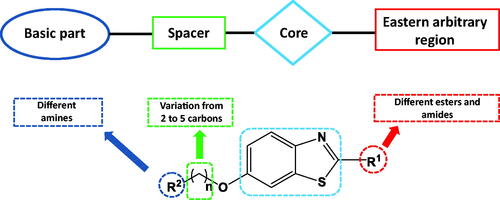
The presented compounds were designed to match the general structure suggested for histamine H3R antagonists/inverse agonists ().Citation32 This pharmacophore blueprint contains a basic moiety (mostly a tertiary amine), linked by a spacer to a central core, which is connected to further affinity-enhancing elements in the eastern arbitrary region which can be another basic moiety or hydrophilic/lipophilic groups as shown in .Citation33–35 The following structural modification strategies were applied to design potent H3R antagonists: (1) employing benzothiazole ring as a central core;Citation36–38 (2) several amines were examined as a basic head including alicyclic amines, cycloalkyl amines and amino acids; (3) the spacer connecting the central benzothiazole core and the basic head was varied from two to five carbons; (4) different esters and amides were added to the eastern arbitrary region of the compound.
Figure 2. Structure modifications adopted in the synthesised compounds.
Chemistry
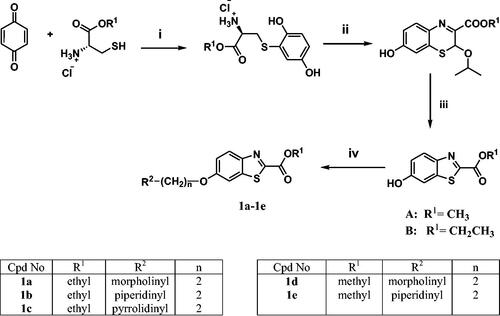
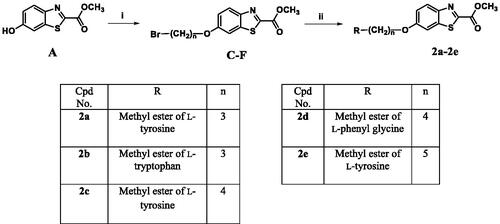
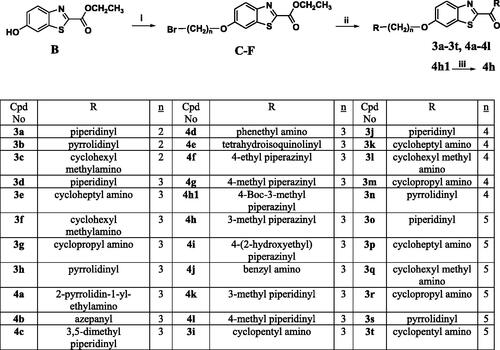
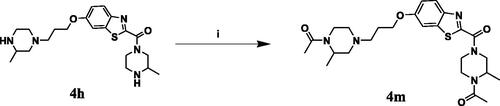
The adopted synthetic pathways for the intended compounds are depicted in Schemes 1–4. The synthesis of methyl/ethyl 6-hydroxybenzothiazole-2-carboxylate core was accomplished through a three-step synthesis (Scheme 1). First, 1,4-benzoquinone was reacted with cysteine methyl/ethyl ester hydrochloride through Michael addition to afford the hydroquinone. Then, the hydroquinone intermediate underwent an oxidation reaction by potassium ferricyanide to give the benzothiazine derivative, which underwent contraction to benzothiazole ring in the presence of an acidic medium to yield compounds A and B. The mechanism of ring contraction was proposed to take place via benzothiazine ring hydrolysis to give a mercaptoaldehyde derivative, intramolecular attack to form the contracted ring, followed by oxidation and decarboxylation of the aldehyde group.Citation39 Compounds A and B were refluxed with different chloroalkyl amines in presence of potassium carbonate to yield compounds 1a–1e. In Scheme 2, the 6-hydroxybenzothiazole-2-carboxylate methyl ester (A) reacted with the appropriate alpha, omega-dibromoalkane to give an ether in presence of potassium carbonate and potassium iodide to produce compounds C–F, which were then refluxed with the methyl ester of different aromatic amino acids to yield compounds 2a–2e. In Scheme 3, the alkyl bromides (C–F) were refluxed with corresponding amines (added in excess) to give compounds 3a–3t and 4a–4l. Compound 4 m was synthesised by direct coupling of acetic acid with compound 4h using EDC in the presence of DMAP that acts as a base (Scheme 4).Citation40
Scheme 1. Reagents and conditions: (i) MeOH, RT, overnight, (ii) K3Fe(CN)6, 4 M NaOH, aq. isopropanol, RT, overnight, (iii) 1 M HCl, EtOH, RT, overnight, (iv) 2 equiv. of chloro alkyl amine hydrochloride, 4 equiv. of K2CO3, KI, acetone, reflux, overnight.
Scheme 2. Reagents and conditions: (i) 3 equiv. of α,ω-dibromoalkane, 4 equiv. of K2CO3, KI, acetone, reflux, overnight, (ii) 3 equiv. of the methyl ester of the aromatic amino acid, MeOH, reflux, overnight.
Scheme 3. Reagents and conditions: (i) 3 equiv. of α,ω-dibromoalkane, 4 equiv. of K2CO3, KI, acetone, reflux, overnight, (ii) 3 equiv. of appropriate amine, MeOH, reflux, overnight, (iii) TFA, DCM, 0 °C, RT, 2 h for Boc-protected derivative.
Scheme 4. Reagents and conditions: (i) 4 equiv. of glacial acetic acid, 4 equiv. of DMAP, 4 equiv. of EDC in DCM and DMF in ratio 1:1, RT, overnight.
Biological evaluation
Initial screening was done to all compounds shown in in which the spacer length was varied against hH1R (10 µM), hH4R (10 µM) and hH3R (1 µM) in a 1-point radioligand displacement assays. Ki values were determined for the most promising compounds. Additionally, the compounds were screened against AChE at 10 µM, compounds that showed percent inhibition above 50 as threshold were selected for IC50 value determination. Compounds from the 2nd stage in which the 3-methylene spacer was only adopted (4a–4m) were screened against H1R (1 µM), H3R and AChE as shown in .
Table 1. Screening at H1R, H3R, H4R, and AChE by the first cluster of compounds (1a–1e).
Table 2. Inhibition of H1R, H3R, H4R and AChE by the 2nd cluster of compounds (2a–2e).
Table 3. Inhibition of H1R, H3R, H4R and AChE by 3rd cluster of compounds (3a–3t). 
Table 4. Inhibition of H3R, and AChE (compounds 4a–4m). 
Human histamine hH3R affinity
We tried attaching an alkyl ether ending with diverse basic heads to the benzothiazole core and an ester or amide in the eastern arbitrary region in the presented series ().
An alicyclic amine as the basic head, a 2-carbon spacer, and short alkyl esters at the eastern arbitrary region were used in the first cluster of compounds. However, these analogues (1a–1e, ) showed weak ligand displacement at H3R at 1 µM of the tested compound (% inhibition 25–63%).
Next, in the 2nd cluster of compounds we tried aromatic l-amino acid esters at the basic head (l-Tyr, l-Trp and l-PhGly), while varying the spacer length between 3–5 carbons and methyl ester at the eastern part of the molecule. Like the first cluster of compounds, none of the adopted changes led to potent compounds at H3R ().
In the 3rd group of compounds, we adopted a synthetic strategy in which the same terminal basic group in the western part of the molecule, was also used in the eastern arbitrary region but incorporated via an amide linkage to the benzothiazole core (). This cluster includes several derivatives with variable spacer length (n = 2–5).
Effect of the spacer length on H3R affinity
Generally, we found that compounds with 3- and 5-carbon spacers between the basic amine nitrogen and the ether oxygen provide higher affinity to H3R compared to compounds with 2- and 4-carbon spacers. For instance, in piperidine and pyrrolidine derivatives, the alkyl spacer with 3 or 5 methylene groups (compounds 3d, 3o, 3h, 3s) was statistically at the same affinity (Ki = 0.074, 0.068, 0.038, and 0.036 μM, respectively) than those with 4 methylene groups (compounds 3j, 3n) with Ki values 0.17 and 0.059 μM, respectively. The 2 methylene spacer (3a, 3b) showed the lowest affinity with Ki values of >1 µM for 3a and 0.64 μM for 3b. Based on this, in our last stage of synthesis, we made more diversification in the basic head focussed only on compounds having a 3-carbon spacer, since it provides the so far best balance in the gain of affinity and lipophilicity ().
The basic amine head
Generally, the 3ry alicyclic amines like pyrrolidine and piperidine demonstrated higher affinity to H3R than the 2ry cycloalkyl amines, which is in accordance with previous investigations.Citation34 It can be protonated for ionic interactions at physiological pH values (cf. ).
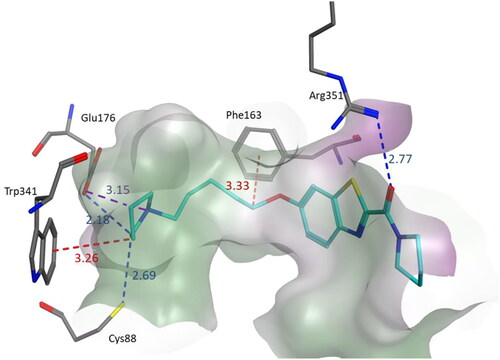
Figure 3. Binding mode of compound 3s within the homology model of H3R. 3s was docked into the active site using MOE. In the binding model, compound 3s formed three hydrogen bond interactions with Glu176, Cys88, and Arg351, a salt bridge with Glu176 and two CH–π interactions involving Trp341 and Phe163. Interactions are indicated by dashed lines and distances between the heavy atoms are given in Å.
Pyrrolidine analogues
The use of pyrrolidine afforded the ligands with the highest affinity to H3R in the present series. Compound 3s (5C spacer) showed the highest affinity with Ki value 0.036 μM. Decreasing the spacer length to 3 and 4 methylene groups led to a close inhibitory activity with Ki values 0.038 μM for 3h and 0.059 μM for 3n. While compound 3b with two methylene groups showed a 10-fold decrease in affinity (Ki = 0.64 μM) (). In compound 4a having the 1-(2-aminoethyl) pyrrolidine group as the basic part, it only slightly reduced the activity (Ki = 0.053 μM, ) compared to the pyrrolidine analogue (3h) indicating that having two basic nitrogen atoms can be tolerated in this part of the molecule.
Piperidine analogues
Piperidine ring was also tried as a basic amine. Compounds 3d, 3j, 3o with 3-, 4- and 5-carbon spacers showed good inhibitory activity with Ki = 0.074, 0.17, 0.068 μM, respectively (). Obeying the general trend, compound 3a with a 2-carbon spacer showed the lowest affinity to H3R with Ki >1 μM. Introduction of a mono methyl substituent at position 3 of the piperidine moiety (4k) seemed to be more tolerated than at position 4 (4l). Compound 4k showed the highest affinity among all compounds bearing the substituted piperidine moiety with Ki value of 0.064 μM (). Also, fusing a benzene ring to the piperidine ring as in the tetrahydro-isoquinoline derivative 4e led to a clear reduction in the affinity with Ki value of 0.34 μM ().
Piperazine analogues
The piperazine ring was tried as a basic head to see the influence of having two basic nitrogen atoms at the western part of the compound. First, the 1-ethyl and 1-methyl piperazine derivatives were used in compounds 4f and 4g. Compound 4f showed moderate inhibitory activity with Ki value of 0.43 µM while compound 4g showed weak inhibitory activity with Ki value of >1 μM (). Also, we tried 3-methyl piperazine and 1-(2-hydroxyethyl) piperazine to yield compounds 4h and 4i respectively. The inhibition by 4h was close to 4f with Ki value of 0.47 µM while 4i showed a 4-fold boost in the affinity with Ki value of 0.12 μM. Moreover, acetylation of 4h was done to give 4m, which showed Ki value of 0.4 μM, similar to the values of 4f and 4h ().
Azepane ring
In compound 4b, the azepane ring was tried as the basic part (with a 3-carbon spacer). This compound showed the highest affinity towards H3R in the present series with a Ki value of 0.012 μM. The increased lipophilicity may contribute to this effect.
Secondary amines
The effect of replacing the 3ry alicyclic amine as a basic centre with different 2ry amines was also examined. Cyclopentyl amino derivatives showed low potency, e.g. 3t, which has a 5-carbon spacer, only showed a marginal Ki value of >1 µM (). The same applies for the cyclohexyl methyl amino derivative 3q (also having 5-carbon spacer) with a Ki value >3 µM. Ring expansion to give cycloheptyl amino (3e, 3k, 3p) and ring contraction to cyclopropyl amino (3 g, 3m, 3r) did not recover the activity towards H3R regardless of the spacer length. The use of aralkyl amines like the phenyl ethyl amino 4d and benzyl amino 4j led to ligands with Ki values >1 and 0.64 μM respectively ().
