
Abstract
We designed and synthesised novel quinazolinone tethered phenyl urea derivatives (6a–p) that triple target the double mutant EGFRL858R/T790M, COX-2, and 15-LOX. Compounds (6e, 6d, 6j, 6m, and 6n) not only had low micromolar IC50 inhibitory activities against the three targets, but they also showed good selectivity for COX-2 over COX-1 and for EGFRL858R/T790M over wild-type EGFR. Except for 6e and 6n, all of the tested compounds inhibited the NO production significantly more potently than celecoxib, diclofenac, and indomethacin. Compounds 6i and 6k reduced ROS levels more effectively than celecoxib and diclofenac. In terms of inhibiting TNF-α production, 6o-treated cells showed TNF-α level, which is ∼10 times lower than celecoxib. Furthermore, 6e and 6j had the highest anticancer activity against the breast cancer cell line BT-459 with growth inhibition percentages of 67.14 and 70.07%, respectively. Docking studies confirm their favoured binding affinity. The proposed compounds could be promising multi-targeted leads.
Graphical Abstract
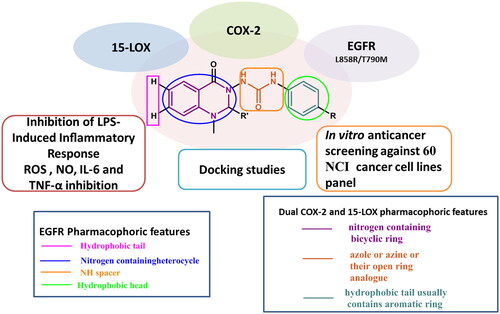
Introduction
Despite the intense efforts in the development of anticancer drugs, cancer still ranks as one of the most lethal diseases worldwideCitation1,Citation2. Moreover, the development of resistance to some highly effective anticancer agents remains a major challenge in cancer therapyCitation3. Consequently, the multifactorial nature of cancer has moved the field towards the design of multi-targeted anticancer agents to overcome the resistance problemCitation4.
Anticancer agents with multi-targeted mechanisms not only provide an effective strategy for the treatment of cancers with a low incidence of resistance, but they also avoid the issues of drug-drug interaction and dose-limiting toxicity that are common in combination therapyCitation4. Many multi-targeted anticancer agents have succeeded to reach preclinical and clinical stagesCitation3. As a result, the multi-target-single-agent strategy has become a mainstream approach for cancer treatment, and researchers have become increasingly interested in developing novel multi-target-single-agent drugs in recent yearsCitation2.
Epidermal growth factor receptor (EGFR) is a protein kinase that is overexpressed in a variety of human solid tumours, including pancreatic, colorectal, non-small cell lung (NSCLC), renal cell carcinoma (RCC), ovarian, breast, and head and neck cancersCitation2,Citation5. Thus EGFR inhibitors have become widely used for the treatment of those cancers. Although many tumours show an initial response to the treatment with EGFR inhibitors, resistance to treatment often occurs due to point mutations in the ATP binding pocket of EGFR, such as T790M, L858R, and C797S. These mutations have resulted in the development of three generations of EGFR tyrosine kinase inhibitors (TKIs)Citation2,Citation6. EGFR classic mutations are in-frame deletions in exon 19 and an L858R point mutation in exon 21. First-generation EGFR inhibitors (4- amino quinazolines) erlotinib (A) and gefitinib (B) were efficacious for these classic mutations (). However, resistance develops after the administration of first-generation EGFR TKIs due to a secondary mutation where a bulkier methionine replaces threonine at position 790 (gate keeper) (T790M). The bulkier methionine group sterically hinders the binding of these reversible inhibitors. Also, the T790M mutation confers an increased affinity for ATP to bind with the receptorCitation6–10.
Figure 1. EGFR inhibitors (first, second, and third generations), some reported compounds as dual COX-2/15-LOX inhibitors, and the rationale for the design of our multi-target directed ligands (MTDLs).
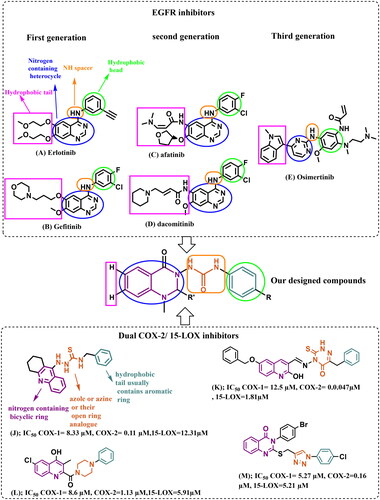
As a result, second-generation irreversible inhibitors have been developed, including afatinib (C) and dacomitinib (D) (). These inhibitors possess the aniline-quinazoline along with a reactive acrylamide moiety, which irreversibly (covalently) binds with C797S residue by undergoing a Michael addition reaction. Unfortunately, their poor selectivity between EGFRT790M mutants and the wild-type (WT) EGFR causes dose-limiting major adverse effects, such as grade 3 diarrhoea, skin rashes, and many othersCitation7–10.
The search for compounds showing selective inhibition against drug-resistant mutant EGFRT790M has led to the design and development of third generation EGFR inhibitors, such as AZD9291 (Osimertinib) (E), with the 2-arylamine pyrimidine scaffold exhibited great selective inhibitory effects on EGFRL858R/T790M over wild-type EGFR (). They bind irreversibly (covalently) with C797. However, resistance to third-generation inhibitors has also been observed as a result of new mutations, specifically the point mutation C797, which is one of the key residues for drug bindingCitation7–10.
The relation between inflammation and cancer progression has been recently supported by many studies. Multiple inflammatory mediators are expressed in the majority of cancer cells and contribute to different stages of tumour growth, starting from tumour initiation till metastasisCitation11,Citation12. Cyclooxygenases (COX-1/2) and lipoxygenases (5/12/15-LOX) are among the major enzymes involved in the formation of eicosanoids inflammatory mediators via the arachidonic acid (AA) cascadeCitation11.
The overexpression of COX-2 in various types of tumours including human colorectal cancer, pancreatic cancers, oesophageal, breast, prostate, lung carcinomas, and melanoma provided evidence for COX-2 role in cancer progression and resistance to radiotherapy and chemotherapy. Moreover, COX-2 inhibitors displayed both chemo-preventive and anticancer effects by synergistically or additively acting with anticancer agentsCitation2,Citation11,Citation12. Interestingly, non-steroidal anti-inflammatory drugs (NSAIDs) with greater COX-2 inhibition selectivity could inhibit angiogenesis and restore normal apoptosis in many types of cancer cellsCitation2,Citation5,Citation13.
Many studies have revealed the role of 15-Lipoxygenase (15-LOX), the other arm of the AA cascade, and its metabolites, in cancer progression and metastasis. The selective 15-LOX inhibitor, PD146176, showed antiproliferative activity against prostatic cancer cellsCitation11,Citation14,Citation15.
Dual COX-2/LOX inhibitors, targeting eicosanoids for anticancer effects, showed superior anticancer activities to their single pathway counterpart inhibitors. This could be because inhibiting only the COX-2 arm increases AA availability and shifts the AA metabolic machinery towards excessive production of downstream LOX inflammatory mediatorsCitation11,Citation16. Furthermore, the combination therapy of licofelone (a dual COX/LOX inhibitor) with gefitinib (EGFR TKI) showed significant tumour growth inhibition in pancreatic cancer (PC) at a dose lower than doses of the individual agentsCitation4,Citation17,Citation18. Accordingly, we herein describe our efforts to design and synthesise novel quinazolinones as multi-target directed ligands (MTDLs) that triple target EGFRL858R/T790M, COX-2, and 15-LOX simultaneously. In vitro EGFR, COX-1/COX-2, and 15-LOX inhibitory activities of the newly synthesised derivatives were evaluated, as well as their anticancer and anti-inflammatory activities. Moreover, in silico studies were performed for those compounds using docking analysis to elucidate a postulated model for their binding with EGFRL858R/T790M, COX-2, and 15-LOX at the molecular level.
The rationale for designing multi-target directed ligands (MTDLs)
Designing multi-target directed ligands (MTDLs) that triple target EGFRL858R/T790M, COX-2, and 15-LOX is very challenging. One of the most critical challenges in designing EGFR TKIs has been the development of secondary mutations. Therefore, there is a critical need to develop reversible non-covalent EGFR inhibitors that are more selective against the double mutant EGFRL858R/T790MCitation6.
The ATP binding site of EGFR consists of five distinct regions, adenine binding site/hinge region, hydrophilic ribose binding pocket, phosphate binding pocket, back hydrophobic region, and front hydrophobic regionCitation19,Citation20. Therefore, the majority of the ATP-mimic EGFR-TKIs share four common pharmacophoric features, including a hydrophobic head (that occupies the back hydrophobic region), a scaffold (primarily a nitrogen-containing heterocycle) capable of interacting via an H-bond with the adenine/hinge region, an NH spacer (hinge region), and a hydrophobic tail (that occupies the front hydrophobic region)Citation19–21 ().
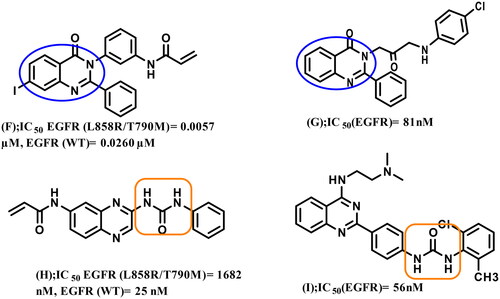
4-Amino-quinazoline represents an important class of first and second generations EGFR-TKIs as it fits into the ATP binding pocket, forming H-bond with the hinge region due to N-1 and N-3 atoms. The substitution of its N-1 and N-3 with carbon atoms decreases the inhibitory activity by 3700 and 200-fold, respectivelyCitation6,Citation22,Citation23. Recently, some studies have validated 2,3‐disubstituted quinazolinones as promising scaffolds for EGFR inhibition and NSCLC management (compounds F and G)Citation7,Citation24, the addition of a lipophilic group at the 2‐position improving overall activityCitation23. Other studies recommended urea functionality as a hydrogen bond domain (compounds H and I)Citation25,Citation26 ().