Selectivity
Within the group of histamine receptors, subtype H3R shows the highest sequence homology to the histamine H4 receptor (H4R). It was therefore essential to test newly developed histamine H3R ligands for their potential interaction with this most closely related subtype of the histamine receptor family (testing was performed at 10 μM), since modulation of H3Rs is more specific towards neurodegenerative diseases while H4Rs mainly have role in inflammation and immune response. Based on the obtained data, it was shown that the active compounds at H3R did not possess remarkable affinities at the H4R. Additionally, to exclude side effects of first-generation sedating H1 antagonists which readily cross the blood–brain barrier, leading to drowsiness, fatigue, and psychomotor disturbances, the affinity at the H1R was screened mostly at a concentration of 10 μM and was, except for one compound (3o), considered negligible ().
Cholinesterase inhibitory activity
AChE inhibition
To achieve our aim to find MTDLs for Alzheimer’s disease, all compounds were tested against human AChE as shown in . The compounds were tested at 10 µM screening concentration then IC50 values were determined for compounds showing higher than 50% inhibition at 10 μM. Similar to the activity towards H3R, pyrrolidine derivatives were among the most potent compounds against AChE regardless of the spacer length. All compounds having pyrrolidine showed % inhibition of more than 50% at 10 µM, however, the strongest inhibitor was found to be compound 3b bearing the 2 methylene groups with an IC50 value of 0.44 µM. Interestingly, and unlike what was observed towards H3R, the 2nd best pyrrolidine derivative is compound 3n with 4-carbon spacer (IC50 = 1.3 µM), indicating that spacer length of two and four methylene groups ( and ) was generally superior for AChE inhibition.
Compound 4b, the most affine compound to H3R, having azepanyl head and 3-carbon spacer showed the ability to inhibit AChE with IC50 = 5.91 µM. This compound showed higher potency towards AChE than the respective pyrrolidine analogue 3h. Piperidine derivatives showed potency only with the 4-carbon spacer in compound 3j with IC50 = 8.56 µM. A remarkable drop in inhibitory activity towards AChE occurred in all the secondary amines except cyclopentyl amino at spacer 3 and 5 (compounds 3i and 3t) which showed IC50 values of 7.31 and 3.55 µM, respectively. Compounds 1c (pyrrolidine derivative) and 1e (piperidine derivative) which have simple ethyl and methyl esters respectively (2 carbon spacer) showed AChE inhibitory activity (IC50 = 7.63 µM, 2.92 µM respectively). This confirms the superiority of the 2-carbon spacer as a favourable requirement for the AChE inhibitory activity.
BuChE inhibition
As for BuChE, only compounds with dual activity towards H3R and AChE were tested for their inhibitory activity towards this enzyme in addition to three compounds (3d, 3o, and 4k) with very good activity towards H3R with Ki values 0.074, 0.068, and 0.064 µM, respectively (). Most of the tested compounds showed inhibitory activity towards BuChE in the micromolar range with IC50 values below 5.5 µM. Interestingly, compound 4a having 1-(2-amino ethyl) pyrrolidine ring and three carbon spacer showed the highest inhibitory activity with IC50 of 0.169 µM.
Table 5. Inhibition of selected compounds at BuChE and MAO-A and B enzymes.
MAO-A and B inhibition
The activity of MAO-B enzyme is increased in the cortical areas and in the hippocampus of the brains of patients with AD. This overexpression of MAO-B leads to stimulation of free radical formation and thus may feasibly advance the neurodegenerative mechanisms occurring in AD. Therefore, many new MAO-B inhibitors are designed and developed as potential disease-modifying AD drugs.Citation41 On the other hand MAO-A was found to increase in the hypothalamus and frontal pole. MAO-A activity appears to be lower in the locus ceruleus in patients with AD, and is accompanied by an approximately 80% decrease in the number of neurons, revealing that activated MAO-A in neurons is involved in the pathology of this disease as a predisposing factor.Citation42 Consequently, the same set of compounds selected for testing against BuChE was further tested against MAO-A and -B enzymes (). Only compounds having piperidine and pyrrolidine as a basic head with 5-carbon spacer (compounds 3o and 3s) displayed significant and selective MAO-B inhibitory activity (>60% inhibition at 10 µM) with IC50 values 3.04 and 1.6 µM, respectively.
Molecular modeling
Docking studies on H3R
Among the four known histamine receptor subtypes, crystal structure only for the histamine H1 receptor was known up to date (PDB ID: 3RZE).Citation43 Therefore, due to the lack of crystal structures, homology modelling methods are used to search for new H3R ligands and to explain SAR. Using the prepared homology model,Citation44 we made an attempt to demonstrate how the tested compounds interacted with the H3R active site. We chose one of the most active H3R ligands 3s (Ki = 0.036 µM), to present the observed interactions. The homology model was kindly provided by Marek Bajda group.Citation44 Compound 3s interacts with the homology model through different types of interactions () described as follows: (1) The protonated pyrrolidine nitrogen interacts with Glu176 through ionic interaction, (2) One of the methylene groups in the spacer interacts with Phe163 through CH–π interaction, (3) The CH groups in the pyrrolidine ring formed two hydrogen bonds involving Glu176 (CH….O interaction) and Cys88 (CH….S interaction) and one CH–π interaction involving Trp341, 4) The carbonyl group in the eastern arbitrary region interacts with Arg351 through hydrogen bond interaction.
The overall binding mode apparently resembles that described for the known H3R inverse agonists such as ABT-239, JNJ5207852, and others.Citation44 These inverse agonists contain in their structure nitrogen atom capable of protonation at physiological pH values. The docking results suggested that the ionic interaction with Glu176 in the active site is essential for efficient binding of non-imidazole H3 antagonists. Compound 3s like the other antagonists showed this type of interaction through the protonated nitrogen atom in the pyrrolidine ring. The overall interactions including ionic, hydrogen bond and hydrophobic interactions with their appropriate distances led to the high affinity of this compound at H3R.
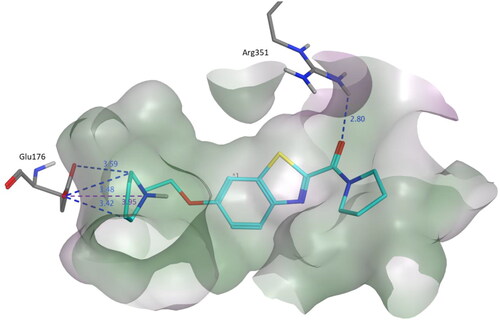
Upon comparing the docking results of the most potent pyrrolidine derivative 3s (Ki = 0.036 µM) with the least potent 3b (Ki = 0.64 µM), it is clear that the number of interactions is lower in 3b than 3s. In 3b, only two amino acids (Glu176 and Arg351) were shown to interact with 3b. In addition, the distance between the amino acids and the compound is larger in case of 3b since the distance of the ionic interaction in 3b was 3.95 Å while in 3s it was shorter (3.15 Å). Also, the distance of the hydrogen bonds formed with Glu176 was greater in case of 3b ().
Figure 4. Binding mode of compound 3b within the homology model of H3R. 3b was docked into the active site using MOE. In the binding model, compound 3b formed three hydrogen bond interactions with Glu176, a salt bridge with Glu176 and one hydrogen bond with Arg351. Dashed lines indicated the interactions and distances between the heavy atoms are given in Å.
Docking studies on AChE enzyme
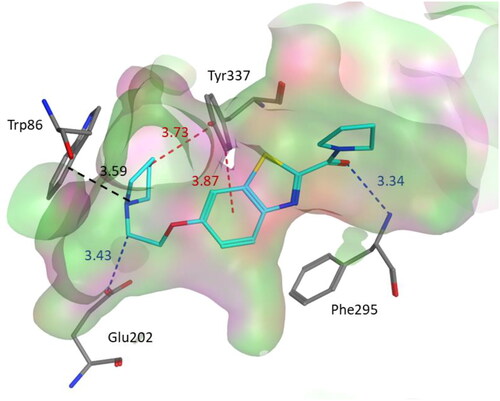
In an attempt to get a better insight into the binding mode of our most potent inhibitor against human AChE 3b (IC50 = 0.44 μM), molecular docking was performed at AChE binding pocket (PDB code: 4EY5) using MOE. As depicted in , 3b binds to AChE binding site in a way analogous to the cocrystallized ligand (Huperzine A) with a score −43.3102 and rmsd 1.4070. Compound 3b was extended along the active gorge. The pyrrolidine ring was located close to the amino acid residues from the anionic site and the catalytic triad.
Figure 5. Binding mode of compound 3b within the AChE active site (PDB code: 4EY5). 3b was docked into the active site of AChE using MOE. In the binding model, compound 3b is anchored between amino acids Phe295, Tyr337, Glu202 and Trp86. It formed cation-pi interaction with Trp86. In addition, we found two CH–π interactions involving Tyr337 and two hydrogen bond interactions with Glu202 and Phe295. Dashed lines indicate the interactions and distances between the heavy atoms are given in Å.
Docking studies on BuChE enzyme
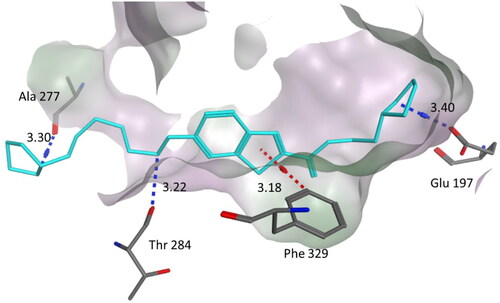
Compound 4a was docked to the active site of human BuChE. The binding mode of the active compound 4a, which revealed significant hBuChE inhibitory activity with IC50 0.169 μM is shown in .
Figure 6. Binding mode of compound 4a within the BuChE active site (PDB code: 1POI). 4a was docked into the active site of BuChE using MOE. In the binding model, compound 4a is anchored between amino acids Phe329, Thr284, Ala277 and Glu197. It formed three hydrogen bond interactions involving Glu197, Ala277 and Thr284. In addition one CH–π interaction with Phe329. Dashed lines indicate the interactions and distances between the heavy atoms are given in Å.
In-silico evaluation of drug-like properties
Since the major indication of these compounds is the treatment of AD, it seemed interesting to assess the ability of the most potent and selective novel analogues to pass BBB. Different physicochemical properties associated with a high probability of CNS penetration have been previously reported in the literature.Citation45,Citation46 CNS-active drug candidates are assumed to have definite attributes exemplified in: molecular weight <450, solubility >60 µg/ml, logP <5, HBA <7 and HBD <3. The molecule topological polar surface area (TPSA) is one of the concepts used to assess drug-likeness and ability to cross BBB where its ideal value is less than 70 Å2. However, the upper limit of PSA for a molecule to penetrate CNS was around 90 Å2.Citation45 Therefore, different physicochemical parameters of several potent inhibitors with dual activity from the current series were calculated using MOE software as shown in to estimate their drug-likeness. The calculated parameters for most of our potent compounds were within the ideal ranges for brain penetration as reported in the literature. In particular, compounds 3b, 3h, and 3n have reduced logP, logD, and low molecular weight. These values would likely allow BBB penetration. Also, those compounds showed an advantageous enhancement of solubility. Altogether, a high possibility for brain penetration for the highly potent and selective compounds in the current work was anticipated.
Table 6. Calculated physicochemical properties of compounds (3b, 3h, 3j, 3n, 3s, 3t, 4a, 4b)*.
Conclusion
In this work, we present benzothiazole derivatives as multi-target ligands to efficiently treat AD. The presented derivatives have a benzothiazole-based central core, which is connected to piperidine, pyrrolidine, azepane and other rings via an alkoxy linker. H3R affinity was determined by radioligand displacements assay against [3H] N-methylhistamine. Several of the presented compounds reached Ki values in the nanomolar range. The most affine compound, the azepane-linked benzothiazole derivative displayed a Ki value of 0.012 μM. The linker length had a slight influence on H3R affinity in that the ethoxy linker was the least favourite linker. Additionally, affinity at the H4 receptor was tested against [3H]-histamine, but no notable receptor inhibition at 10 µM was detected. To exclude side effects, the affinity at the H1 receptor was determined against [3H]-pyrilamine and was considered negligible. Upon examining the inhibitory activity of all our compounds towards AChE, some of them showed good potency towards the inhibition of these enzymes with IC50 values in the low or sub-micromolar range. Moreover, some of the compounds were tested towards BuChE and showed inhibitory activity towards the enzyme in the micromolar range with IC50 values below 5.5 µM. Among the 42 synthesised compounds, we have 6 compounds (3b, 3j, 3n, 3s, 3t, and 4a) that can be described as MTDL with affinities for hH3R and inhibitory activity for AChE and BuChE. These structures using the benzothiazole moiety as a privileged scaffold serve as a starting point for further structural optimizations to improve MTDL pharmacological properties and to yield potential drugs for the treatment of neurodegenerative diseases.
Experimental section
Chemistry
Solvents and reagents were purchased from commercial suppliers and used as received. 1H NMR and 13C NMR spectra were recorded by a Bruker DRX 500 spectrometer, Bruker Fourier 300 as well as Varian 400 spectrometer. 1H shifts are referenced to the residual protonated solvent signal (δ 2.50 for DMSO-d6), (δ 7.24 for CDCl3), (δ 4.87 for CD3OD) and 13C shifts are referenced to the deuterated solvent signal (δ 39.5 for DMSO-d6), (δ 77.0 for CDCl3), (δ 49.5 for CD3OD). Chemical shifts are given in parts per million (ppm), and all coupling constants (J) are given in Hz. All final compounds have a percentage purity of at least 95%, and this was verified utilising HPLC coupled with mass spectrometry. Mass spectrometric analysis (UHPLC-ESI-MS) was done utilising Waters ACQUITY Xevo TQD framework, which consisted of an ACQUITY UPLC H-Class system and XevoTM TQD triple-quadrupole tandem mass spectrometer with an electrospray ionisation (ESI) interface (Waters Corp., Milford, MA, USA). Acquity BEH C18 50 mm × 2.1 mm column (particle size, 1.7 µm) was utilised to separate analytes (Waters, Ireland). Two LC methods were used. Method I: The percentage of B started at an initial of 5% and maintained for 1 min, then increased up to 100% during 10 min, kept at 100% for 2 min, and flushed back to 5% in 3 min then kept at 5% for 1 min. Method II: The solvent system consisted of water containing 0.1% formic acid (A) and 0.1% formic acid in acetonitrile (B). HPLC was used for purity determination, HPLC-technique: flow rate 400 μL/min. The percentage of B started at an initial of 5% and maintained for 0.5 min, then increased up to 100% for 4 min, kept at 100% for 2 min, and flushed back to 5% in 1 min. The MS scan was carried out at the accompanying conditions: capillary voltage 3.5 kV, cone voltage 20 V, radio frequency (RF) lens voltage 2.5 V, source temperature 150 °C and desolvation gas temperature 500 °C. Nitrogen was used as the desolvation and cone gas at a flow rate of 1000 and 20 L/h, respectively. System operation and data acquisition were controlled using Mass Lynx 4.1 software (Waters). Full-scan HRMS experiments were done with a dual electrospray interface (ESI) and a quadrupole time-of-flight mass spectrometer (Q-TOF, Agilent 6530 Series, Agilent Technologies, Cernusco s. N., Italy). Spectra were recorded in the mass/charge (m/z) range 50 − 3000 Da. Melting points were obtained using a Buchi B-540 melting point apparatus and are uncorrected.
General synthetic methods and experimental details
General procedure for the synthesis of 6-hydroxybenzothiazole-2-carboxylic acid ethyl and methyl ester (A, B)
A solution of 1, 4-benzoquinone (5 mmol) in MeOH (20 ml) was added dropwise to a solution of L-cysteine ethyl or methyl ester hydrochloride (5 mmol) in MeOH (20 ml) with stirring at room temperature overnight. The reaction was monitored by TLC (petroleum ether/EtOAc, 6/4) and upon the reaction completion, the solvent was evaporated. The residue was dissolved in isopropanol (50 ml) then an aqueous solution of 1 M K3Fe(CN)6 (20 ml) and 4 M NaOH (2.5 ml) were added, respectively. The mixture was stirred at room temperature overnight. At the end of the reaction (known by TLC monitoring), the solid was removed by filtration and the solution was extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4 and concentrated in vacuo. To a solution of the obtained benzothiazine in EtOH (20 ml), HCl (1 M, 12 ml) was added. After stirring for 24 h at room temperature, H2O was added and the mixture was extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure, and the resulting residue was purified by column chromatography (CC) using (EtOAc/petroleum ether, 4:6) as a solvent system to give compounds A and B.
6-Hydroxybenzothiazole-2-carboxylic acid methyl ester (a)
Synthesised according to the method outlined above using L-cysteine methyl ester hydrochloride; brown solid; yield: 50.87%; mp 90.2–90.8 °C; MS (ESI): m/z = 210 (M + H)+.Citation39
6-Hydroxybenzothiazole-2-carboxylic acid ethyl ester (B)
Synthesised according to the method outlined above using L-cysteine ethyl ester hydrochloride. The product was purified by CC (EtOAc: Petroleum ether, 4:6); yellow solid; yield: 60%; mp 90.6–90.9 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.00 (d, J = 8.9 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 7.09 (dd, J = 8.9, 2.4 Hz, 1H), 4.39 (q, J = 7.1 Hz, 2H), 1.33 (t, J = 7.1 Hz, 3H), 1.20 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 160.63, 158.19, 154.42, 146.82, 138.60, 126.21, 118.20, 107.04, 62.77, 14.48; MS (ESI): m/z = 224.03 (M + H)+.
General procedure for the synthesis of compounds (1a–1e)
Different derivatives of chloro ethyl amine hydrochloride (4 mmol) were added to a solution of 6-hydroxybenzothiazole ester (0.446 g, 2 mmol) in acetone (20 ml). Then (1.1 g, 8 mmol) potassium carbonate and a few specs of potassium iodide were added to the mixture and heated to reflux for 6 h. Afterward, the mixture was concentrated in vacuo. The residue was partitioned between ethyl acetate and distilled water, the aqueous layer was re-extracted by ethyl acetate. The organic layers were collected and the volume was reduced under reduced pressure. Afterward, the product was purified by CC to yield compounds (1a–1e).
Ethyl 6-(2-morpholinoethoxy)benzo[d]thiazole-2-carboxylate (1a)
The title compound was synthesised according to the procedure in Section “General procedure for the synthesis of compounds (1a–1e)”, using 4–(2-chloroethyl) morpholine hydrochloride. The product was purified by CC (DCM:MeOH:TEA, 100:1:1); light brown solid; yield: 46.3%; mp 128.4–128.8 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.10 (d, J = 9.1 Hz, 1H), 7.82 (d, J = 2.4 Hz, 1H), 7.26 (dd, J = 9.1, 2.5 Hz, 1H), 4.43 (q, J = 7.1 Hz, 2H), 4.22 (t, J = 5.7 Hz, 2H), 3.64 − 3.56 (m, 4H), 2.77 (t, J = 5.6 Hz, 2H), 2.58 − 2.47 (m, 4H), 1.37 (t, J = 7.1 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δ 160.56, 158.83, 155.65, 147.69, 138.56, 126.08, 118.50, 105.67, 66.56, 66.44, 62.94, 57.25, 54.00, 14.51; MS (ESI): m/z = 337.11 (M + H)+.
Ethyl 6-(2-(piperidin-1-yl)ethoxy)benzo[d]thiazole-2-carboxylate (1b)
The title compound was synthesised according to the procedure in Section “General procedure for the synthesis of compounds (1a–1e)”, using 1–(2-chloroethyl) piperidine hydrochloride. The product was purified by CC (DCM:MeOH:TEA, 100:3:1); yellow semisolid; yield: 35.2%; 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 9.1 Hz, 1H), 7.36 (d, J = 2.3 Hz, 1H), 7.15 (dd, J = 9.1, 2.4 Hz, 1H), 4.52 (q, J = 7.1 Hz, 2H), 4.30 (t, J = 5.3 Hz, 2H), 3.01 (s, 2H), 2.74 (s, 4H), 1.77 − 1.70 (m, 4H), 1.51 (d, J = 4.7 Hz, 2H), 1.46 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 160.67, 158.23, 155.95, 147.99, 138.61, 126.19, 117.80, 104.35, 65.74, 62.92, 57.34, 54.84, 24.96, 23.44, 14.29; MS (ESI): m/z = 335.14 (M + H)+.
Ethyl 6-(2-(pyrrolidin-1-yl)ethoxy)benzo[d]thiazole-2-carboxylate (1c)
The title compound was synthesised according to the procedure in Section “General procedure for the synthesis of compounds (1a–1e)”, using 1–(2-chloroethyl) pyrrolidine hydrochloride. The product was purified by CC (DCM:MeOH:TEA, 100:4:1); yellow semisolid; yield: 23.5%; 1H NMR (300 MHz, DMSO-d6) δ 7.59 (d, J = 8.9 Hz, 1H), 7.27 (d, J = 2.4 Hz, 1H), 6.87 (dd, J = 8.9, 2.5 Hz, 1H), 4.47 − 4.40 (m, 2H), 4.29 (t, J = 4.5 Hz, 2H), 3.59 (s, 4H), 3.52 − 3.41 (m, 4H), 2.17 − 1.92 (m, 5H); 13C NMR (75 MHz, DMSO-d6) δ 168.34, 163.00, 156.18, 148.22, 138.15, 124.47, 115.96, 105.09, 64.71, 54.51, 54.18, 53.35, 23.10, 14.20; MS (ESI): m/z = 335.14 (M + H)+.
Methyl 6-(2-morpholinoethoxy)benzo[d]thiazole-2-carboxylate (1d)
The title compound was synthesised according to the procedure in Section “General procedure for the synthesis of compounds (1a–1e)”, using 4-(2-chloroethyl) morpholine hydrochloride. The product was purified by CC (DCM:MeOH:TEA, 100:3:1); brown solid; yield: 44.8%; mp 139.2–139.5 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.09 (d, J = 9.1 Hz, 1H), 7.81 (d, J = 2.5 Hz, 1H), 7.26 (dd, J = 9.1, 2.5 Hz, 1H), 4.21 (t, J = 5.7 Hz, 2H), 3.97 (s, 3H), 3.63 − 3.56 (m, 4H), 2.77 (t, J = 5.6 Hz, 2H), 2.51 (t, J = 3.7 Hz, 4H); 13C NMR (75 MHz, DMSO-d6) δ 160.94, 158.86, 155.23, 147.65, 138.54, 126.07, 118.54, 105.65, 66.57, 66.47, 57.25, 54.00, 53.81; MS (ESI): m/z = 323.1 (M + H)+.
Methyl 6-(2-(piperidin-1-yl)ethoxy)benzo[d]thiazole-2-carboxylate (1e)
The title compound was synthesised according to the procedure in Section “General procedure for the synthesis of compounds (1a–1e)”, using 1-(2-chloroethyl) piperidine hydrochloride. The product was purified by CC (DCM:MeOH:TEA, 100:3:1); yellowish white solid; yield: 51.6%; mp 121–121.5 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.07 (d, J = 9.0 Hz, 1H), 7.80 (d, J = 2.0 Hz, 1H), 7.23 (dd, J = 8.9, 2.3 Hz, 1H), 4.17 (t, J = 5.2 Hz, 2H), 3.94 (s, 3H), 2.72 (s, 2H), 2.47 − 2.44 (m, 4H), 1.49 (s, J = 50.0 Hz, 4H), 1.36 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 160.91, 158.88, 155.19, 147.60, 138.52, 126.03, 118.53, 105.66, 66.68, 57.50, 54.73, 53.77, 25.87, 24.23; MS (ESI): m/z = 321.1 (M + H)+.
General procedure for preparing compounds (2a–2e)
Preparation of the amino acid methyl ester
A 250 ml round bottom flask containing methanol (30 ml), was cooled in an ice bath, then the solid amino acid (10 mmol) was added in one portion. The solution was stirred for 5 min, then acetyl chloride (4 ml) was added dropwise using a dropping funnel over 15 min, the solution was then heated to reflux for 5 h, allowed to cool to room temperature and the solvent was removed under reduced pressure to give the methyl ester hydrochloride of the amino acid. The free base was obtained by adding dil. NH4OH and extraction with CH2Cl2.The organic layer was dried over anhydrous MgSO4, evaporated under reduced pressure and was used in the next step without further purification.
Preparation of compounds (C–F)
A suspension of methyl 6-hydroxybenzothiazole-2-carboxylate (compound A) (2 mmol), appropriate alpha, omega-dibromoalkanes (6 mmol), anhydrous K2CO3 (8 mmol) and few specs of potassium iodide in acetone (30 ml) was stirred at 80 °C for 6 h. The suspension was partitioned between ethyl acetate and distilled water. The combined organic phase was dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography over silica gel using methylene chloride as eluent to give the product a solid (C–F). The LC-MS confirmed the purity and the masses of these compounds and they were used in the next step without further elucidation.
Preparation of compounds (2a–2e)
To a solution of the compounds (C–F) dissolved in methanol (20 ml), the methyl ester of the amino acid (3 equiv.) was added and the reaction mixture was left to reflux overnight. The solvent was evaporated in vacuo and extraction was done using methylene chloride and water. The organic layer was dried over anhydrous Na2SO4, concentrated in vacuo and the residue was purified by CC.