Figure 2. Reported 2,3‐disubstituted quinazolinones or urea-containing compounds with significant inhibitory activity against EGFR TK.
The common pharmacophoric structural features of the previously reported dual COX-2/15-LOX inhibitors include three regions: nitrogen-containing bicyclic ring, a central five or six-membered nitrogen-containing heterocycle (azole or azine), and a hydrophobic terminal usually containing an aromatic ring. Replacement of the central azole or azine with an open ring analogue has also been reported as observed in compounds (J–M)Citation16.
Herein, we attempted to design compounds that match the pharmacophoric features of both EGFR-TKIs and COX-2/15-LOX dual inhibitors by (I) using a quinazoline ring (a well-known scaffold of EGFR-TKIs that occupies the adenine/hinge region, and also presents a nitrogen-containing bicyclic ring in dual COX-2/15-LOX inhibitors pharmacophore), (II) incorporating urea functionality spacer (instead of NH spacer in EGFR-TKIs to trigger extra H-bond interactions, also represents open ring analogue of azole or azine in the dual COX-2/15-LOX inhibitor pharmacophore), and (III) attaching different substituted phenyl rings (as the hydrophobic head and the hydrophobic terminal in the pharmacophores of EGFR-TKIs and dual COX-2/15-LOX inhibitors, respectively).
Moreover, we removed the large hydrophobic tail of EGFR-TKIs, which had little effect on their efficacyCitation22,Citation23, making the molecules smaller and easier to shift to tolerate the bulkier methionine inside the T790M binding pocket. We avoided the acrylamide moiety (Michael acceptor) responsible for irreversible covalent bond formation with C797 at the ATP‐binding site of EGFR‐TK, which caused toxicity in second generation therapies, as well as the C797 mutation responsible for resistance in third generation therapiesCitation7–10,Citation23. We also added an extra hydrophobic moiety at position 2 of the quinazoline ring in some compounds to investigate extra hydrophobic interactions with our three targets and the effect of achieving the famous V-shape characteristic of selective COX-2 inhibitorsCitation27.
Materials and methods
Chemistry
All reagents were commercially obtained with the highest percent of purity available, especially for synthesis, unless otherwise mentioned. 1H and 13C-NMR spectra were recorded on a Bruker Avance III spectrometer operating at 400 and 100 MHz, respectively. The NMR solvent used was DMSO-d6 ((CD3)2SO). Chemical shifts are reported in parts per million (ppm) relative to the internal standard tetramethylsilane (Me4Si). Coupling constants (J-value) were calculated in hertz (Hz). All NMR characterisations were made by comparison with previous NMR spectra of the appropriate structure class and/or predictions from ChemDraw Ultra™. NMR analyses were performed at the Applied Nucleic Acids Research Centre & Chemistry, Faculty of Science, Zagazig University, Zagazig, Egypt or the Microanalytical Unit–FOPCU–NMR laboratory, Faculty of Pharmacy, Cairo University, Egypt.
Analytical thin layer chromatography (TLC) was carried out on precoated silica plates (ALUGRAM® SIL G/UV254) and visualised with UV light (254 nm) and/or ninhydrin stain.
Melting points were determined using a Gallenkamp (London, UK) melting point apparatus and are uncorrected. Mass spectra were collected using a GC/MS Shimadzu Qp-2010 plus instrument (Shimadzu Corporation, Tokyo, Japan) at Micro Analytical Centre, Cairo University, Egypt.
Elemental analyses were performed using a Vario EL-III (Elementar) CHN analyser (Hanau, Germany) at Micro Analytical Centre, Cairo University, Egypt, or the Regional Centre for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt.
General method for the synthesis of N-phenylhydrazinecarboxamides (3a–c)
N-(4-un/substitiutedphenyl)hydrazine carboxamides (3a, 3b, 3c) were prepared according to previously reported proceduresCitation28,Citation29. Trimethylamine (27.64 mmol) was added to a solution of the appropriate aniline 1a–c (13.82 mmol) in dichloromethane (50 ml). The reaction mixture was cooled to 0 °C, followed by the addition of ethyl chloroformate (20.73 mmol) dropwise. Then the reaction mixture was slowly warmed to room temperature and stirred for 2 h. After reaction completion, water (25 ml) was added to quench excess ethyl chloroformate. The layers were separated, and the aqueous layer was extracted twice with dicholoromethane (15 ml). The combined organic layers were dried over anhydrous MgSO4, filtered, and evaporated under reduced pressure to obtain the respective crude carbamates 2a–c.
A mixture of the appropriate carbamate 2a–c (24.66 mmol) and hydrazine hydrate (73.98 mmol) in ethanol (20 ml) was stirred under reflux for 5 h. After completion of the reaction, the reaction mixture was cooled, and the formed crystals were filtered to obtain the corresponding carboxamides 3a–c. The crystals were washed with water followed by petroleum ether. The carboxamides 3a–c were pure enough to be used for the subsequent reactions.
N-phenylhydrazinecarboxamide (3a)
White microcrystals, 85% yield, M. p. 149–151 °C [Lit.Citation29 M. p. 146–148 °C].
N-(4-methoxyphenyl)hydrazinecarboxamide (3b)
White microcrystals, 80% yield, M. p. 152–154 °C.
N-(4-fluorophenyl)hydrazinecarboxamide (3c)
White microcrystals, 79% yield, M. p. 178–180 °C [Lit.Citation29 (M. p. 182–184 °C].
General method for the synthesis of 2-(2-(methylamino)benzoyl)-N-phenylhydrazine-1-carboxamide (5a) and N-(4-substitutedphenyl)-2-(2-(methylamino)benzoyl)hydrazine-1-carboxamide (5b, 5c)
A mixture of N-methyl isatoic anhydride (4, 0.82 g, 4.63 mmol) and hydrazine carboxamide (3a, 3b, or 3c, 4.63 mmol) in ethanol (20 ml) containing a catalytic amount of glacial acetic acid (5 drops) was refluxed for 4 h. After cooling, the formed crystals were isolated by filtration under vacuum to provide the required intermediates 5a–c with a 78–85% yield and sufficient purity to be used in the subsequent step without further purification.
2-(2-(Methylamino)benzoyl)-N-phenylhydrazine-1-carboxamide (5a)
Pale-yellow crystals, 85% yield, M. p. 205–207 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H, NHCONHNH, exch.), 8.84 (s, 1H, NHCONHNH, exch.), 8.01 (s, 1H, NHCONHNH, exch.), 7.66 (d, J = 7.6 Hz, 1H, benzohydrazide-C3-H), 7.54 (s, 1H, NHCH3, exch.), 7.46 (d, J = 7.6 Hz, 2H, phenyl-C2,6-H), 7.30 (m, 3H, phenyl-C3,4,5-H), 6.95 (t, J = 7.9 Hz, 1H, benzohydrazide-C5-H), 6.67 (d, J = 8.0 Hz, 1H, benzohydrazide-C6-H), 6.58 (t, J = 7.5 Hz, 1H, benzohydrazide-C4-H), 2.79 (s, 3H, NHCH3).
N-(4-methoxyphenyl)-2-(2-(methylamino)benzoyl)hydrazine-1-carboxamide (5b)
Yellow crystals, 80% yield, M. P. 152–154 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.98 (s, 1H, NHCONHNH, exch.), 8.65 (s,1H,NHCONHNH, exch.), 7.92 (s, 1H, NHCONHNH, exch.), 7.66 (d, J = 7.5 Hz, 1H, benzohydrazide-C3-H), 7.55 (s, 1H, NHCH3, exch.), 7.39–7.28 (m, 3H, benzohydrazide-C5-H and phenyl-C2,6-H), 6.84 (d, J = 9.1 Hz, 2H, phenyl-C3,5-H), 6.66 (d, J = 8.1 Hz, 1H, benzohydrazide-C6-H), 6.57 (t, J = 7.9 Hz, 1H, benzohydrazide-C4-H), 3.70 (s, 3H, OCH3), 2.78 (s, 3H, NHCH3).
N-(4-fluorophenyl)-2-(2-(methylamino)benzoyl)hydrazine-1-carboxamide (5c)
White fluffy crystals, 78% yield, M. p. 178–180 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H, NHCONHNH, exch.), 8.88 (s, 1H, NHCONHNH, exch.), 8.04 (s, 1H, NHCONHNH, exch.), 7.66 (d, J = 7.7 Hz, 1H, benzohydrazide-C3-H), 7.57–7.43 (m, 3H, NHCH3, exch. + phenyl-C2,6-H), 7.41–7.29 (m, 1H, benzohydrazide-C5-H), 7.09 (t, J = 8.9 Hz, 2H, phenyl-C3,5-H), 6.66 (d, J = 8.4 Hz, 1H, benzohydrazide-C6-H), 6.58 (t, J = 8.0 Hz, 1H, benzohydrazide-C4-H), 2.78 (s, 3H, NHCH3).
General methods for the synthesis of compounds (6a-p)
a- Synthesis of compounds (6a, b, g, k, and l)
To a suspension of benzoylhydrazine-1-carboxamide (5a, 5b, or 5c, 2.81 mmol) in ethanol (25 ml), 37% formalin or acetaldehyde (4.215 mmol) and 5 drops glacial acetic acid were added. The reaction mixture was heated under reflux for 2 h and then concentrated to its half volume under vacuum evaporator. The mixture was cooled, and a precipitate was allowed to be formed. The obtained product was collected under vacuum filtration and crystallised from ethanol to produce the title compounds 6a, b, g, k, and l.
b- Synthesis of compounds (6c–f, 6h–j, 6m–p)
A mixture of benzoylhydrazine-1-carboxamide (5a, 5b, or 5c, 2.81 mmol) and the appropriate aromatic aldehyde (2.81 mmol) in glacial acetic acid (20 ml) was heated under reflux for 4 h. The reaction mixture was concentrated to its half volume and then poured into cold water (50 ml). The separated solid was isolated by filtration, dried, and crystallised from ethanol to give the title compounds 6c–f, 6h–j, 6m–p.