Methyl (S)-6-(3-((3-(4-hydroxyphenyl)-1-methoxy-1-oxopropan-2-yl)amino)propoxy)benzo[d]thiazole-2-carboxylate (2a)
The title compound was synthesised according to the procedure in Section “General Procedure for preparing compounds (2a–2e)”, using methyl ester of L-tyrosine amino acid and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:1); yellow semisolid; yield: 47.9%; 1H NMR (500 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.08 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.20 (dd, J = 8.5, 2.1 Hz, 1H), 6.94 (d, J = 7.8 Hz, 2H), 6.62 (d, J = 8.5 Hz, 2H), 5.76 (s, 1H), 4.08 (t, J = 6.3 Hz, 2H), 3.96 (s, 3H), 3.53 (s, 3H), 3.37 (t, J = 6.7 Hz, 1H), 2.78 − 2.65 (m, 3H), 2.55 (dd, J = 12.4, 5.8 Hz, 1H), 1.84 (dd, J = 13.0, 6.5 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ 174.53, 160.48, 158.58, 155.78, 154.66, 147.08, 138.09, 129.97, 127.67, 125.55, 118.01, 114.90, 105.01, 66.51, 62.89, 53.30, 51.13, 43.80, 37.93, 28.95; MS (ESI): m/z = 445.12 (M + H)+.
Methyl (S)-6-(3-((3-(1H-indol-2-yl)-1-methoxy-1-oxopropan-2-yl)amino)propoxy)benzo[d]thiazole-2-carboxylate (2b)
The title compound was synthesised according to the procedure in Section “General Procedure for preparing compounds (2a–2e)”, using methyl ester of L-tryptophan amino acid and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:1.5); orange semisolid; yield: 8.3%; 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.05 (d, J = 9.0 Hz, 1H), 7.68 (d, J = 2.2 Hz, 1H), 7.45 (d, J = 7.9 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.18 − 7.15 (m, 1H), 7.09 (d, J = 1.5 Hz, 1H), 7.02 (d, J = 7.6 Hz, 1H), 6.93 (t, J = 7.3 Hz, 1H), 4.08 (t, J = 6.2 Hz, 2H), 3.95 (s, 3H), 2.98 (d, J = 6.6 Hz, 3H), 2.72 − 2.65 (m, 1H), 2.61 − 2.54 (m, 1H), 1.88 − 1.81 (m, 3H), 1.22 (s, 3H); MS (ESI): m/z = 468.12 (M + H)+.
Methyl 2-(6-(3-(((R)-2-methoxy-2-oxo-1-phenylethyl)amino)propoxy)benzo[d]thiazole-2-carboxamido)-2-phenylacetate (2c)
The title compound was synthesised according to the procedure in Section “General Procedure for preparing compounds (2a–2e)”, using methyl ester of L-tyrosine amino acid and 1,4-dibromobutane. The product was purified by CC (DCM:MeOH, 100:1); yellow semisolid; yield: 74.8%; 1H NMR (500 MHz, DMSO-d6) δ 9.18 (s, 1H), 8.08 (d, J = 9.1 Hz, 1H), 7.77 (d, J = 2.5 Hz, 1H), 7.23 (dd, J = 9.0, 2.6 Hz, 1H), 6.95 − 6.93 (m, 2H), 6.64 − 6.62 (m, 2H), 4.05 (t, J = 6.5 Hz, 2H), 3.96 (s, 3H), 3.53 (s, 3H), 3.38 (dd, J = 9.1, 4.8 Hz, 1H), 2.75 − 2.70 (m, 2H), 2.55 (dt, J = 11.5, 7.1 Hz, 1H), 2.48 − 2.42 (m, 1H), 1.78 − 1.72 (m, 2H), 1.55 − 1.49 (m, 2H), 1.23 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 169.66, 160.47, 158.61, 155.78, 154.64, 147.07, 138.11, 129.95, 127.62, 125.57, 118.05, 114.92, 105.01, 68.16, 62.87, 53.30, 51.14, 46.73, 37.97, 26.19, 25.85; MS (ESI): m/z = 459.14 (M + H)+.
Methyl (R)-6-(4-((3-(4-hydroxyphenyl)-1-methoxy-1-oxopropan-2-yl)amino)butoxy)benzo[d]thiazole-2-carboxylate (2d)
The title compound was synthesised according to the procedure in Section “General Procedure for preparing compounds (2a–2e)”, using methyl ester of L-phenyl glycine amino acid and 1,4-dibromobutane. The product was purified by CC (DCM:MeOH, 100:2); brown semisolid; yield: 13.5%; 1H NMR (400 MHz, DMSO-d6) δ 8.07 (dd, J = 9.1, 2.8 Hz, 1H), 7.74 (d, J = 2.4 Hz, 1H), 7.59 (d, J = 7.3 Hz, 1H), 7.40 − 7.25 (m, 5H), 4.39 (dd, J = 13.3, 6.1 Hz, 1H), 4.05 (t, J = 6.3 Hz, 2H), 3.94 (s, 3H), 3.68 (s, 1H), 3.58 (s, 2H), 2.01 − 1.94 (m, 1H), 1.60 − 1.54 (m, 1H), 1.35 (t, J = 7.1 Hz, 1H), 1.22 (s, 4H); MS (ESI): m/z = 429.12 (M + H)+.
Methyl (R)-6-(4-((2-methoxy-2-oxo-1-phenylethyl)amino)butoxy)benzo[d]thiazole-2-carboxylate (2e)
The title compound was synthesised according to the procedure in Section “General Procedure for preparing compounds (2a–2e)”, using methyl ester of L-tyrosine amino acid and 1,5-dibromopentane. The product was purified by CC (DCM:MeOH, 100:2); yellow semisolid; yield: 21%; 1H NMR (500 MHz, DMSO-d6) δ 9.18 (s, 1H), 8.08 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 2.5 Hz, 1H), 7.23 (dd, J = 9.1, 2.5 Hz, 1H), 6.95 − 6.93 (m, 2H), 6.66 − 6.62 (m, 2H), 4.05 (t, J = 6.5 Hz, 2H), 3.96 (s, 3H), 3.53 (s, 3H), 3.36 (t, J = 5.9 Hz, 2H), 2.76 − 2.69 (m, 2H), 2.43 − 2.37 (m, 1H), 1.76 − 1.70 (m, 2H), 1.45 − 1.39 (m, 4H), 1.23 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 174.58, 160.48, 158.64, 155.78, 154.64, 147.07, 138.11, 129.94, 127.65, 125.57, 118.06, 114.91, 104.99, 68.24, 62.90, 53.30, 51.11, 47.04, 37.99, 29.05, 28.31, 23.13; MS (ESI): m/z = 473.15 (M + H)+.
General procedure for preparing compounds (3a–3t, 4a–4l)
A suspension of 6-hydroxy benzothiazole-2-carboxylic acid ester B (0.446 g, 2 mmol), 1, 2-dibromoalkanes (6 mmol), anhydrous K2CO3 (1.1 g, 8 mmol) and few specs of KI in acetone (30 ml) was stirred at 80 °C for 6 h. The suspension was partitioned between ethyl acetate and distilled water. The combined organic phase was dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography over silica gel using methylene chloride as an eluent to give the product as a solid. To a solution of this product dissolved in methanol (20 ml), the appropriate amine (3 equiv.) was added and the reaction mixture was left to reflux overnight. The solvent was evaporated in vacuo and extraction was done using methylene chloride and water. The organic layer was dried over anhydrousNa2SO4, concentrated in vacuo and the residue was purified by CC.
Piperidin-1-yl-(6-(2-(piperidin-1-yl)ethoxy)benzo[d]thiazol-2-yl)methanone (3a)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using piperidine and 1,2-dibromoethane. The product was purified by CC (DCM:MeOH, 100:7); white semisolid; yield: 15.7%; 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.16 (dd, J = 9.0, 2.6 Hz, 1H), 4.20 (s, 2H), 4.14 (t, J = 5.9,3.4 Hz, 2H), 3.63 (s, 2H), 2.67 (dd, J = 10.6, 4.7 Hz, 2H), 2.45 − 2.40 (m, 4H), 1.59 (d, J = 4.4 Hz, 4H), 1.48 (m, 5H), 1.40 − 1.35 (m, 2H), 1.21 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 160.48, 158.58, 155.78, 129.97, 125.55, 118.01, 114.90, 105.01, 66.51, 62.89, 53.30, 51.13, 43.80, 37.93, 28.95; MS (ESI): m/z = 374.2 (M + H)+.
Pyrrolidin-1-yl-(6-(2-(pyrrolidin-1-yl)ethoxy)benzo[d]thiazol-2-yl)methanone (3b)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using pyrrolidine and 1,2-dibromoethane. The product was purified by CC (DCM:MeOH, 100:7); white solid; yield: 21.4%; mp 79.1–79.7 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.00 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 2.5 Hz, 1H), 7.19 (dd, J = 9.0, 2.6 Hz, 1H), 4.19 (t, J = 5.8 Hz, 2H), 4.09 (t, J = 6.8 Hz, 2H), 3.56 (t, J = 6.9 Hz, 2H), 2.89 (m, 2H), 2.60 (m, 4H), 2.00 − 1.93 (m, 2H), 1.90 − 1.83 (m, 2H), 1.71 (m, 4H); 13C NMR (126 MHz, DMSO-d6) δ 163.04, 158.27, 157.65, 147.54, 137.28, 125.11, 117.10, 105.03, 54.05, 53.94, 48.53, 47.50, 26.02, 23.08; MS (ESI): m/z = 346.2 (M + H)+; HRMS (ESI): calculated for (C18H23N3O2S + Na+) 368.1404, found 368.1403 (M + Na)+.
N-(Cyclohexylmethyl)-6-(2-((cyclohexylmethyl)amino)ethoxy)benzo[d]thiazole-2-carboxamide (3c)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cyclohexyl methyl amine and 1,2-dibromoethane. The product was purified by CC (DCM:MeOH, 100:6); yellow semisolid; yield:10.4%; 1H NMR (500 MHz, DMSO-d6) δ 9.00 (t, J = 6.2 Hz, 1H), 7.99 (d, J = 9.0 Hz, 1H), 7.76 (d, J = 2.5 Hz, 1H), 7.21 (dd, J = 9.0, 2.6 Hz, 1H), 4.12 (t, J = 5.7 Hz, 2H), 3.13 (t, J = 6.6 Hz, 2H), 2.92 (t, J = 5.7 Hz, 2H), 2.44 (d, J = 6.7 Hz, 2H), 1.76 − 1.57 (m, 11H), 1.43 − 1.35 (m, 1H), 1.24 − 1.10 (m, 7H), 0.96 − 0.82 (m, 4H); 13C NMR (126 MHz, DMSO-d6) δ 162.02, 159.52, 157.70, 147.08, 137.85, 124.49, 117.36, 105.47, 67.96, 55.90, 48.18, 45.26, 37.40, 30.94, 30.39, 26.25, 26.01, 25.57, 25.32; MS (ESI): m/z = 430.24 (M + H)+.
Piperidin-1-yl-(6-(3-(piperidin-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (3d)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using piperidine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH:TEA, 100:5:1); brown solid; yield: 52.4%; mp 90.1–90.6 °C; 1H NMR (300 MHz, DMSO-d6) δ 7.98 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.17 (dd, J = 9.0, 2.6 Hz, 1H), 4.23 (t, J = 6.2 Hz, 2H), 4.11 (t, J = 6.3 Hz, 2H), 3.66 (t, J = 5.3 Hz, 2H), 2.64 − 2.53 (m, 4H), 2.02 − 1.91 (m, 2H), 1.69 − 1.51 (m, 11H), 1.42 (d, J = 5.1 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δ 162.80, 159.17, 158.23, 147.34, 137.63, 125.40, 117.57, 105.38, 66.93, 55.18, 54.18, 44.51, 26.83, 25.44, 24.42, 24.02; MS (ESI): m/z = 388.2 (M + H)+.
N-Cycloheptyl-6-(3-(cycloheptylamino)propoxy)benzo[d]thiazole-2-carboxamide (3e)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cycloheptyl amine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:6); brown solid; yield: 33%; mp 177.1–177.7 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.87 (d, J = 8.4 Hz, 1H), 8.02 (d, J = 9.0 Hz, 1H), 7.79 (d, J = 2.5 Hz, 1H), 7.22 (dd, J = 9.0, 2.5 Hz, 1H), 4.19 (t, J = 5.9 Hz, 2H), 4.02 − 3.92 (m, 1H), 3.27 − 3.18 (m, 1H), 3.15 − 3.06 (m, 2H), 2.21 − 2.00 (m, 4H), 1.85 (dd, J = 12.6, 5.5 Hz, 2H), 1.61 (m, 21H); 13C NMR (75 MHz, DMSO-d6) δ 162.89, 158.71, 157.87, 147.60, 138.29, 125.07, 117.82, 106.04, 65.91, 58.68, 51.31, 34.51, 30.65, 28.10, 27.85, 24.39, 23.80; MS (ESI): m/z = 444.4 (M + H)+.
N-(Cyclohexylmethyl)-6-(3-((cyclohexylmethyl)amino)propoxy)benzo[d]thiazole-2-carboxamide (3f)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cyclohexyl methyl amine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:7); white solid; yield: 26.2%; mp 227.2–228.2 °C; 1H NMR (300 MHz, DMSO-d6) δ 9.03 (t, J = 6.2 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 2.5 Hz, 1H), 7.20 (dd, J = 9.0, 2.5 Hz, 1H), 4.16 (t, J = 6.1 Hz, 2H), 3.14 (t, J = 6.5 Hz, 2H), 2.86 (t, J = 7.1 Hz, 2H), 2.56 (d, J = 6.8 Hz, 2H), 2.07 − 1.96 (m, 2H), 1.80 − 1.45 (m, 13H), 1.28 − 1.07 (m, 6H), 0.93 (dd, J = 23.3, 15.3 Hz, 4H); 13C NMR (75 MHz, DMSO-d6) δ 162.54, 159.99, 158.08, 147.57, 138.35, 125.01, 117.80, 105.90, 66.53, 55.11, 46.00, 45.76, 37.82, 36.57, 31.03, 27.75, 26.50, 26.43, 25.81; MS (ESI): m/z = 444.4 (M + H)+.
N-Cyclopropyl-6-(3-(cyclopropylamino)propoxy)benzo[d]thiazole-2-carboxamide (3g)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cyclopropyl amine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:6); brown semisolid; yield: 10.2%; 1H NMR (300 MHz, DMSO-d6) δ 8.89 (d, J = 4.8 Hz, 1H), 7.73 (d, J = 9.0 Hz, 1H), 7.53 (d, J = 2.5 Hz, 1H), 6.97 (dd, J = 9.0, 2.6 Hz, 1H), 3.92 (t, J = 6.1 Hz, 2H), 3.16 (d, J = 33.9 Hz, 2H), 2.84 − 2.63 (m, 2H), 1.77 (m, 2H), 0.49 − 0.36 (m, 8H), 0.13 (d, J = 41.7 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.35, 161.18, 158.00, 147.64, 138.30, 125.02, 117.82, 105.97, 66.24, 45.53, 30.24, 26.93, 23.61, 6.13, 4.44; MS (ESI): m/z = 332.12 (M + H)+.
Pyrrolidin-1-yl-(6-(3-(pyrrolidin-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (3h)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using pyrrolidine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:6); brown solid; yield: 22.6%; mp 190.2–190.9 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.03 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 2.5 Hz, 1H), 7.20 (dd, J = 9.0, 2.5 Hz, 1H), 4.19 (t, J = 6.0 Hz, 2H), 4.09 (t, J = 6.7 Hz, 2H), 3.57 (t, J = 6.8 Hz, 2H), 3.19 − 3.05 (m, 2H), 2.51 (dd, J = 3.6, 1.8 Hz, 2H) 2.21 (dd, J = 15.2, 6.0 Hz, 2H), 2.02 − 1.82 (m, 10H); 13C NMR (75 MHz, DMSO-d6) δ 163.71, 158.71, 157.91, 148.17, 137.73, 125.66, 117.55, 105.64, 65.99, 53.61, 51.72, 49.04, 48.02, 26.52, 25.67, 23.66, 23.16; MS (ESI): m/z = 360.2 (M + H)+; HRMS (ESI): calculated for (C19H25N3O2S + Na+) 382.1560, found 382.1591 (M + Na)+.
N-(2-(Pyrrolidin-1-yl)ethyl)-6-(3-((2-(pyrrolidin-1-yl)ethyl)amino)propoxy)benzo[d]thiazole-2-carboxamide (4a)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using 1-(2-aminoethyl) pyrrolidine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH:TEA, 100:0.5:0.5); white solid; yield: 21.2%; mp 120.5–120.8 °C; 1H NMR (400 MHz, CDCl3) δ 8.42 (t, J = 5.7 Hz, 1H), 8.15 (d, J = 7.6 Hz, 1H), 7.33 (d, J = 2.4 Hz, 1H), 7.13 (dd, J = 9.0, 2.5 Hz, 1H), 3.95 (q, J = 6.1 Hz, 2H), 3.57 (q, J = 7.3 Hz, 4H), 3.42 (m, 6H), 3.22 (d, J = 4.6 Hz, 5H), 2.10 (dd, J = 8.1, 5.3 Hz, 5H), 1.43 (dd, J = 9.2, 5.2 Hz, 7H), 1.22 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 158.03, 147.58, 138.55, 125.51, 118.15, 117.56, 106.71, 104.86, 69.36, 63.98, 54.48, 53.01, 46.00, 40.15, 23.30, 8.62, 7.99; MS (ESI): m/z = 446.2 (M + H)+; HRMS (ESI): calculated for (C23H35N5O2S + H+) 446.2582, found 446.2572 (M + H)+.
Azepan-1-yl-(6-(3-(azepan-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (4b)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using azepane and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:5); white solid; yield: 57.5%; mp 130.6–131.1 °C; 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 9.0 Hz, 1H), 7.32 (d, J = 2.3 Hz, 1H), 7.06 (dd, J = 9.0, 2.4 Hz, 1H), 4.21 − 4.16 (m, 2H), 4.14 (t, J = 5.2 Hz, 2H), 3.81 − 3.65 (m, 4H), 3.29 − 3.23 (m, 2H), 2.46 (d, J = 3.9 Hz, 2H), 2.10 − 1.59 (m, 18H); 13C NMR (101 MHz, CDCl3) δ 163.16, 160.95, 157.24, 148.22, 137.86, 125.38, 116.27, 104.44, 65.52, 64.15, 55.1, 54.66, 48.96, 48.43, 29.42, 27.1, 26.99, 26.90, 26.72, 24.44, 23.39; MS (ESI): m/z = 416.23 (M + H)+; HRMS (ESI): calculated for (C23H33N3O2S + Na+) 438.2184, found 438.2209 (M + Na)+.
(3,5-Dimethylpiperidin-1-yl)(6-(3-(3,5-dimethylpiperidin-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (4c)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using 3,5-dimethyl piperidine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:5); white solid; yield: 42%; mp 182.3–183.1 °C; 1H NMR (400 MHz, CDCl3) δ 7.97 − 7.93 (d, J = 2.6 Hz, 1H), 7.34 (d, J = 2.4 Hz, 1H), 7.07 (dd, J = 9.0, 2.5 Hz, 1H), 4.15 (t, J = 5.6 Hz, 2H), 3.75 (dd, J = 11.2, 3.6 Hz, 1H), 3.68 (dd, J = 11.2, 5.6 Hz, 1H), 3.41 (d, J = 9.7 Hz, 2H), 3.15 (s, 2H), 2.95 (s, 2H), 2.87 (s, 2H), 2.45 (s, 2H), 2.30 (dd, J = 26.7, 14.4 Hz, 3H), 2.14 (s, 1H), 1.90 (dd, J = 12.0, 10.4 Hz, 2H), 1.76 (d, J = 4.0 Hz, 2H), 0.94 (dd, J = 14.9, 6.6 Hz, 12H); 13C NMR (101 MHz, CDCl3) δ 162.90, 159.54, 157.32, 147.89, 137.82, 125.33, 116.35, 104.42, 65.67, 64.21, 53.35, 50.72, 42.43, 32.41, 31.32, 19.16, 18.79; MS (ESI): m/z = 444.26 (M + H)+.
N-Phenethyl-6-(3-(phenethylamino)propoxy)benzo[d]thiazole-2-carboxamide (4d)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using phenethyl amine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:1); grey solid; yield: 63.2%; mp 90.5–91 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.90 (t, J = 5.0 Hz, 1H), 7.77 (d, J = 9.0 Hz, 1H), 7.52 (s, 1H), 7.11 − 6.95 (m, 11H), 3.91 (t, J = 5.6 Hz, 2H), 3.36 − 3.30 (m, 2H), 3.08 (dd, J = 10.6, 5.6 Hz, 3H), 2.68 (t, J = 7.2 Hz, 2H), 2.30 (s, 3H), 1.67 (dd, J = 13.5, 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 162.16, 159.90, 158.26, 147.42, 140.88, 139.60, 138.32, 129.05, 128.80, 126.22, 117.83, 105.81, 72.93, 63.52, 51.52, 46.17, 36.30, 35.29, 29.43; MS (ESI): m/z = 460.2 (M + H)+.
(3,4-Dihydroisoquinolin-2(1H)-yl)(6-(3-(3,4-dihydroisoquinolin-2(1H)-yl)propoxy)benzo[d]thiazol-2-yl)methanone (4e)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using tetrahydroisoquinoline and 1,3 -dibromopropane. The product was purified by CC (DCM:MeOH, 100:1); white solid; yield: 76.6%; mp 134.2–134.7 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.02 (d, J = 8.4 Hz, 1H), 7.74 (s, 1H), 7.22 (d, J = 22.2 Hz, 5H), 7.05 (d, J = 14.3 Hz, 4H), 5.46 (s, 1H), 4.82 (s, 1H), 4.47 (s, 1H), 4.14 (t, J = 5.6 Hz, 2H), 3.88 (s, 1H), 3.56 (s, 2H), 2.94 (d, J = 17.3 Hz, 2H), 2.79 (s, 2H), 2.69 − 2.58 (m, 4H), 2.04 − 1.99 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 159.89, 158.44, 147.38, 137.72, 135.33, 134.88, 134.61, 133.14, 130.17, 128.94, 128.81, 127.05, 126.83, 126.73, 126.35, 125.86, 125.58, 125.51, 117.71, 105.37, 67.08, 56.05, 54.60, 50.96, 45.85, 44.22, 29.43, 29.16, 26.77; MS (ESI): m/z = 484.2 (M + H)+.
(4-Ethylpiperazin-1-yl)(6-(3-(4-ethylpiperazin-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (4f)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using 1-ethyl piperazine and 1,3-dibromopropane. The product was purified by adding a small amount of DCM followed by the addition of petroleum ether; beige solid; yield: 48.1%; mp 114.6–114.9 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 2.5 Hz, 1H), 7.15 (dd, J = 9.0, 2.5 Hz, 1H), 4.29 (s, 4H), 4.08 (t, J = 6.4 Hz, 2H), 3.67 (s, 2H), 2.47 − 2.33 (m, 16H), 1.94 − 1.86 (m, 2H), 1.00 (td, J = 7.2, 4.3 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 162.47, 159.14, 158.34, 147.29, 137.66, 125.43, 117.66, 105.34, 72.92, 66.98, 63.52, 54.59, 53.20, 52.44, 51.84, 26.45, 12.28; MS (ESI): m/z = 446.25 (M + H)+.
(4-Methylpiperazin-1-yl)(6-(3-(4-methylpiperazin-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (4 g)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using 1-methyl piperazine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH:TEA, 100:5:0.5); beige solid; yield: 28.1%; mp 99.2–99.7 °C; 1H NMR (400 MHz, CDCl3) δ 7.95 − 7.91 (d, J = 8.0 Hz, 1H), 7.37 (d, J = 11.1 Hz, 1H), 7.10 (d, J = 8.8 Hz, 1H), 4.48 (s, 2H), 4.08 (s, 2H), 3.85 (s, 2H), 2.67 − 2.50 (m, 14H), 2.38 (s, 3H), 2.34 (s, 3H), 2.02 (d, J = 5.4 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 162.12, 159.66, 158.23, 147.55, 137.90, 125.19, 117.09, 104.04, 66.64, 55.49, 54.66, 52.41, 45.96, 43.46, 29.66, 26.50; MS (ESI): m/z = 418.2 (M + H)+.
(3-Methylpiperazin-1-yl)(6-(3-(3-methylpiperazin-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (4h)
The title compound was synthesised according to procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using 1-boc-2-methyl piperazine and 1,3-dibromopropane to give compound 4h1, which was deprotected using TFA in DCM. The product was purified by CC (DCM:MeOH:TEA, 100:5:0.5); white solid; yield: 27.6%; mp 114.3–114.8 °C; 1H NMR (400 MHz, CD3OD) δ 8.02 (d, J = 9.0 Hz, 1H), 7.64 (d, J = 2.4 Hz, 1H), 7.25 (dd, J = 9.1, 2.4 Hz, 1H), 4.24 (t, J = 5.9 Hz, 2H), 3.72 − 3.42 (m, 9H), 3.35 − 3.31 (m, 7H), 2.39 (s, 1H), 2.31 − 2.24 (m, 2H), 1.43 (dt, J = 13.6, 6.6 Hz, 7H); 13C NMR (101 MHz, CD3OD) δ 160.89, 159.77, 158.50, 147.41, 137.95, 125.05, 117.34, 104.07, 65.60, 54.59, 54.22, 52.19, 51.13, 50.09, 48.29, 46.48, 41.67, 24.63, 14.79, 7.79, 6.22; MS (ESI): m/z = 418.2 (M + H)+.