1-(1-Methyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)-3-phenylurea (6a)
White fluffy crystals, 91% yield, M. p. 240–241 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.99 (s, 1H, NNHCONH, exch), 8.58 (s, 1H, NNHCONH, exch), 7.76 (d, J = 7.6 Hz, 1H, quinazolinone-C5-H), 7.47–7.43 (m, 3H, quinazolinone-C7-H, phenyl-C2,6-H), 7.28–7.24 (m, 2H, phenyl-C3,5-H), 6.97 (t, J = 7.5 Hz, 1H, quinazolinone-C6-H), 6.87 (m, 2H, quinazolinone-C8-H and phenyl-C4-H), 4.70 (s, 2H, quinazolinone-C2-H), 2.87 (s, 3H, NCH3).13C NMR (101 MHz, DMSO-d6) δ 163.29 (C), 154.73 (C), 149.58 (C), 139.40 (C), 134.01 (C), 128.70 (2 × Ar-CH), 128.25 (Ar-CH), 122.09 (Ar-CH), 118.43 (2 × Ar-CH), 118.22 (Ar-CH), 115.91 (Ar-CH), 112.64 (Ar-CH), 69.35 (CH2), 35.28 (NCH3). MS, m/z: 296.20 (M+). Analysis calcd. for C16H16N4O2: C, 64.85; H, 5.44; N, 18.91. Found: C, 64.60; H, 5.08; N, 18.58.
1-(1,2-Dimethyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)-3-phenylurea (6b)
Brown crystals, 87% yield, M. p. 161–163 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.91 (s, 1H, NNHCONH, exch.), 8.61 (s, 1H, NNHCONH, exch.), 7.73 (dd, J = 7.7, 1.6 Hz, 1H, quinazolinone-C5-H), 7.53–7.39 (m, 3H, quinazolinone-C7-H and phenyl-C2,6-H), 7.34–7.22 (m, 2H, phenyl-C3,5-H), 6.97 (t, J = 7.4 Hz, 1H, quinazolinone-C6-H), 6.84–6.74 (m, 2H, quinazolinone-C8-H and phenyl-C4-H), 4.92 (q, J = 5.9 Hz, 1H, CHCH3), 2.89 (s, 3H, NCH3), 1.24 (d, J = 5.9 Hz, 3H, CHCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.48 (C), 154.65 (C), 146.83 (C), 139.44 (C), 134.22 (C), 128.70 (2 × Ar-CH), 127.89 (Ar-CH), 122.12 (Ar-CH), 118.56 (2 × Ar-CH), 117.47 (Ar-CH), 115.06 (Ar-CH), 112.74 (Ar-CH), 75.71 (CHCH3), 34.74 (NCH3), 14.08 (CHCH3). MS, m/z: 310.10 (M+), 311.10 (M++1). Analysis calcd. for C17H18N4O2: C, 65.79; H, 5.85; N, 18.05. Found: C, 65.55; H, 5.46; N, 18.06.
1-(1-Methyl-4-oxo-2-phenyl-1,4-dihydroquinazolin-3(2H)-yl)-3-phenylurea (6c)
Yellow crystals, 89% yield, M. p. 180–181 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (s, 1H, NNHCONH, exch.), 8.34 (s, 1H, NNHCONH, exch.), 7.79 (dd, J = 7.7, 1.6 Hz, 1H, quinazolinone-C5-H), 7.49–7.22 (m, 10H, 2 × (phenyl-C2,3,4,5,6-H)), 6.97 (t, J = 7.4 Hz, 1H, quinazolinone-C7-H), 6.84 (t, J = 7.5 Hz, 1H, quinazolinone-C6-H), 6.69 (d, J = 8.2 Hz, 1H, quinazolinone-C8-H), 5.90 (s, 1H, quinazolinone-C2-H), 2.83 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.59 (C), 154.29 (C), 146.70 (C), 139.27 (C), 137.30 (C), 134.62 (C), 129.05 (2 × Ar-CH), 128.76 (d, J = 8.4 Hz, 4 × Ar-CH), 127.95 (Ar-CH), 126.50 (2 × Ar-CH), 118.18 (2 × Ar-CH), 117.65 (Ar-CH), 114.50 (Ar-CH), 112.25 (Ar-CH), 80.38 (quinazolinone-C2-H), 35.02 (NCH3). MS, m/z: 372.20 (M+), 374.20 (M++2). Analysis calcd. for C22H20N4O2: C, 70.95; H, 5.41; N, 15.04. Found: C, 70.59; H, 5.09; N, 15.30.
1-(2-(4-Fluorophenyl)-1-methyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)-3-phenylurea (6d)
Off-white crystals, 86% yield, M. p. 230–232 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (s, 1H, NNHCONH, exch.), 8.39 (s, 1H, NNHCONH, exch.), 7.80 (d, J = 9.1 Hz, 1H, 1H, quinazolinone-C5-H), 7.40 (m, 5H, quinazolinone-C7-H, phenyl-C2,6-H and 4-fluorophenyl-C2,6-H), 7.24 (m, 4H, phenyl-C3,5-H and 4-fluorophenyl-C3,5-H), 6.98 (t, J = 7.3 Hz, 1H, phenyl-C4-H), 6.86 (t, J = 7.4 Hz, 1H, quinazolinone-C6-H), 6.71 (d, J = 8.3 Hz, 1H, quinazolinone-C8-H), 5.94 (s, 1H, quinazolinone-C2-H), 2.82 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 164.11 (C), 161.93 (C), 161.68 (C), 154.76 (C), 147.06 (C), 139.71 (C), 135.15 (C), 134.12 (C), 129.21 (2 × Ar-CH), 128.44 (2 × Ar-CH), 122.63 (Ar-CH), 118.71 (Ar-CH), 118.25 (Ar-CH), 116.10 (Ar-CH), 115.88 (2 × Ar-CH), 114.90 (2 × Ar-CH), 112.79 (Ar-CH), 80.11 (quinazolinone-C2-H quinazolinone-C2-H), 35.37 (NCH3). MS, m/z: 390.20 (M+). Analysis calcd. for C22H19FN4O2: C, 67.68; H, 4.91; N, 14.35. Found: C, 67.90; H, 4.66; N, 14.28.
1-(1-Methyl-2-(4-nitrophenyl)-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)-3-phenylurea (6e)
Yellow fluffy crystals, 79% yield, M. p. 241–243 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.83 (s, 1H, NNHCONH, exch.), 8.52 (s, 1H, NNHCONH, exch.), 8.23 (d, J = 8.8 Hz, 2H, 4-nitrophenyl-C3,5-H), 7.80 (d, J = 7.7, 1H, quinazolinone-C5-H), 7.59 (d, J = 8.8 Hz, 2H, -nitrophenyl-C2,6-H), 7.52–7.38 (m, 3H, phenyl-C3,4,5-H), 7.33–7.21 (m, 2H, phenyl-C2,6-H), 6.97 (t, J = 7.4 Hz, 1H, quinazolinone-C7-H), 6.88 (t, J = 7.5 Hz, 1H, quinazolinone-C6-H), 6.73 (d, J = 8.2 Hz, 1H, quinazolinone-C8-H), 6.12 (s, 1H, quinazolinone-C2-H), 2.86 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.26 (C), 154.33 (C), 147.90 (C), 146.39 (C), 144.40 (C), 139.24 (C), 134.77 (C), 128.75 (2 × Ar-CH), 128.09 (2 × Ar-CH), 128.05 (Ar-CH),123.87 (2 × Ar-CH), 122.20 (Ar-CH), 118.36 (Ar-CH), 118.17 (2 × Ar-CH), 114.58 (Ar-CH), 112.60 (Ar-CH), 79.31 (quinazolinone-C2-H), 35.12 (NCH3). MS, m/z: 418.40 (M++1). Analysis calcd. for C22H19N5O4: C, 63.30; H, 4.59; N, 16.78. Found: C, 63.40; H, 4.34; N, 17.05.
1-(2-(4-Methoxyphenyl)-1-methyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)-3-phenylurea (6f)
White fluffy crystals, 79% yield, M. p. 198–200 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.75 (s, 1H, NNHCONH, exch.), 8.29 (s, 1H, NNHCONH, exch.), 7.78 (dd, J = 7.7, 1.6 Hz, 1H, quinazolinone-C5-H), 7.50–7.37 (m, 3H, phenyl-C3,4,5-H), 7.33–7.18 (m, 4H, 4-methoxyphenyl-C2,6-H and phenyl-C2,6-H), 7.02–6.79 (m, 4H, quinazolinone-C7,6-H and 4-methoxyphenyl-C3,5-H), 6.68 (d, J = 8.3 Hz, 1H, quinazolinone-C8-H), 5.83 (s, 1H, quinazolinone-C2-H), 3.72 (s, 3H, OCH3), 2.79 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.62 (C), 159.80 (d, J = 3.8 Hz, C), 154.26 (C), 146.75 (C), 139.29 (C), 134.57 (C), 129.35 (2 × Ar-CH), 128.80 (2 × Ar-CH), 127.92 (Ar-CH), 127.87 (Ar-CH), 122.11 (Ar-CH), 118.15 (2 × Ar-CH), 117.85 (d, J = 60.9 Hz, Ar-CH), 114.23 (d, J = 41.9 Hz, Ar-CH), 114.02 (2 × Ar-CH), 113.11 (d, J = 183.1 Hz, Ar-CH), 80.05 (quinazolinone-C2-H), 55.14 (OCH3), 34.86 (NCH3). MS, m/z: 402.20 (M+). Analysis calcd. for C23H22N4O3: C, 68.64; H, 5.51; N, 13.92. Found: C, 68.36; H, 5.19; N, 14.02.
1-(4-Methoxyphenyl)-3-(1-methyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)urea (6g)
White fluffy crystals, 87% yield, M. p. 219–221 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.80 (s, 1H, NNHCONH, exch.), 8.50 (s, 1H, NNHCONH, exch.), 7.75 (d, J = 7.7 Hz, 1H, quinazolinone-C5-H), 7.49–7.40 (m, 1H, quinazolinone-C7-H), 7.35 (d, J = 9.1 Hz, 2H, phenyl-C2,6-H), 6.89–6.83 (m, 4H, phenyl-C3,5-H and quinazolinone-C6,8-H), 4.69 (s, 2H, CH2), 3.70 (s, 3H, OCH3), 2.88 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 163.34 (C), 154.95 (C), 154.62 (C), 149.59 (C), 134.01 (C), 132.42 (C), 128.26 (Ar-CH), 120.28 (2 × Ar-CH), 118.23 (Ar-CH), 115.98 (Ar-CH), 113.90 (2 × Ar-CH), 112.64 (Ar-CH), 69.43 (CH2), 55.16 (OCH3), 35.29 (NCH3). MS, m/z: 326.10 (M+). Analysis calcd. for C17H18N4O3: C, 62.57; H, 5.56; N, 17.17. Found: C, 62.62; H, 5.31; N, 16.81.