(4-(2-Hydroxyethyl)piperazin-1-yl)(6-(3-(4-(2-hydroxyethyl)piperazin-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (4i)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using 1-(2-hydroxyethyl) piperazine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH:TEA, 100:5:0.5); red solid; yield: 22.5%; mp 173.5–174 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J = 9.3 Hz, 1H), 7.70 (s, 1H), 7.15 (d, J = 9.5 Hz, 1H), 4.27 (s, 2H), 4.07 (s, 2H), 3.65 (s, 1H), 3.48 (d, J = 21.4 Hz, 3H), 2.43 − 2.32 (m, 18H), 1.87 (s, 4H), 1.20 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 162.40, 159.17, 158.35, 147.25, 137.64, 125.42, 117.67, 105.31, 72.89, 63.49, 60.33, 58.87, 53.63, 53.23, 26.51; MS (ESI): m/z = 477.24 (M + H)+.
N-Benzyl-6-(3-(benzylamino)propoxy)benzo[d]thiazole-2-carboxamide (4j)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using benzyl amine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:6); white solid; yield: 57.4%; mp 90.1–90.7 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.59 (t, J = 6.4 Hz, 1H), 7.97 (d, J = 9.0 Hz, 1H), 7.74 (d, J = 2.5 Hz, 1H), 7.34 − 7.17 (m, 11H), 4.46 (d, J = 6.3 Hz, 2H), 4.13 (t, J = 6.4 Hz, 2H), 3.71 (s, 2H), 2.67 (t, J = 6.8 Hz, 2H), 1.94 − 1.87 (m, 2H), 1.29 − 1.20 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 162.00, 160.06, 158.31, 147.44, 139.40, 138.38, 128.74, 128.52, 128.43, 127.90, 127.35, 127.03, 124.99, 117.89, 105.84, 67.02, 53.30, 45.63, 43.12, 29.28; MS (ESI): m/z = 431.17 (M + H)+.
(3-Methylpiperidin-1-yl)(6-(3-(3-methylpiperidin-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (4k)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using 3-methyl piperidine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:6); white solid; yield: 76.4%; mp 161.1–161.6 °C; 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.8 Hz, 1H), 7.34 (s, 1H), 7.08 (d, J = 8.9 Hz, 1H), 4.51 (d, J = 12.4 Hz, 1H), 4.11 (t, 2H), 3.20 (dd, J = 23.8, 9.7 Hz, 2H), 2.88 (s, 2H), 2.25 (s, 3H), 1.88 (ddd, J = 41.8, 24.6, 11.8 Hz, 9H), 1.67 − 1.58 (m, 1H), 1.25 (d, J = 11.0 Hz, 2H), 0.94 (dd, J = 18.5, 4.9 Hz, 8H); 13C NMR (101 MHz, CDCl3) δ 162.67, 159.72, 157.69, 147.70, 137.79, 125.18, 116.62, 104.30, 66.25, 60.62, 55.43, 53.57, 50.99, 47.01, 33.08, 31.79, 29.77, 26.16, 25.23, 23.84, 19.29; MS (ESI): m/z = 415.23 (M + H)+.
(4-Methylpiperidin-1-yl)(6-(3-(4-methylpiperidin-1-yl)propoxy)benzo[d]thiazol-2-yl)methanone (4 l)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using 4-methyl piperidine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH, 100:6); white solid; yield: 80.2%; mp 162.2–162.5 °C; 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.9 Hz, 1H), 7.34 (s, 1H), 7.08 (d, J = 9.0 Hz, 1H), 4.66 (d, J = 12.6 Hz, 1H), 4.12 (s, 2H), 3.32 (d, J = 9.8 Hz, 2H), 3.20 (t, J = 12.7 Hz, 1H), 2.87 (dd, J = 31.4, 18.9 Hz, 3H), 2.45 (s, 2H), 2.31 (s, 2H), 1.70 (dd, J = 62.5, 34.6 Hz, 9H), 1.34 − 1.23 (m, 2H), 0.99 (t, J = 5.7 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 162.71, 159.75, 157.60, 147.75, 137.80, 125.19, 116.55, 104.34, 66.15, 64.19, 53.39, 46.79, 44.15, 34.90, 33.92, 31.10, 21.65; MS (ESI): m/z = 415.23 (M + H)+.
N-Cyclopentyl-6-(3-(cyclopentylamino)propoxy)benzo[d]thiazole-2-carboxamide (3i)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cyclopentyl amine and 1,3-dibromopropane. The product was purified by CC (DCM:MeOH:TEA, 100:3:1); grey solid; yield: 15.6%; mp 169.4–171.3 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.96 (d, J = 7.9 Hz, 1H), 8.02 (d, J = 9.0 Hz, 1H), 7.79 (d, J = 2.5 Hz, 1H), 7.22 (dd, J = 9.0, 2.5 Hz, 1H), 4.22 (dt, J = 11.6, 6.6 Hz, 2H), 3.53 − 3.45 (m, 1H), 3.35 (s, 1H), 3.12 − 3.04 (m, 1H), 2.16 − 2.07 (m, 1H), 2.03 − 1.83 (m, 4H), 1.78 − 1.47 (m, 14H), 1.24 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 162.85, 159.78, 157.74, 147.96, 138.57, 125.22, 117.89, 106.36, 65.87, 58.91, 51.49, 43.71, 32.28, 29.79, 24.12, 24.06; MS (ESI): m/z = 388.2 (M + H)+.
Piperidin-1-yl-(6-(4-(piperidin-1-yl)butoxy)benzo[d]thiazol-2-yl)methanone (3j)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using piperidine and 1,4-dibromobutane. The product was purified by CC (DCM:MeOH, 100:9); yellowish white solid; yield: 44.2%; mp 169.7–171.5 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.00 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.17 (dd, J = 9.0, 2.5 Hz, 1H), 4.22 (t, J = 5.1 Hz, 2H), 4.09 (t, J = 5.8 Hz, 2H), 3.65 (t, J = 5.2 Hz, 2H), 3.34 (s, 1H), 2.72 (s, 5H), 1.77 (d, J = 2.9 Hz, 4H), 1.64 (m, 10H), 1.45 (m, 2H); 13C NMR (126 MHz, DMSO-d6) δ 162.28, 158.69, 157.74, 146.85, 137.13, 124.92, 117.09, 104.92, 67.78, 52.81, 46.72, 44.01, 26.33, 26.11, 25.47, 24.49, 23.92; MS (ESI): m/z = 402.21 (M + H)+; HRMS (ESI): calculated for (C22H31N3O2S + Na+) 424.2038, found 424.2040 (M + Na)+.
N-Cycloheptyl-6-(4-(cycloheptylamino)butoxy)benzo[d]thiazole-2-carboxamide (3k)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cycloheptyl amine and 1,4-dibromobutane. The product was purified by CC (DCM:MeOH, 100:6); white solid; yield: 22%; mp 193.3–194.1 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.83 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 2.5 Hz, 1H), 7.19 (dd, J = 9.0, 2.5 Hz, 1H), 4.11 (t, J = 6.0 Hz, 2H), 3.99 − 3.91 (m, 1H), 3.20 − 3.12 (m, 1H), 3.01 − 2.95 (m, 2H), 2.05 − 1.97 (m, 2H), 1.87 − 1.74 (m, 6H), 1.72 − 1.63 (m, 6H), 1.60 − 1.37 (m, 15H); 13C NMR (126 MHz, DMSO-d6) δ 162.25, 158.23, 157.58, 147.07, 137.86, 124.56, 117.26, 105.42, 67.52, 58.14, 50.82, 43.89, 34.02, 30.25, 27.61, 27.34, 23.90, 23.31; MS (ESI): m/z = 458.4 (M + H)+.
N-(Cyclohexylmethyl)-6-(4-((cyclohexylmethyl)amino)butoxy)benzo[d]thiazole-2-carboxamide (3l)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cyclohexyl methyl amine and 1,4-dibromobutane. The product was purified by CC (DCM:MeOH, 100:7); white solid; yield: 22%; mp 234.8–235.6 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.02 (t, J = 6.1 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.77 (d, J = 2.4 Hz, 1H), 7.20 (dd, J = 9.0, 2.5 Hz, 1H), 4.11 (t, J = 5.5 Hz, 2H), 3.13 (t, J = 6.5 Hz, 2H), 2.99 − 2.92 (t, J = 5.9 Hz, 2H), 2.75 (t, J = 6.9 Hz, 2H), 1.68 (m, J = 35.4, 23.9, 15.6 Hz, 17H), 1.26 − 1.09 (m, 6H), 0.93 (d, J = 7.9 Hz, 4H); 13C NMR (126 MHz, DMSO-d6) δ 162.01, 159.49, 157.58, 147.09, 137.87, 124.56, 117.29, 105.45, 67.56, 52.81, 47.19, 45.27, 37.32, 30.39, 29.97, 26.00, 25.56, 25.32, 24.98; MS (ESI): m/z = 458.28 (M + H)+.
N-Cyclopropyl-6-(4-(cyclopropylamino)butoxy)benzo[d]thiazole-2-carboxamide (3m)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”,using cyclopropyl amine and 1,4-dibromobutane. The product was purified by CC (DCM:MeOH, 100:9); reddish semisolid; yield: 20.5%; 1H NMR (500 MHz, DMSO-d6) δ 9.11 (d, J = 4.9 Hz, 1H), 7.96 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 2.5 Hz, 1H), 7.18 (dd, J = 9.0, 2.6 Hz, 1H), 4.08 (t, J = 6.4 Hz, 2H), 3.58 (t, J = 6.5 Hz, 1H), 2.91 (m, 1H), 2.81 (t, J = 6.8 Hz, 1H), 2.63 − 2.56 (m, 1H), 2.16 (d, J = 6.6 Hz, 2H), 1.82 − 1.79 (m, 1H), 1.68 − 1.63 (m, 1H), 0.72 − 0.69 (m, 3H), 0.58 − 0.56 (m, 2H), 0.52 − 0.50 (m, 1H), 0.48 (m, 1H), 0.37 − 0.34 (m, 2H); 13C NMR (126 MHz, DMSO-d6) δ 171.72, 161.72, 157.73, 147.04, 137.86, 124.50, 117.34, 105.37, 67.92, 57.56, 29.85, 26.12, 23.11, 22.15, 5.57, 4.70; MS (ESI): m/z = 346.1 (M + H)+.
Pyrrolidin-1-yl(6-(4-(pyrrolidin-1-yl)butoxy)benzo[d]thiazol-2-yl)methanone (3n)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using pyrrolidine and 1,4-dibromobutane. The product was purified by CC (DCM:MeOH, 100:9); white solid; yield: 37.8%; mp 234.8–235.1 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.01 (d, J = 9.0 Hz, 1H), 7.74 (d, J = 2.5 Hz, 1H), 7.19 (dd, J = 9.0, 2.6 Hz, 1H), 4.09 (dt, J = 10.4, 6.4 Hz, 4H), 3.56 (t, J = 6.9 Hz, 2H), 3.10 − 2.98 (m, 4H), 2.00 (m, 14H); 13C NMR (126 MHz, DMSO-d6) δ 163.05, 158.25, 157.73, 147.55, 137.28, 125.12, 117.09, 105.04, 67.57, 53.53, 52.80, 48.53, 47.51, 26.02, 23.16, 22.69; MS (ESI): m/z = 374.2 (M + H)+; HRMS (ESI): calculated for (C20H27N3O2S + H+) 374.1896, found 374.1900 (M + H)+.
Piperidin-1-yl(6-((5-(piperidin-1-yl)pentyl)oxy)benzo[d]thiazol-2-yl)methanone (3o)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using piperidine and 1,5-dibromopentane. The product was purified by CC (DCM:MeOH:TEA, 100:8.5:0.5); yellow solid; yield: 52%; mp 76.2–77.1 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.00 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 2.5 Hz, 1H), 7.17 (dd, J = 9.0, 2.5 Hz, 1H), 4.23 (s, 2H), 4.07 (t, J = 6.4 Hz, 2H), 3.66 (s, 2H), 3.46 (d, J = 12.0 Hz, 2H), 2.62 (s, 4H), 1.79 (dd, J = 14.0, 7.0 Hz, 2H), 1.60 (dd, J = 16.2, 11.4 Hz, 12H), 1.49 − 1.37 (m, 4H); 13C NMR (75 MHz, DMSO-d6) δ 162.73, 159.17, 158.32, 147.30, 137.65, 125.40, 117.57, 105.32, 68.50, 57.87, 53.69, 47.22, 44.51, 28.77, 26.83, 25.96, 24.42, 23.69; MS (ESI): m/z = 416.23 (M + H)+.