1-(4-Methoxyphenyl)-3-(1-methyl-4-oxo-2-phenyl-1,4-dihydroquinazolin-3(2H)-yl)urea (6h)
Grey powder, 77% yield, M. p. 196–198 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (s, 1H, NNHCONH, exch), 8.26 (s, 1H, NNHCONH, exch), 7.79 (d, J = 7.6 Hz, 1H, quinazolinone-C5-H), 7.42–7.17 (m, 8H, quinazolinone-C7-H, 4-methoxyphenyl-C2,6-H and phenyl-C2,3,4,5,6-H), 6.86–6.81 (m, 3H, quinazolinone-C6-H and 4-methoxyphenyl-C3,5-H), 6.68 (d, J = 8.3 Hz, 1H, quinazolinone-C8-H), 5.89 (s, 1H, quinazolinone-C2-H), 3.70 (s, 3H, OCH3), 2.82 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.62 (C), 154.60 (C), 152.96 (C), 146.69 (C), 137.34 (C), 134.58 (C), 132.95 (C), 129.02 (Ar-CH), 128.70 (2 × Ar-CH), 127.93 (Ar-CH), 126.50 (Ar-CH), 119.92 (2 × Ar-CH), 117.62 (s), 114.55 (Ar-CH), 113.97 (2 × Ar-CH), 112.22 (Ar-CH), 80.42 (quinazolinone-C2-H), 55.15 (OCH3), 35.02 (NCH3). MS, m/z: 402.20 (M+), 403.20 (M++1). Analysis calcd. for C23H22N4O3: C, 68.64; H, 5.51; N, 13.92. Found: C, 68.85; H, 5.67; N, 14.20.
1-(4-Methoxyphenyl)-3-(1-methyl-2-(4-nitrophenyl)-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)urea (6i)
Yellow powder, 83% yield, M. p. 217–219 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.66 (s, 1H, NNHCONH, exch.), 8.44 (s, 1H, NNHCONH, exch.), 8.22 (d, J = 8.8 Hz, 2H, 4-nitrophenyl-C3,5-H), 7.80 (dd, J = 7.7, 1.6 Hz, 1H, quinazolinone-C5-H), 7.59 (d, J = 8.7 Hz, 2H, 4-nitrophenyl-C2,6-H), 7.45 (t, J = 7.8 Hz, 1H, quinazolinone-C7-H), 7.32 (d, J = 9.0 Hz, 2H, 4-methoxyphenyl-C2,6-H), 6.89–6.83 (m, 3H, quinazolinone-C6-H and 4-methoxyphenyl-C3,5-H), 6.72 (d, J = 8.3 Hz, 1H, quinazolinone-C8-H), 6.11 (s, 1H, quinazolinone-C2-H), 3.70 (s, 3H, OCH3), 2.86 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.32 (2 × C), 154.66 (C), 154.55 (C), 147.89 (C), 146.39 (C), 144.45 (C), 134.75 (C), 132.25 (Ar-CH), 128.08 (Ar-CH), 128.05 (2 × Ar-CH), 123.87 (2 × Ar-CH), 120.18 (2 × Ar-CH), 118.15 (Ar-CH), 114.63 (Ar-CH), 113.93 (2 × Ar-CH), 112.58 (Ar-CH), 79.36 (quinazolinone-C2-H), 55.16 (OCH3), 35.13 (NCH3). MS, m/z: 448.10 (M++1). Analysis calcd. for C23H21N5O5: C, 61.74; H, 4.73; N, 15.65. Found: C, 62.04; H, 4.68; N, 15.71.
1-(4-Methoxyphenyl)-3-(2-(4-methoxyphenyl)-1-methyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)urea (6j)
White microcrystals, 86% yield, M. p. 205–207 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (s, 1H, NNHCONH, exch.), 8.21 (s, 1H, NNHCONH, exch.), 7.79 (d, J = 7.7 Hz, 1H, quinazolinone-C5-H), 7.43 (t, J = 8.5 Hz, 1H, quinazolinone-C7-H), 7.33 (d, J = 9.0 Hz, 2H, 4-methoxphenyl-C2,6-H), 7.22 (d, J = 8.7 Hz, 2H, 4-methoxyphenyl-C3,5-H), 6.94–6.79 (m, 5H, 4-methoxyphenyl-C2,3,5,6-H and quinazolinone-C6-H), 6.68 (d, J = 8.3 Hz, 1H, quinazolinone-C8-H), 5.84 (s, 1H, quinazolinone-C2-H), 3.71 (s, 6H, 2 × OCH3), 2.80 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 162.11 (2 × C), 160.22 (C), 155.05 (C), 154.93 (C), 147.20 (C), 135.00 (C), 132.78 (Ar-CH), 129.85 (2 × Ar-CH), 128.36 (Ar-CH), 128.33 (2 × Ar-CH), 120.38 (2 × Ar-CH), 117.97 (Ar-CH), 114.95 (Ar-CH), 114.46 (2 × Ar-CH), 114.43(Ar-CH), 112.63 (Ar-CH), 80.55 (quinazolinone-C2-H), 55.60 (2 × OCH3), 35.33 (NCH3). MS, m/z: 432.30 (M+). Analysis calcd. for C24H24N4O4: C, 66.65; H, 5.59; N, 12.96. Found: C, 66.66; H, 5.26; N, 12.99.
1-(4-Fluorophenyl)-3-(1-methyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)urea (6k)
White fluffy crystals, 88% yield, M.p. 246–248 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H, NNHCONH, exch.), 8.61 (s, 1H, NNHCONH, exch.), 7.75 (dd, J = 7.7, 1.6 Hz, 1H, quinazolinone-C5-H), 7.49–7.43 (m, 3H, quinazolinone-C7-H and phenyl-C2,6-H), 7.10 (t, J = 8.9 Hz, 2H, phenyl-C3,5-H), 6.96–6.81 (m, 2H, quinazolinone-C6,8-H), 4.69 (s, 2H, CH2), 2.88 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 163.36 (C), 158.67 (C), 156.31 (C), 154.85 (C), 149.61 (C), 135.79 (C), 134.06 (C), 128.28 (Ar-CH), 120.34 (2 × Ar-CH), 118.26 (Ar-CH), 115.93 (Ar-CH), 115.32 (Ar-CH), 115.10 (Ar-CH), 112.67 (Ar-CH), 69.37 (CH2), 35.30 (NCH3). MS, m/z: 314.30 (M+), 315.30 (M++1). Analysis calcd. for C16H15FN4O2: C, 61.14; H, 4.81; N, 17.83. Found: C, 60.86; H, 4.78; N, 17.99.
1–(1,2-Dimethyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)-3-(4-fluorophenyl)urea (6l)
White crystals, 86% yield, M. p. 187–190 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.95 (s, 1H, NNHCONH, exch.), 8.64 (s, 1H, NNHCONH, exch.), 7.72 (dd, J = 7.7, 1.5 Hz, 1H, quinazolinone-C5-H), 7.58–7.38 (m, 3H, quinazolinone-C7-H and phenyl-C2,6-H), 7.10 (t, J = 8.9 Hz, 2H, phenyl-C3,5-H), 6.87–6.71 (m, 2H, quinazolinone-C6,8-H), 4.91 (q, J = 5.8 Hz, 1H, CHCH3), 2.88 (s, 3H, NCH3), 1.24 (d, J = 5.9 Hz, 3H, CHCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.63 (C), 158.74 (C), 156.37 (C), 154.83 (C), 146.88 (C), 135.81 (C), 134.29 (C), 127.95 (Ar-CH), 120.50 (2 × Ar-CH), 117.52 (Ar-CH), 115.33 (2 × Ar-CH), 115.11 (Ar-CH), 112.79 (Ar-CH), 75.77 (CHCH3), 34.78 (NCH3), 14.10 (CHCH3). MS, m/z: 328.20 (M+), 329.20 (M++1). Analysis calcd. for C17H17FN4O2: C, 62.19; H, 5.22; N, 17.06. Found: C, 62.33; H, 5.49; N, 17.32.
1-(4-Fluorophenyl)-3-(1-methyl-4-oxo-2-phenyl-1,4-dihydroquinazolin-3(2H)-yl)urea (6m)
Pale-yellow crystals, 79% yield, M. p. 220–222 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.81 (s, 1H NNHCONH, exch.), 8.38 (s, 1H NNHCONH, exch.), 7.78 (d, J = 7.7 Hz, 1H, quinazolinone-C5-H), 7.50–7.29 (m, 8H, quinazolinone-C7-H, 4-fluorophenyl-C2,6-H and pheyl-C2,3,4,5,6-H), 7.10 (t, J = 8.8 Hz, 2H, 4-fluorophenyl-C3,5-H), 6.84 (t, J = 7.5 Hz, 1H, quinazolinone-C6-H), 6.69 (d, J = 8.4 Hz, 1H, quinazolinone-C8-H), 5.89 (s, 1H, quinazolinone-C2-H), 2.82 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.58 (C), 158.62 (C), 156.26 (C), 154.39 (C), 146.70 (C), 137.26 (C), 135.64 (C), 134.59 (C), 129.02 (Ar-CH), 128.67 (2 × Ar-CH), 127.93 (Ar-CH), 126.52 (2 × Ar-CH), 119.94 (2 × Ar-CH), 117.63 (Ar-CH), 115.38 (2 × Ar-CH), 115.16 (Ar-CH), 112.24 (Ar-CH), 80.37 (quinazolinone-C2-H), 35.00 (NCH3). MS, m/z: 390.20 (M+). Analysis calcd. for C22H19FN4O2: C, 67.68; H, 4.91; N, 14.35. Found: C, 67.72; H, 4.71; N, 14.06.
1-(4-Fluorophenyl)-3-(2–(4-fluorophenyl)-1-methyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)urea (6n)
Off-white crystals, 83% yield, M. p. 214–216 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H, NNHCONH, exch.), 8.44 (s, 1H, NNHCONH, exch.), 7.80 (d, J = 9.1 Hz, 1H, quinazolinone-C5-H), 7.50–7.39 (m, 3H, quinazolinone-C7-H and 4-fluorophenyl-C2,6-H), 7.39–7.27 (m, 2H, 4-fluorophenyl-C2,6-H), 7.15 (m, 4H, 2 × 4-fluorophenyl-C3,5-H), 6.86 (t, J = 7.4 Hz, 1H, quinazolinone-C6-H), 6.71 (d, J = 8.3 Hz, 1H, quinazolinone-C8-H), 5.93 (s, 1H, quinazolinone-C2-H), 2.81 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 164.11 (C), 161.99 (C), 161.67 (C), 159.14 (C), 156.77 (C), 154.90 (C), 147.08 (C), 136.02 (C), 135.18 (C), 134.04 (C), 129.27 (Ar-CH), 129.19 (Ar-CH), 128.45 (Ar-CH), 120.64 (Ar-CH), 120.56 (Ar-CH), 118.27 (Ar-CH), 116.06 (Ar-CH), 115.84 (2 × Ar-CH), 115.62 (Ar-CH), 114.89 (Ar-CH), 112.80 (Ar-CH), 80.15 (quinazolinone-C2-H), 35.38 (NCH3). MS, m/z: 408.20 (M+), 409.20 (M++1). Analysis calcd. for C22H18F2N4O2: C, 64.70; H, 4.44; N, 13.72. Found: C, 65.01; H, 4.22; N, 14.01.
1-(4-Fluorophenyl)-3–(1-methyl-2-(4-nitrophenyl)-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)urea (6o)
Yellow powder, 78% yield, M. p. 250–252 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H, NNHCONH, exch.), 8.58 (s, 1H, NNHCONH, exch.), 8.22 (d, J = 8.8 Hz, 2H, 4-nitrophenyl-C3,5-H), 7.80 (dd, J = 7.7, 1.6 Hz, 1H, quinazolinone-C5-H), 7.59 (d, J = 8.8 Hz, 2H, nitrophenyl-C2,6-H), 7.54–7.40 (m, 3H, quinazolinone-C7-H and 4-fluorophenyl-C2,6-H), 7.10 (t, J = 8.9 Hz, 2H, 4-fluorophenyl-C3,5-H), 6.88 (t, J = 7.5 Hz, 1H, quinazolinone-C6-H), 6.73 (d, J = 8.3 Hz, 1H, quinazolinone-C8-H), 6.11 (s, 1H, quinazolinone-C2-H), 2.86 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.28 (C), 158.68 (C), 156.31 (C), 154.46 (C), 147.89 (C), 146.40 (C), 144.39 (C), 135.62 (C), 135.60 (C), 134.76 (C), 128.08 (2 × Ar-CH), 123.84 (2 × Ar-CH), 120.24 (Ar-CH), 120.16 (Ar-CH), 118.16 (Ar-CH), 115.34 (2 × Ar-CH), 115.12 (Ar-CH), 114.87 (Ar-CH), 112.60 (Ar-CH), 79.32 (quinazolinone-C2-H), 35.11 (NCH3). MS, m/z: 436.30 (M++1); 437.30 (M++2). Analysis calcd. for C22H18FN5O4: C, 60.69; H, 4.17; N, 16.08. Found: C, 60.56; H, 3.97; N, 16.40.
1-(4-Fluorophenyl)-3-(2–(4-methoxyphenyl)-1-methyl-4-oxo-1,4-dihydroquinazolin-3(2H)-yl)urea (6p)
Pale-yellow crystals, 85% yield, M. p. 224–226 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.80 (s, 1H, NNHCONH, exch.), 8.32 (s, 1H, NNHCONH, exch.), 7.77 (dd, J = 7.6, 1.1 Hz, 1H, quinazolinone-C5-H), 7.54–7.35 (m, 3H, quinazolinone-C7-H and 4-fluorophenyl-C2,6-H), 7.21 (d, J = 8.6 Hz, 2H, 4-methoxyphenyl-C2,6-H), 7.10 (t, J = 8.9 Hz, 2H, 4-fluorophenyl-C3,5-H), 6.95–6.78 (m, 3H, quinazolinone-C6-H and 4-methoxyphenyl-C3,5-H), 6.68 (d, J = 8.3 Hz, 1H, quinazolinone-C8-H), 5.81 (s, 1H, quinazolinone-C2-H), 3.71 (s, 3H, OCH3), 2.78 (s, 3H, NCH3). 13C NMR (101 MHz, DMSO-d6) δ 161.58 (C), 159.75 (C), 158.61 (C), 156.24 (C), 154.36 (C), 146.75 (C), 135.66 (C), 134.54 (C), 129.32 (Ar-CH), 127.89 (2 × Ar-CH), 119.98 (Ar-CH), 119.91 (Ar-CH), 117.52 (Ar-CH), 115.37 (Ar-CH), 115.15 (Ar-CH), 114.46 (Ar-CH), 113.98 (2 × Ar-CH), 112.19 (Ar-CH), 80.05 (quinazolinone-C2-H), 55.12 (OCH3), 34.85 (NCH3). MS, m/z: 420.30 (M+). Analysis calcd. for C23H21FN4O3: C, 65.71; H, 5.03; N, 13.33. Found: C, 66.00; H, 4.85; N, 13.20.
Biological activity
In vitro COX-1 and COX-2 inhibitory assay
All final synthesised compounds (6a–p) were tested with an in vitro COX-1/COX-2 inhibition assay kit (Cayman, Ann Arbour, MI, USA) using ovine COX-1 or human recombinant COX-2 and compared to celecoxib, indomethacin, and diclofenac sodium as reference drugs. The half-maximal inhibitory concentration (IC50, μM) was determined, and the COX-2 selectivity index (SI) was calculated as IC50 (COX-1)/IC50 (COX-2).
In vitro 15-LOX inhibition assay
Evaluation of 15-LOX inhibitory activities of all the synthesised compounds 6a–p was carried out using soybean 15-LOX inhibitor screening assay (Cayman, Ann Arbour, MI) and nordihydroguaiaretic acid (NDGA) as reference according to the manufacturer’s instructions.
In vitro EGFR inhibition activity
We performed in vitro EGFR inhibition assay for all our newly synthesised compounds (6a–p) against both wild-type EGFR and the double mutant EGFRL858R/T790M purchased from AssayQuant Technologies, Inc. (Marlboro, MA, USA) as per the manufacturer’s instructions. Osimertinib and afatinib were purchased from MedChemExpress LLC (Monmouth Junction, NJ, USA) and used as references.
Effects on NO, ROS, and cytokines production in LPS-activated RAW 264.7 macrophage cells
TNF-α and IL-6 production were detected in RAW 264.7 macrophage cell culture supernatants following the induction of inflammation by LPS. Moreover, the probes of oxidative species 2,7-dichlorofluorescein diacetate (DCFH-DA) (Molecular Probes) and NO, 4-amino-5-methylamino-2,7-difluorofluorescein diacetate (DAF-FM diacetate) (Molecular Probes) were used to investigate the antioxidant and NO production inhibitory potential of the test compounds. Briefly, RAW 264.7 cells were cultured in black 96 well plates (200 000 cells/mL, 100 µl/well) for 24 h followed by the incubation with the individual test compounds or the reference drugs at different concentrations (12.5, 25, 50, and 100 µM) for 2 h at 37 °C. LPS was then added at a final concentration of 1 µg/mL for an additional 18 h. The cell culture supernatant was collected from cells treated with the different compounds at 25 µM to measure TNF-α and IL-6 levels (1:10 dilution) using DuoSet ELISA kits (R&D Systems, Minneapolis, MN) following the manufacturer’s instructions. Then, the plates were washed to perform ROS and NO detection assays as detailed in our previous studiesCitation16,Citation27.
Measurement of anticancer activity against a panel of 60 cell lines
Screening of the anticancer activity of the newly synthesised compounds (6a, 6c, 6d, 6e, 6j, 6l, 6m, 6n, 6o, and 6p) was performed by the US National Cancer Institute (NCI) using 60 different human tumour cell lines in accordance with standard procedures formerly reportedCitation30.
Cell viability assay against normal cell line
To evaluate the possible non-selective cytotoxic effect of the investigated compounds (6a, 6c, 6d, 6e, 6j, 6l, 6m, 6n, 6o, and 6p) on normal cells, their effect on RAW 264.7 cells viability was assessed at different concentrations (12.5, 25, 50, and 100 µM). Cell viability was measured using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4 sulfophenyl)-2H-tetrazolium (MTS) assay (Promega, Madison, Wisconsin) according to the manufacturer’s instructions.
Molecular modelling
The X-ray crystal structures of COX-1 (PDB code: 1EQG/2.61 Å), COX-2 (PDB code: 1CX2/3.00 Å), 15-LOX (PDB code: 4NRE/2.63 Å), EGFRWT (PDB code: 1M17/2.60 Å), and EGFRT790M/L858R (PDB code: 5EDQ/2.80 Å) were retrieved from the Protein Data Bank (http://www.rcsb.org)Citation31. The molecular docking studies were implemented using the Molecular Operating Environment; MOE 2019.0102 (Chemical Computing Group, Montreal, CA)Citation32. The crystal structures were individually prepared using the quick preparation protocol through the Amber10: EHT forcefield. The compounds were drawn using the Chemdraw® program, then transferred to the MOE as smiles format. The compound energy was minimised with a root mean square (RMS) gradient of 0.0001 kcal/mol. The co-crystallised ligands were re-docked into the active site for validation of the docking results by measuring the root mean square deviations (RMSD). The docking protocol is a triangle matcher, using London dG as the initial scoring approach and GBVI/WSA dG as the final scoring approach. The docking energy scores (S; Kcal/mol) and visual inspection of 2D and 3D planes of the ligand–enzyme interactions were used for the analysis of the docking results. A flexible alignment study was performed on compound 6e and the co-crystallised substrate mimic (C8E) of 15-LOX, using MOE 2019.0102. The obtained conformations were evaluated according to the score of the configuration alignment (S; Kcal/mol).