N-Cycloheptyl-6-((5-(cycloheptylamino)pentyl)oxy)benzo[d]thiazole-2-carboxamide (3p)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cycloheptyl amine and 1,5-dibromopentane. The product was purified by CC (DCM:MeOH, 100:6); yellowish white solid; yield: 42.6%; mp 80.2–80.4 °C; 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 9.0 Hz, 1H), 7.34 (d, J = 2.4 Hz, 1H), 7.28 (d, J = 8.5 Hz, 1H), 7.10 (dd, J = 9.0, 2.4 Hz, 1H), 4.14 (m, 1H), 4.00 (t, J = 6.2 Hz, 2H), 3.21 − 3.13 (m, 1H), 2.99 − 2.93 (m, 2H), 2.24 − 2.17 (m, 1H), 2.08 − 1.99 (m, 3H), 1.86 − 1.78 (m, 4H), 1.69 − 1.43 (m, 18H), 1.29 − 1.18 (m, 4H), 0.87 − 0.81 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 161.80, 158.80, 158.05, 147.33, 138.71, 124.66, 117.33, 104.65, 68.03, 59.74, 50.96, 44.96, 34.98, 31.34, 28.56, 28.05, 27.54, 26.17, 24.07, 23.61; MS (ESI): m/z = 472.2 (M + H)+.
N-(Cyclohexylmethyl)-6-((5-((cyclohexylmethyl)amino)pentyl)oxy)benzo[d]thiazole-2-carboxamide (3q)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cyclohexyl methyl amine and 1,5-dibromopentane. The product was purified by CC (DCM:MeOH, 100:8); white solid; yield: 32.8%; mp 149–149.4 °C; 1H NMR (300 MHz, DMSO-d6) δ 9.03 (t, J = 6.1 Hz, 1H), 8.00 (d, J = 9.0 Hz, 1H), 7.78 (d, J = 2.5 Hz, 1H), 7.20 (dd, J = 9.0, 2.5 Hz, 1H), 4.09 (t, J = 6.2 Hz, 2H), 3.14 (t, J = 6.5 Hz, 2H), 2.93 − 2.85 (m, 2H), 2.73 (d, J = 6.8 Hz, 2H), 1.87 − 1.43 (m, 18H), 1.29 − 0.86 (m, 11H); 13C NMR (75 MHz, DMSO-d6) δ 162.49, 160.00, 158.19, 147.52, 138.37, 125.02, 117.81, 105.85, 68.36, 53.30, 47.86, 45.75, 37.82, 35.02, 30.88, 30.51, 28.52, 26.50, 26.07, 25.82, 25.50, 23.17; MS (ESI): m/z = 472.29 (M + H)+.
N-Cyclopropyl-6-((5-(cyclopropylamino)pentyl)oxy)benzo[d]thiazole-2-carboxamide (3r)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cyclopropyl amine and 1,5-dibromopentane. The product was purified by CC (DCM:MeOH, 100:6); brown solid; yield: 70%; mp 141.8–142.5 °C; 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.7 Hz, 1H), 7.39 (s, 1H), 7.32 (s, 1H), 7.09 (d, J = 8.9 Hz, 1H), 4.00 (s, 2H), 2.96 − 2.86 (m, 3H), 2.32 (d, J = 2.3 Hz, 1H), 1.81 (dd, J = 17.9, 7.3 Hz, 4H), 1.55 (d, J = 6.1 Hz, 2H), 1.24 (s, 1H), 0.89 (d, J = 6.5 Hz, 2H), 0.73 (d, J = 21.6 Hz, 4H), 0.61 (d, J = 5.8 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 161.28, 161.00, 158.24, 147.22, 138.76, 124.67, 117.45, 104.52, 68.24, 48.98, 30.30, 28.79, 27.70, 23.65, 22.77, 6.65, 4.98; MS (ESI): m/z = 360.17 (M + H)+.
Pyrrolidin-1-yl-(6-((5-(pyrrolidin-1-yl)pentyl)oxy)benzo[d]thiazol-2-yl)methanone (3s)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using pyrrolidine and 1,5-dibromopentane. The product was purified by CC (DCM:MeOH, 100:7); light brown solid; yield: 24%; mp 151.2–151.8 °C; 1H NMR (300 MHz, DMSO-d6) δ 8.01 (d, J = 9.0 Hz, 1H), 7.75 (d, J = 2.5 Hz, 1H), 7.18 (dd, J = 9.0, 2.5 Hz, 1H), 4.11 − 4.07 (m, 4H), 3.56 (t, J = 6.8 Hz, 2H), 3.20 − 3.12 (m, 4H), 2.05 − 1.65 (m, 14H), 1.55 − 1.44 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 163.49, 158.73, 158.30, 147.98, 137.79, 125.62, 117.57, 105.43, 68.35, 54.30, 53.69, 49.03, 48.01, 28.45, 26.52, 25.43, 23.66, 23.04; MS (ESI): m/z = 388.2 (M + H)+; HRMS (ESI): calculated for (C21H29N3O2S + Na+) 410.1872, found 410.1894 (M + Na)+.
N-Cyclopentyl-6-((5-(cyclopentylamino)pentyl)oxy)benzo[d]thiazole-2-carboxamide (3t)
The title compound was synthesised according to the procedure in Section “General procedure for preparing compounds (3a–3t, 4a–4l)”, using cyclopentyl amine 1,5-dibromopentane. The product was purified by CC (DCM:MeOH:TEA, 100:8.5:0.5); grey semisolid; yield: 34%; 1H NMR (300 MHz, DMSO- 6) δ 8.93 (d, J = 7.9 Hz, 1H), 7.99 (dd, J = 9.0, 3.8 Hz, 1H), 7.76 (d, J = 2.5 Hz, 1H), 7.19 (dd, J = 9.0, 2.5 Hz, 1H), 4.23 (m, 1H), 4.07 (t, J = 6.2 Hz, 2H), 3.35 (m, 1H), 2.88 − 2.74 (m, 2H), 1.95 − 1.83 (m, 4H), 1.81 − 1.56 (m, 13H), 1.50 (dd, J = 11.6, 5.9 Hz, 6H); 13C NMR (75 MHz, DMSO-d6) δ 162.52, 159.58, 158.20, 147.51, 138.36, 124.98, 117.78, 105.79, 68.42, 58.79, 51.48, 46.34, 32.29, 29.88, 28.59, 26.32, 24.05, 23.32; MS (ESI): m/z = 416.23 (M + H)+.
Procedure for preparing compound (4m)
Compound (4h) (1 mmol, 1 equiv.) was dissolved in a mixture of 10 ml DCM and 10 ml DMF, then glacial acetic acid (4 equiv.), DMAP (4 equiv.) and EDC (4 equiv.) were added. The reaction was stirred at room temperature overnight. After the completion of the reaction, the solvent was evaporated under vacuum then the compound was extracted using ethyl acetate and distilled water. The combined organic phase was dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography over silica gel.
1-(4-(6-(3-(4-Acetyl-3-methylpiperazin-1-yl)propoxy)benzo[d]thiazole-2-carbonyl)-2-methylpiperazin-1-yl)ethan-1-one (4m)
The title compound was synthesised according to the procedure in Section “Procedure for preparing compound (4m)”, using compound (4h). The product was purified by CC (DCM:MeOH, 100:8); white semisolid; yield: 34%; 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.37 (s, 1H), 7.12 (d, J = 8.7 Hz, 1H), 4.53 (s, 1H), 4.17 − 3.95 (m, 3H), 3.57 (dd, J = 107.2, 45.2 Hz, 3H), 3.17 − 2.30 (m, 6H), 2.14 (dd, J = 30.7, 21.1 Hz, 10H), 1.65 (s, 2H), 1.35 − 1.16 (m, 7H); 13C NMR (101 MHz, CDCl3) δ 169.26, 150.73, 138.05, 134.27, 132.92, 125.51, 117.11, 106.48, 104.06, 63.33, 53.43, 50.00, 47.65, 46.21, 43.71, 35.90, 33.63, 29.65, 24.18, 21.70, 21.22, 16.43, 16.21; MS (ESI): m/z = 502.2 (M + H)+.
Biological assays
Cell culture and membrane preparations
HEK-293 cells stably expressing the human H3R were cultured in DMEM (10% FBS, 1% penicillin/streptomycin, 1% L-glutamine, 1.1% HEPES buffer (1M)). CHO-K1 cells stably expressing the human H1R were cultivated in DMEM (10% FBS, 1% L-glutamine, 1% penicillin/streptomycin, 1% NEAA). For human H4R assays, Sf-9 insect cells were cultured in Spodopan (5% FBS, 1% penicillin/streptomycin) and co-infected with baculoviruses containing G-protein Gαi2, Gβ1γ2 and human H4R. Cell membrane preparations were obtained according to Bautista et al. and stored at −80 °C until use.Citation14
Radioligand displacement assay
Human H3R
The displacement assay for H3R was performed according to the protocol reported by Bautista et al.Citation14 Briefly, membrane fractions (20 µg/200 µL binding buffer) were co-incubated with [3H]-Nα-methylhistamine (2 nM) and test ligand for 90 min at room temperature while shaking. Assays for Ki value determination were run in duplicates with appropriate concentrations between 100 µM and 0.01 nM of the tested compound. Assays for one-point inhibition were performed in triplicates at a concentration of 1 µM test ligand, calculating percent inhibition relative to total radioligand binding. Non-specific binding was determined in the presence of 10 µM pitolisant. Data were analysed with GraphPad Prism 6.1 (San Diego, CA, USA) using non-linear regression fit (one-site competition with a logarithmic scale). The Ki values were calculated from the IC50 values using the Cheng-Prusoff equation.Citation47 Statistical calculations were performed on –log (Ki).
Human H1R
The displacement assay for H1R was performed according to the protocol reported by Bautista et al.Citation14 Briefly, membrane fractions (40 µg/200 µL binding buffer) were co-incubated with [3H]histamine (1 nM) and test ligand for 120 min at room temperature while shaking. Assays for one-point inhibition were performed in triplicates at a concentration of 10 µM test ligand, calculating percent inhibition relative to total radioligand binding. Non-specific binding was determined in the presence of 10 µM chlorpheniramine. Data were analysed with GraphPad Prism 6.1 (San Diego, CA, USA).
Human H4R
The displacement assay for H4R was performed according to the protocol reported by Bautista et al.Citation14 Briefly, membrane fractions (40 µg/200 µL binding buffer) were co-incubated with [3H]histamine (10 nM) and test ligand for 60 min at room temperature while shaking. Assays for one-point inhibition were performed in triplicates at a concentration of 1 µM test ligand, calculating percent inhibition relative to total radioligand binding. Non-specific binding was determined in the presence of 100 µM JNJ7777120. Data were analysed with GraphPad Prism 6.1 (San Diego, CA, USA).
Inhibition of AChE
The AChE assay was performed using an AChE inhibitor screening kit (BioVision, #K197-100, San Francisco, USA). According to the manufacturer’s protocol, tested compounds, donepezil, or DMSO (1%, as solvent control) were incubated with AChE for 15 min in dark at 25 °C, and then the probe and substrate were added for 30 min. The activity of AChE was obtained using a spectrophotometer at 412 nm. The assays were performed in triplicates.
Inhibition of BuChE
The hBuChE assay was performed using the Ellman’s spectrophotometric method adapted to a 96-well plate procedure as previously described. Briefly, BuChE from human serum (50 U/mg), substrate butyrylthiocoline iodide and chromophoric reagent 5,5′-dithiobis(2-nitrobenzoic acid) (all from Sigma Aldrich, Italy) were incubated with test compounds in clear flat-bottomed 96-well plates (Greiner Bio-One, Austria) in duplicate. Final concentrations where 0.09 U/mL (BuChE), 0.5 mM (butyrylthiocoline iodide), 0.33 mM (DTNB), while compounds were initially tested at 10 µM concentration. Only for compounds showing >60% inhibition, IC50 values were measured in the range 30-0.01 µM. The increase in absorbance was read at 412 nm for 5 min with a Tecan Infinite M1000 Pro multiplate reader (Tecan, Italy). Inhibition values were calculated with GraphPad Prism 5.0 software as the mean of three independent experiments.Citation48
Inhibition of monoamine oxidases
Inhibition of hMAOs was evaluated using human recombinant MAO-A and MAO-B (microsomes from baculovirus-infected insect cells) and kynuramine as non-selective MAO-A/B substrate (all from Sigma Aldrich Italy) through a previously described fluorescence-based method.Citation49 Experiments were performed in triplicates in black, round-bottomed polystyrene 96-well microtiter plates (Greiner). Fluorescence was measured at excitation/emission wavelengths of 310/400 nm in a 96-well microplate fluorescence reader (Tecan Infinite M1000 Pro) and inhibition values were calculated with GraphPad Prism 5.0 software as the mean of three independent experiments.
Molecular docking
All procedures were performed using the Molecular Operating Environment (MOE)® software package (version 2019, Chemical Computing Group). During the molecular modelling studies on H3R a previously prepared homology model was used.Citation44 The ligands (3b, 3s) were prepared by MOE (washing, calculating partial charges, adjusting tautomeric states and minimising the energy). The binding site was defined using Site Finder and the docking was performed using Amber10: EHT as a force field and the “triangle matcher” method (number of return poses set to 200), Induced fit as refinement and London dG as the scoring function. The number of returned poses was set to 20.