Results and discussion
Chemistry
The synthesis of compounds (6a–p) was carried out in four steps as illustrated in Scheme 1. First, appropriate anilines (1a–c) were reacted with ethyl chloroformate in dichloromethane and trimethylamine to yield the corresponding carbamates (2a–c). Carboxamides (3a–c) were then synthesised by hydrazinolysis of the ethyl ester of the carbamates (2a–c).
Scheme 1. Reagents and conditions: (i) ethyl chloroformate/triethylamine/dichloromethane/0 °C—rt., 2h (ii) hydrazine hydrate 98%, ethanol, reflux, 5h (iii) ethanol/glacial acetic acid, reflux, 4 h; (iv) 37% formaline or acetaldehyde, ethanol/glacial acetic acid, reflux, 2 h; and (v) appropriate aromatic aldehyde, glacial acetic acid, reflux, 4 h.
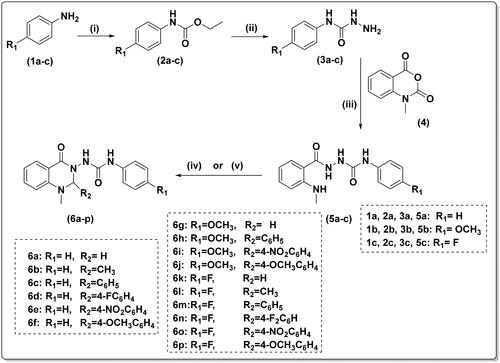
To obtain the urea linker, N-methyl isatoic anhydride (4) was reacted with the appropriate carboxamides (3a–c) in refluxing ethanol with a catalytic amount of glacial acetic acid, yielding key intermediates 5a–c. The compounds 5a–c were identified as having four NH groups in 1H-NMR revealed at δ 10.01–7.54 ppm.
Finally, cyclization of the key intermediate compounds (5a–c) to the target quinazolinones (6a–p) was achieved using different aldehydes. 1H-NMR and 13C-NMR spectroscopy, mass spectrometry, and elemental analysis were used to confirm the final compounds’ structures. In 1H-NMR, singlet signals of both NHCONHNH at δ 10.01–9.98 ppm and NHCH3 at 7.57–5.54 ppm disappeared. The NCHN signal of quinazolinone-C2-H appeared at 6.11–4.69 ppm. Moreover, 13C-NMR spectra revealed the appearance of NCN signal of quinazolinone-C2-H at 80.42–69.35 ppm.
Biological activity
COX-1 and COX-2 inhibitory activities
The new quinazolines 6i and 6n had more potent COX-2 inhibitory profiles (IC50 = 0.47 and 0.65 µM) than the two reference drugs diclofenac sodium and indomethacin (IC50 = 0.76 and 1.57 µM, respectively); compounds 6e and 6l had greater COX-2 inhibitory activity than indomethacin, with IC50 values 0.83 and 1.49 µM, respectively, but were less potent than the reference drug celecoxib (IC50 = 0.03 µM). On the other hand, compounds 6a, 6e, 6d, 6f, 6g, 6i, 6m, and 6n showed moderate COX-1 inhibitory activity (IC50 = 3.05–12.46 µM) in comparison to the references celecoxib and diclofenac sodium (IC50 = 7.03 and 4.11 µM, respectively). In terms of COX-2 selectivity, compounds 6e, 6h, 6i, 6k, 6o, and 6n were 2–5 times more selective than diclofenac sodium (the reference drug) towards COX-2. The selectivity of the new compounds was significantly lower than that of celecoxib, which could be considered an advantage in avoiding the cardiovascular side effects of highly selective COX-2 inhibitors ().
Table 1. In vitro COX-1, COX-2, COX-2 selectivity index, and 15-LOX inhibitory activities of the final synthesised compounds (6a–p) and references.
In general, the preference of substitution of phenyl urea ring arranged as F > OCH3 > unsubstituted considering COX-2 selectivity. It is also notable that quinazolines with p-nitro phenyl substitution at 2-position (6e, 6i, 6o) showed superior COX-2 selectivity than their counterparts carrying the same phenyl urea derivative ().
In vitro 15-LOX inhibition activity
Nearly all the new quinazolines 6a–o revealed potent inhibitory activities against 15-LOX (IC50 = 1.36–2.32 µM) that showed comparable potency to that of the reference NDGA (IC50 = 1.33 µM) except for compound 6p that showed lower potency (IC50 = 3.87 µM) ().
Among the unsubstituted phenyl urea derivatives (6a–f), compounds 6a, 6c, and 6e carrying no substitution, phenyl and p-nitrophenyl at quinazoline C2, respectively, showed the most potent 15-LOX inhibitory activities (IC50 = 1.48, 1.36, and 1.76 µM). For the p-methoxyphenyl urea derivatives (6g–j), compound 6g with 2-unsubstituted quinazoline was the most potent one (IC50 = 1.61 µM). Among the p-fluorophenyl urea derivatives (6k–p), compounds 6l and 6o carrying methyl and p-nitrophenyl at quinazoline C2 showed superior 15-LOX inhibitory potencies (IC50 = 1.58 and 1.42 µM).
In vitro EGFR inhibition activity
We performed in vitro EGFR inhibition assay for all our newly synthesised compounds (6a–p) against both wild-type EGFR and the double mutant EGFRL858R/T790M using osimertinib and afatinib as references. Compounds 6a, 6c, 6d, 6e, 6g, 6j, and 6p showed micromolar anti-EGFR activity that was less potent than the used references but was more selective against the double mutant EGFRL858R/T790M (SI = >217–8.9) than the two reference drugs osimertinib and afatinib (SI = 8.4 and 0.11, respectively).
Moreover, compounds 6h, 6i, 6l, 6m, 6n, and 6o were more selective against the double mutant EGFRL858R/T790M (SI = >7.4–1.5) than afatinib (SI = 0.11). Compounds 6b, 6f, and 6k did not show any inhibitory activity against both EGFR types.
Except for compounds 6e and 6o, which had IC50 values of 48.69 and 52.44 µM, respectively, none of our compounds showed significant activity against EGFRWT. Compound 6a exhibited the greatest EGFRL858R/T790M inhibitory activity, with IC50 values 0.46 µM and no significant activity against EGFRWT.
Among the unsubstituted phenyl urea derivatives (6a–f), compounds 6a, 6d, and 6e carrying unsubstituted, p-fluorophenyl, and p-nitrophenyl at quinazoline C2, respectively, showed the best EGFRL858R/T790M inhibitory activities (IC50 = 0.46, 5.57, and 5.48 µM). Compounds 6j with p-methoxy at quinazoline C2 were the most potent of the p-methoxyphenyl urea derivatives (6g–j) (IC50 = 4.03 µM). Among the p-fluorophenyl urea derivatives (6k–p), compounds 6m and 6p carrying p-fluorophenyl and p-methoxyophenyl at quinazoline C2 showed superior EGFRL858R/T790M inhibitory potencies (IC50 = 4.07 and 5.41 µM) ().
Table 2. In vitro inhibitory activities of the final synthesised compounds (6a–p) and references against wild-type EGFR and the double mutant EGFRL858R/T790M.
Despite being less potent than the references, our compounds demonstrated greater selectivity against EGFRL858R/T790M than the EGFRWT. Moreover, compound 6o has a selectivity profile comparable to osimertinib and nearly 80-fold greater selectivity than afatinib. Interestingly, 6o had the strongest anti-inflammatory effect against cytokine production, as well as a strong antioxidant and NO release inhibitory effects.
Effects on NO and ROS production in LPS-activated RAW 264.7 macrophage cells
LPS-activated RAW 264.7 macrophage cells are a popular in vitro model for studying various inflammatory responses and screening the mechanism of action of new anti-inflammatory candidates. The bacterial toxin LPS causes a strong inflammatory response in RAW 264.7 cells, resulting in increased production of several inflammatory mediators, including COX-2Citation33, and nitric oxide (NO) via the induction of the inducible isoform of nitric oxide synthaseCitation34. Oxidative stress is the imbalance between oxidants and antioxidants, and occurs as a result of excessive generation of reactive oxygen species (ROS) leading to cellular injuryCitation35. ROS plays an important role in the inflammatory response, including LPS-mediated inflammation, and can stimulate the production of a wide range of inflammatory cytokinesCitation36. Moreover, increased ROS production is involved in the pathophysiology of multiple pathological conditions, including cancerCitation37. As a result, targeting ROS with candidates with anti-oxidant potential is a successful strategy for treating both inflammation and cancer, as evidenced by numerous in vivo and in vitro studiesCitation36,Citation38,Citation39. Compounds 6a–p were evaluated for their ability to inhibit NO and ROS production in LPS-activated RAW 264.7 macrophage cells.
All the tested compounds 6a–p inhibited the production of the inflammatory mediator NO (IC50 = 0.97–13.54 µM) significantly more potent than the three reference drugs celecoxib, diclofenac, and indomethacin (IC50 = 14.39, 24.08, and 46.45 µM, respectively) with the exception of compound 6n that showed a comparable potency to celecoxib (IC50 = 19.85 µM) and compound 6e that did not show any significant NO inhibitory activity. The best compounds were 6a, 6b, 6c, 6g, and 6o (IC50 = 0.97–1.97 µM), which showed ∼7–14-fold better IC50 values than that of celecoxib (IC50 = 14.39 µM) and 12–24-fold better IC50 values than that of diclofenac (IC50 = 24.08 µM) ().
Table 3. In vitro inhibitory activities of all final synthesised compounds (6a–p) and references on LPS-induced ROS, NO, TNF-α, and IL-6 production in RAW 264.7 cells.
Except for nitro-containing compounds, unsubstituted phenyl urea derivatives (6a–f) outperform their corresponding counterparts with p-methoxyphenyl urea derivatives (6g–j) or p-fluorophenyl urea derivatives (6k–p).
Regarding ROS production, the majority of the tested compounds exhibited potent inhibitory activity, comparable to the two references diclofenac and indomethacin. Compounds 6i and 6k were the most effective at lowering ROS levels, with IC50 values of 5.90 and 5.04 µM, respectively (more potent than that of celecoxib, 7.57 µM, and ≈8-fold more potent than diclofenac, 43.78 µM). Unfortunately, neither compound 6e nor compound 6g inhibited ROS production significantly.
Effects on TNF-α and IL-6 production in LPS-activated RAW 264.7 macrophages cells
LPS activates macrophages to induce the production of inflammatory cytokines, including TNF-α and IL-6Citation40. Excessive cytokine production following LPS injection in vivo can result in an uncontrolled inflammatory response that leads to severe systemic inflammatory response, tissue damage, and sepsisCitation41. Our findings showed that compounds 6i, 6k, and 6o had IL-6 levels (84.60, 88.53, and 37.23 pg/mL) that are ∼2–4.5-fold lower than celecoxib (176.4 pg/mL). Compounds 6a, 6e, 6f, 6h, 6j, 6m, and 6p (164.6–328.9 pg/mL) were significantly more potent than diclofenac (342.90 pg/mL).
In terms of inhibiting the production of the inflammatory mediator TNF-α, compound 6o showed TNF-α level, which is ∼10 times lower than celecoxib (39.37 vs. 383.3 pg/mL) and nearly 17 times lower than diclofenac (666.6 pg/mL). Compounds 6b, 6h, and 6j exhibited comparable inhibitory activities (779.60, 720.60, and 757.30 pg/mL, respectively) to that of diclofenac (666.6 pg/mL). Unfortunately, the other compounds showed lower activities compared to the two references, celecoxib and diclofenac ().
Interestingly, the p-fluorophenyl urea derivative carrying p-nitrophenyl at quinazoline C2 6o showed the highest activity in terms of inhibiting the production of either TNF-α or IL-6.
In vitro anticancer screening against 60 NCI cancer cell lines panel
Ten of the synthesised compounds that showed the best inhibitory activities, namely (6a, 6c, 6d, 6e, 6j, 6l, 6m, 6n, 6o, and 6p), were sent for testing to the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP), division of cancer treatment and diagnosis, NIH, Bethesda, Maryland, USA (www.dtp.nci.nih.gov)Citation30. In vitro anticancer screening was carried out for the selected compounds at a single dose (10−5 M) against 60 different human tumour cell lines of nine different cancer cell types: leukaemia, lung, colon, CNS, melanoma, ovarian, renal, prostate, and breast cancers.
The screening results revealed that compound 6n (the p-fluorophenyl urea derivative carrying p-fluorophenyl at quinazoline C2) showed the broadest anticancer activity against 32 out of the 60 NCI cell lines with growth inhibition percentages in the 10.09–57.11% range. Also, the p-fluorophenyl urea derivatives 6m and 6o showed anticancer activities against 19 (10.52–40.1%) and 21 (10.66–41.34%) cell lines, respectively.
Compounds 6e (the unsubstituted phenyl urea derivative carrying p-nitrophenyl at quinazoline C2) and 6j (p-methoxyphenyl urea derivative carrying p-methoxyphenyl at quinazoline C2) exhibited the most potent anticancer activity against breast cancer cell line BT-459 with growth inhibition percentages of 67.14 and 70.07%, respectively. In addition, compound 6e exhibited good anticancer activity against colon cancer cell line HCT-116 and melanoma cell line M14 with cell growth inhibition percentages of 53.14 and 67.23%, respectively ().
Table 4. The growth inhibition percentages for in vitro panel/cell lines from the single dose (10−5 M) test of compounds (6a, 6c–e, 6j, 6l–p).
It is worth mentioning, that 8 out of the 10 tested compounds showed inhibitory activity against the ovarian cancer cell line OVCAR-3 and the renal cancer cell line UO-31 with cell growth inhibition ranging between 10.24–41.94 and 19.47–57.11%, respectively.
Unfortunately, compound 6a (the unsubstituted phenyl urea derivative with no substituent at quinazoline C2) did not show any anticancer activities.
Cell viability assay against normal cell line
Cell viability testing for the newly synthesised compounds (6a, 6c, 6d, 6e, 6j, 6l, 6m, 6n, 6o, and 6p) on RAW 264.7 cells showed that all the compounds were non-toxic and had a cell viability percentage that exceeded 90% of the control values when tested at concentrations up to 50 μM. Moreover, most of the compounds showed a cell viability percentage in the 83–100% range at a concentration of 100 μM, indicating their safety and selectivity (Supplementary Material Figure 1).
Molecular modelling
Docking study into COX-1/COX-2
The docking studies into COX-1 (PDB: 1EQG)Citation42 and COX-2 (PDB: 1CX2)Citation43 were initiated by re-docking the co-crystallised ligands; ibuprofen and SC-558 [1-phenylsulfonamide-3-trifluoromethyl-5-parabromophenylpyrazole] into the active sites, respectively. The results revealed that the re-docked ibuprofen and SC-558 showed 0.7825 and 0.5734 Å for RMSD values, respectively, validating the docking procedure.
The targeted compounds 6a–p were docked into both COX-1 and COX-2. In order to understand the biological results; the binding modes, docking energy, and interactions of the compounds were assessed against the co-crystallised ligands, ibuprofen, and SC-588. The docking energy for the compounds 6a–p ranged from −8.4697 to −5.4624 Kcal/mol for COX-2 and −6.5881 to −5.3863 Kcal/mol for COX-1 (Supplementary Material Table 1).
Within the 6k–p series bearing p-fluorophenyl urea, we found that compounds 6k, 6n–p have better COX-2 selectivity indices, revealed by the docking studies; as the p-fluorophenyl moiety (at urea linker) occupied a pocket surrounded by hydrophobic residues; Val116, Val349, Leu359, Trp387, Ile517, Phe518, and Ala527. Compounds 6k, 6o, and 6p showed arene-H bonds with Tyr355, Leu531, and Ser353, respectively. Compound 2n exhibited numerous H-bonds with Ser353, Val349, and Ala527 in addition to arene-H interaction with Tyr355, Trp387, and Leu531.
On the contrary, binding modes for the compounds 6k, 6n–p showed no interaction within the COX-1 active site except 6p, which formed arene-H bond with Tyr355. Their p-fluorophenyl moiety (at urea linker) was surrounded with hydrophilic amino acid residues (Glu524 and Ser353), which might result in repulsion by the fluoro atom.
Within 6g–j series bearing p-methoxyphenyl urea, methoxy group of the compounds 6h–i showed hydrophobic contact with His90, Ser353, and Ala 516 in the COX-2 active site. In addition to arene-H and arene-cation formation with the quinazolinonyl moiety of the compound 6i. The compounds were deeply oriented within the COX-2 pocket with S scores of −7.5751 and −7.1059 Kcal/mol, respectively. However, in COX-1, the quinazolinonyl moiety of the compounds 6h–i protruded out from the pocket with S scores −6.1801 and −5.9169 Kcal/mol, respectively.
Within 6a–f series (unsubstituted phenyl urea derivatives), compound 6e showed better COX-2 selectivity (S score −8.4697 Kcal/mol) as p-nitrophenyl ring allowed the compound to occupy the side pocket and exhibited H-bond with Tyr385 (). Moreover, the quinazolinonyl moiety of the compound 6e was stabilised by H-bond and arene-cation formation with Val349 and Arg120, respectively. On the other hand, compound 6e exhibited a different binding orientation with COX-1, as the urea linker formed a conventional H-bond with Arg120 (S score −6.1528 Kcal/mol) ().
Figure 3. (A) 3D Interaction diagram of compound 6e (thick green sticks) in the molecular surface of COX-2 (PDB: 1CX2) binding site. (B) 2D Interaction diagram of compound 6e with amino acid residues of COX-2.
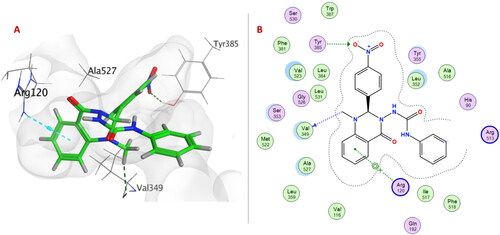
Figure 4. (A) 3D Interaction diagram of compound 6e (thick green sticks) in the molecular surface of COX-1 (PDB: 1EQG) binding site. (B) 2D Interaction diagram of compound 6e with amino acid residues of COX-1.
These results showed that the aforementioned compounds would have higher affinities to COX-2 over COX-1, in concordance with their selectivity towards COX-2.
Docking study into 15-LOX
The docking study into 15-LOX (PDB: 4NRE)Citation44 was initiated by re-docking the co-crystallised ligand; C8E [hydroxyethyloxy)tri(ethyloxy)octane] into the active site. The results revealed that the re-docked ligand showed 1.3448 Å for RMSD value.
Docking simulations of the targeted compounds 6a–p into 15-LOX active site afforded good binding energy ranging from −7.0847 to −5.8295 Kcal/mol (Supplementary Material Table 2).
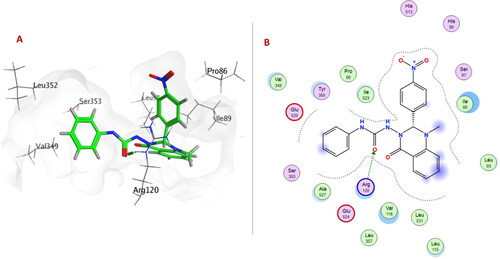
Compound 6c exhibited the best docking binding energy (−7.0847 Kcal/mol); matching the in vitro 15-LOX binding assay (1.36 µM). As the compound 6c showed several hydrophobic interactions with Phe184, Tyr185, Ala188, Phe192, Leu419, Gln425, Arg429 and Leu609 residues. Moreover, compound 6c was stabilised through H-bond formation with Leu419.
Quinazolinone ring in compounds 6a, 6e (), 6f, 6h–j, 6m, 6o, 6p formed either arene-arene, arene-H, or H-bond interactions with one or two of the following amino acid residues: Phe184, Phe192, Leu420, Arg429, Gln425, or Ala606.
Figure 5. (A) 3D Interaction diagram of compound 6e (thick green sticks) in the molecular surface of 15-LOX (PDB: 4NRE) binding site. (B) 2D Interaction diagram of compound 6e with amino acid residues of 15-LOX.
Urea linker in compounds 6b–d, 6h, 6i, 6k–n, or 6p formed H-bond with one of the following amino acid residues: Leu419, Val426, Arg429, Asp602, or Ala606.
All compounds revealed stabilising non-covalent interactions could be responsible for the inhibitory effect against 15-LOX by blocking its binding pocket.