For the docking simulations on human AChE, PDB code 4EY5 (rhAChE cocrystallized with Huperzine A) was used. Molecular docking simulation with 3b as a ligand was performed using the Amber10: EHT as a force field and the “triangle matcher” method (number of return poses set to 200), Induced fit as refinement and London dG as the scoring function. The number of returned poses was set to 20. Similarly, compound 4a was docked in the active site of human BuChE using PDB code 1POI.
Supplemental Material
Download PDF (1.7 MB)Acknowledgement
Thanks to Assoc. Prof. Dr. Marek Bajda, Jagiellonian University Medical College in Cracow, Faculty of Pharmacy, Department of Physicochemical Drug Analysis for providing us with the homology model of H3R to be able to perform the docking studies. Partial support by the EU COST project CA15135 is acknowledged (to HS). Also funding by Ministry of Science and Technology, Taiwan, Chang Gung Memorial Hospital, Taiwan is acknowledged.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Kamer AR, Craig RG, Niederman R, Fortea J, de Leon MJ. Periodontal disease as a possible cause for Alzheimer’s disease. Periodontol 2000. 2020;83(1):242–271.
- Lopes FB, Aranha CMSQ, Fernandes JPS. Histamine H3 receptor and cholinesterases as synergistic targets for cognitive decline: Strategies to the rational design of multitarget ligands. Chem Biol Drug Des. 2021;98(2):212–225.
- Abeysinghe AADT, Deshapriya RDUS, Udawatte C. Alzheimer’ s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci. 2020;256:117996.
- Sharma K. Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol Med Rep. 2019;20(2):1479–1487.
- Kumar B, Thakur A, Dwivedi AR, Kumar R, Kumar V. Multi-target-directed ligands as an effective strategy for the treatment of Alzheimer’s disease. Curr Med Chem. 2022;29(10):1757–1803.
- Pasieka A, Panek D, Malawska B. Multifunctional ligand approach: search for effective therapy against Alzheimer’s disease. Exon Publications. 2020:181–203.
- Nikolic K, Mavridis L, Bautista-Aguilera OM, Marco-Contelles J, Stark H, do Carmo Carreiras M, Rossi I, Massarelli P, Agbaba D, Ramsay RR, et al. Predicting targets of compounds against neurological diseases using cheminformatic methodology. J Comput Aided Mol Des. 2015;29(2):183–198.
- de Freitas Silva M, Dias KST, Gontijo VS, Ortiz CJC, Viegas C. Jr Multi-target directed drugs as a modern approach for drug design towards Alzheimer’s disease: an update. Curr Med Chem. 2018;25(29):3491–3525.
- Maramai S, Benchekroun M, Gabr MT, Yahiaoui S. Multitarget therapeutic strategies for Alzheimer’s disease: review on emerging target combinations. Biomed Res Int. 2020;2020.
- Mishra P, Kumar A, Panda G. Anti-cholinesterase hybrids as multi-target-directed ligands against Alzheimer’s disease (1998–2018). Bioorganic Med Chem. 2019;27(6):895–930.
- Khanfar MA, Affini A, Lutsenko K, Nikolic K, Butini S, Stark H. Multiple targeting approaches on histamine H3 receptor antagonists. Front Neurosci. 2016;10:201.
- Ghamari N, Zarei O, Arias-Montaño J-A, Reiner D, Dastmalchi S, Stark H, Hamzeh-Mivehroud M. Histamine H3 receptor antagonists/inverse agonists: where do they go? Pharmacol Ther. 2019;200:69–84.
- Harwell V, Fasinu PS. Pitolisant and other histamine-3 receptor antagonists—an update on therapeutic potentials and clinical prospects. Medicines. 2020;7(9):55.
- Bautista-Aguilera ÓM, Hagenow S, Palomino-Antolin A, Farré-Alins V, Ismaili L, Joffrin P-L, Jimeno ML, Soukup O, Janočková J, Kalinowsky L, et al. Multitarget-directed ligands combining cholinesterase and monoamine oxidase inhibition with histamine H3R antagonism for neurodegenerative diseases. Angew Chem Int Ed Engl. 2017;56(41):12765–12769.
- Godyń J, Zaręba P, Łażewska D, Stary D, Reiner-Link D, Frank A, Latacz G, Mogilski S, Kaleta M, Doroz-Płonka A, et al. Cyanobiphenyls: novel H3 receptor ligands with cholinesterase and MAO B inhibitory activity as multitarget compounds for potential treatment of Alzheimer’s disease. Bioorg Chem. 2021;114:105129.
- Zhou Y, Hu Y, Lu X, Yang H, Li Q, Du C, Chen Y, Hong KH, Sun H. Discovery of a selective 6-hydroxy-1, 4-diazepan-2-one containing butyrylcholinesterase inhibitor by virtual screening and MM-GBSA rescoring. Dose Response. 2020;18(2):1559325820938526.
- Darvesh S. Butyrylcholinesterase as a diagnostic and therapeutic target for Alzheimer’s disease. Curr Alzheimer Res. 2016;13(10):1173–1177.
- El-Sayed NA-E, Farag AE-S, Ezzat MAF, Akincioglu H, Gülçin İ, Abou-Seri SM. Design, synthesis, in vitro and in vivo evaluation of novel pyrrolizine-based compounds with potential activity as cholinesterase inhibitors and anti-Alzheimer’s agents. Bioorg Chem. 2019;93:103312.
- Jończyk J, Lodarski K, Staszewski M, Godyń J, Zaręba P, Soukup O, Janockova J, Korabecny J, Sałat K, Malikowska-Racia N, et al. Search for multifunctional agents against Alzheimer’s disease among non-imidazole histamine H3 receptor ligands. In vitro and in vivo pharmacological evaluation and computational studies of piperazine derivatives. Bioorg Chem. 2019;90:103084.
- Gill RK, Rawal RK, Bariwal J. Recent advances in the chemistry and biology of benzothiazoles. Arch Pharm. 2015;348(3):155–178.
- Sharma PC, Sinhmar A, Sharma A, Rajak H, Pathak DP. Medicinal significance of benzothiazole scaffold: an insight view. J Enzyme Inhib Med Chem. 2013;28(2):240–266.
- Keri RS, Patil MR, Patil SA, Budagumpi SA. comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur J Med Chem. 2015;89:207–251.
- Pathak N, Rathi E, Kumar N, Kini SG, Rao CM. A review on anticancer potentials of benzothiazole derivatives. Mini Rev Med Chem. 2020;20(1):12–23.
- Rouf A, Tanyeli C. Bioactive thiazole and benzothiazole derivatives. Eur J Med Chem. 2015;97:911–927.
- Aboushady Y, Gabr M, ElHady AK, Salah M, Abadi AH, Wilms G, Becker W, Abdel-Halim M, Engel M. Discovery of hydroxybenzothiazole urea compounds as multitargeted agents suppressing major cytotoxic mechanisms in neurodegenerative diseases. ACS Chem Neurosci. 2021;12(22):4302–4318.
- AlNajjar YT, Gabr M, ElHady AK, Salah M, Wilms G, Abadi AH, Becker W, Abdel-Halim M, Engel M. Discovery of novel 6-hydroxybenzothiazole urea derivatives as dual Dyrk1A/α-synuclein aggregation inhibitors with neuroprotective effects. Eur J Med Chem. 2022;227:113911.
- Salah M, Abdel-Halim M, Engel M. Design and synthesis of conformationally constraint Dyrk1A inhibitors by creating an intramolecular H-bond involving a benzothiazole core. Medchemcomm. 2018;9(6):1045–1053.
- Rosales Hernández MC, Fragoso Morales LG, Correa Basurto J, Olvera Valdez M, García Báez EV, Román Vázquez DG, Anaya García AP, Cruz A. In silico and in vitro studies of benzothiazole-isothioureas derivatives as a multitarget compound for Alzheimer’s disease. IJMS. 2022;23(21):12945.
- Matthews DC, Mao X, Dowd K, Tsakanikas D, Jiang CS, Meuser C, Andrews RD, Lukic AS, Lee J, Hampilos N, et al. Riluzole, a glutamate modulator, slows cerebral glucose metabolism decline in patients with Alzheimer’s disease. Brain. 2021;144(12):3742–3755.
- Behl T, Kaur D, Sehgal A, Singh S, Sharma N, Zengin G, Andronie-Cioara FL, Toma MM, Bungau S, Bumbu AG, et al. Role of monoamine oxidase activity in Alzheimer’s disease: an insight into the therapeutic potential of inhibitors. Molecules. 2021;26(12):3724.
- Łażewska D, Bajda M, Kaleta M, Zaręba P, Doroz-Płonka A, Siwek A, Alachkar A, Mogilski S, Saad A, Kuder K, et al. Rational design of new multitarget histamine H3 receptor ligands as potential candidates for treatment of Alzheimer’s disease. Eur J Med Chem. 2020;207:112743.
- Lipp R, Stark H, Schunack W. Pharmacochemistry of H3-receptors. 1992:57–72. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Pharmacochemistry+of+H3-receptors+stark&btnG=
- Sylvain C, Maikel W, Patrice T, Rob L, Iwan JPE. Histamine H3 receptor antagonist reach out for the clinics. Drug Disc Today. 2005;10:1613–1627.
- Wingen K, Stark H. Scaffold variations in amine warhead of histamine H3 receptor antagonists. Drug Discov Today Technol. 2013;10(4):e483–e489.
- Meier G, Apelt J, Reichert U, Grassmann S, Ligneau X, Elz S, Leurquin F, Ganellin CR, Schwartz JC, Schunack W, et al. Influence of imidazole replacement in different structural classes of histamine H3-receptor antagonists. Eur J Pharm Sci. 2001;13(3):249–259.
- Walczynski K, Guryn R, Zuiderveld OP, Timmerman H. Non‐imidazole histamine H3 ligands, part 2: new 2‐substituted benzothiazoles as histamine H3 antagonists. Arch Pharm Pharm Med Chem. 1999;332(11):389–398.
- Walczyński K, Guryn R, Zuiderveld OP, Timmerman H. Non-imidazole histamine H3 ligands. Part I. Synthesis of 2-(1-piperazinyl)-and 2-(hexahydro-1H-1, 4-diazepin-1-yl) benzothiazole derivatives as H3-antagonists with H1 blocking activities. Farm. 1999;54(10):684–694.
- Bordi F, Mor M, Morini G, et al. QSAR study on H3-receptor affinity of benzothiazole derivatives of thioperamide. Farm. 1994;49:153–166.
- Löwik DWPM, Tisi LC, Murray JAH, Lowe CR. Synthesis of 6-hydroxybenzothiazole-2-carboxylic acid. Synthesis. 2001;2001(12):1780–1783.
- Ghosh AK, Shahabi D. Synthesis of amide derivatives for electron deficient amines and functionalized carboxylic acids using EDC and DMAP and a catalytic amount of HOBt as the coupling reagents. Tetrahedron Lett. 2021;63(152719):152719.
- Wang K, Yu L, Shi J, Liu W, Sang Z. Multifunctional indanone–chalcone hybrid compounds with anti-β-amyloid (Aβ) aggregation, monoamine oxidase B (MAO-B) inhibition and neuroprotective properties against Alzheimer’s disease. Med Chem Res. 2019;28(11):1912–1922.
- Cai Z. Monoamine oxidase inhibitors: promising therapeutic agents for Alzheimer’s disease. Mol Med Rep. 2014;9(5):1533–1541.
- Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, et al. Structure of the human histamine H 1 receptor complex with doxepin. Nature. 2011;475(7354):65–70.
- Jończyk J, Malawska B, Bajda M. Hybrid approach to structure modeling of the histamine H3 receptor: Multi-level assessment as a tool for model verification. PLOS One. 2017;12(10):e0186108.
- Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2(4):541–553.
- Darwish SS, Abdel-Halim M, Salah M, Abadi AH, Becker W, Engel M. Development of novel 2, 4-bispyridyl thiophene–based compounds as highly potent and selective Dyrk1A inhibitors. Part I: benzamide and benzylamide derivatives. Eur J Med Chem. 2018;157:1031–1050.
- Yung-Chi C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108.
- Pisani L, Catto M, De Palma A, Farina R, Cellamare S, Altomare CD. Discovery of potent dual binding site acetylcholinesterase inhibitors via homo‐and heterodimerization of coumarin‐based moieties. ChemMedChem. 2017;12(16):1349–1358.
- Pisani L, Iacobazzi RM, Catto M, Rullo M, Farina R, Denora N, Cellamare S, Altomare CD. Investigating alkyl nitrates as nitric oxide releasing precursors of multitarget acetylcholinesterase-monoamine oxidase B inhibitors. Eur J Med Chem. 2019;161:292–309.