The co-crystallised ligand (C8E) has a U-shaped conformation in the active siteCitation44. Compound 6e was examined for its conformational similarity with C8E through the flexible alignment approach in MOE 2019.0102.
As indicated from the 3D flexible alignment result, compound 6e showed similar conformations to C8E and this was verified and approved by the docking study. The score of the configuration alignment (S) is −70.4956 Kcal/mol; indicating excellent better alignment ().
Figure 6. Flexible alignment of compound 6e (thick green sticks) with the co-crystallised ligand; C8E [hydroxyethyloxy)tri(ethyloxy)octane] (thick blue sticks) of 15-LOX (PDB: 4NRE).
![Figure 6. Flexible alignment of compound 6e (thick green sticks) with the co-crystallised ligand; C8E [hydroxyethyloxy)tri(ethyloxy)octane] (thick blue sticks) of 15-LOX (PDB: 4NRE).](/cms/asset/dd02051f-14a8-48e3-815a-32c163cc6969/ienz_a_2199166_f0006_c.jpg)
Docking study into EGFRWT and EGFRL858R/T790M
The docking studies into EGFR wild-type (EGFRWT, PDB: 1M17)Citation45 and EGFR double mutant type (EGFRL858R/T790M, PDB: 5EDQ)Citation46 were initiated by re-docking the co-crystallised ligands; erlotinib and 5N3 [N-(7-chloro-1H-indazol-3-yl)-7,7-dimethyl-2-(1H-pyrazol-4-yl)-5H-furo[3,4-d]pyrimidin-4-amine] into the active sites, respectively. The results revealed that the re-docked erlotinib and 5N3 showed 0.8681 and 1.3783 Å for RMSD values, respectively. The binding modes of the compounds were evaluated by docking into the ATP binding sites of both EGFRWT and EGFRL858R/T790M. The docking energy scores for the compounds 6a–p ranged from −7.4186 to −6.6860 Kcal/mol for EGFRWT and −8.1780 to −6.5123 Kcal/mol for EGFRL858R/T790M (Supplementary Material Table 3).
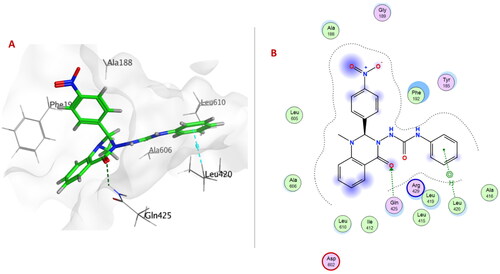
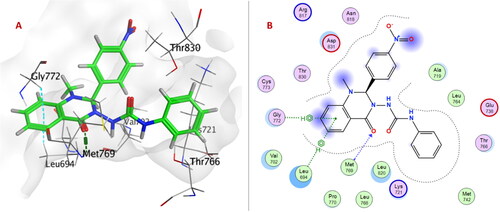
Within EGFRWT, the quinazolinone ring in compound 6e formed H-bond with Met769 and arene-H interactions with both Leu694 and Gly772. One NH group of the urea linker of compound 6o showed H-bond with the acidic Asp831 in addition to an H-bond between the nitro group and Met769.
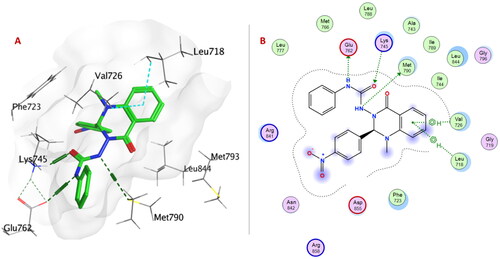
Within EGFRL858R/T790M; urea linker allowed the compounds 6a–p to be oriented into the ATP binding site and exposed to the key amino acid residues. Urea linker in compounds 6a, 6c–e, 6g, 6h, 6j, and 6m–o formed H-bond with one or two amino acid residues: Lys745, Glu762, Met790, and Thr854. The formation of a non-covalent bond with Met790 showed EGFRL858R/T790M selectivity over EGFRWT. As Met790 is considered a gatekeeper residue of EGFRL858R/T790MCitation47,Citation48.
Urea linker in compound 6e is considered an anchor for positioning the rest of the compound in the ATP-binding pocket within EGFRT790M/L858R. Two NH groups exhibited H-bonds with Glu762 and Met790, and carbonyl group formed H-bonds with Lys745. Moreover, quinazolinone ring showed two arene-H interactions with Leu718 and Val726 ().
Figure 7. (A) 3D Interaction diagram of compound 6e (thick green sticks) in the molecular surface of EGFRL858R/T790M (PDB: 5EDQ) binding site. (B) 2D Interaction diagram of compound 6e with amino acid residues EGFRL858R/T790M.
On the other hand, only the quinazolinone ring of compound 6e showed H-bond with Met769 and arene-H interactions with Leu694 and Gly772 within the ATP-binding pocket of EGFRWT ().
Figure 8. (A) 3D Interaction diagram of compound 6e (thick green sticks) in the molecular surface of EGFRWT (PDB: 1M17) binding site. (B) 2D Interaction diagram of compound 6e with amino acid residues of EGFRWT.
A summary of the crucial amino acid residues involved in hydrogen bonds, arene-arene, arene-cation, arene-H interactions, and remarkable docking energy scores of compounds 6a–p with COX-1, COX-2, 15-LOX, EGFRWT, and EGFRT790M/L858R are presented in Supplementary Material Tables 1–3. These results supported the in vitro biological results.
Conclusion
Sixteen novel quinazolinone tethered phenyl urea derivatives (6a–p) were designed to triple target EGFR, COX-2, and 15-LOX. The Novel compounds (6a–p) were synthesised and their inhibitory activity evaluated against COX-1, COX-2, 15-LOX, wild-type EGFR, and the double mutant EGFRL858R/T790M. Their anti-inflammatory activities were also evaluated by testing their inhibitory effect of LPS-stimulated NO, ROS, TNF-α, and IL-6 production in RAW 264.7 macrophages. Moreover, the ten most active compounds were submitted to NCI for an anticancer activity assessment. Five compounds (6e, 6d, 6j, 6m, and 6n) showed moderate inhibitory activities against the three targets with low micromolar IC50 values (0.83–5.04, 1.79–2.23, and 4.03–13.52 µM for COX-2, 15-LOX, and the double mutant EGFRL858R/T790M, respectively). They also showed good selectivity for COX-2 over COX-1 and for EGFR L858R/T790M over wild-type EGFR. All the tested compounds 6a–p inhibited the production of the inflammatory mediator NO (IC50 = 0.97–13.54 µM) significantly more potent compared to the three reference drugs celecoxib, diclofenac, and indomethacin (IC50 = 14.39, 24.08, and 46.45 µM, respectively) with the exception of compounds 6n and 6e. The most potent compounds in reducing ROS levels were compounds 6i and 6k, which showed IC50 values of 5.90 and 5.04 µM (≈1.5-fold more potent than that of celecoxib, 7.57 µM, ≈8-fold more potent than that of diclofenac, 43.78 µM). Interestingly, in terms of inhibiting the production of the inflammatory mediator TNF-α, compound 6o-treated raw 264.7 cells showed TNF-α level, which is ∼10 times lower than that of celecoxib (39.37 vs. 383.3 pg/mL). Compound 6n showed the broadest anticancer activity against 32 out of the 60 NCI cell lines with growth inhibition percentages in the 10.09–57.11% range. Compounds 6e and 6j exhibited the most potent anticancer activity against breast cancer cell line BT-459 with growth inhibition percentages of 67.14 and 70.07%, respectively.
These results were complemented by molecular docking studies, which identify the ability of the target compounds to make essential key interactions, known to be pivotal for EGFR, COX-2, and 15-LOX inhibitors. Our compounds could offer new structural insights into the understanding and development of triple-target EGFRL858R/T790M, COX-2, and 15-LOX inhibitors for anticancer outcomes.
Supplemental Material
Download PDF (8.5 MB)Acknowledgements
We thank the developmental Therapeutics Program at the Division of cancer treatment and diagnosis of the National Cancer Institute of Health for the screening using the NCI60 cancer cell line panel. We are also thankful to the Faculty of Pharmacy at Zagazig University for the purchase of MOE license version 2019 (Chemical Computing Group, Montreal, CA, USA).
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Abdellatif KR, Fadaly WA, Mostafa YA, Zaher DM, Omar HA. Thiohydantoin derivatives incorporating a pyrazole core: design, synthesis and biological evaluation as dual inhibitors of topoisomerase-i and cycloxygenase-2 with anti-cancer and anti-inflammatory activities. Bioorg Chem. 2019;91:103132.
- Hu L, Fan M, Shi S, Song X, Wang F, He H, Qi B. Dual target inhibitors based on EGFR: promising anticancer agents for the treatment of cancers (2017). Eur J Med Chem. 2022;227:113963.
- Abdelazeem AH, El-Saadi MT, Said EG, Youssif BG, Omar HA, El-Moghazy SM. Novel diphenylthiazole derivatives with multi-target mechanism: synthesis, docking study, anticancer and anti-inflammatory activities. Bioorg Chem. 2017;75:127–138.
- Gouda AM, Abdelazeem AH, Omar HA, Abdalla AN, Abourehab MA, Ali HI. Pyrrolizines: design, synthesis, anticancer evaluation and investigation of the potential mechanism of action. Bioorg Med Chem. 2017;25(20):5637–5651.
- El-Sayed NA, Nour MS, Salem MA, Arafa RK. New oxadiazoles with selective-COX-2 and EGFR dual inhibitory activity: design, synthesis, cytotoxicity evaluation and in silico studies. Eur J Med Chem. 2019;183:111693.
- Bhatia P, Sharma V, Alam O, Manaithiya A, Alam P, Alam MT, Imran M. Kahksha Novel quinazoline-based EGFR kinase inhibitors: a review focussing on SAR and molecular docking studies (2015–2019). Eur J Med Chem. 2020;204:112640.
- Pawara R, Ahmad I, Nayak D, Wagh S, Wadkar A, Ansari A, Belamkar S, Surana S, Nath Kundu C, Patil C, et al. Novel, selective acrylamide linked quinazolines for the treatment of double mutant EGFR-l858R/T790M non-small-cell lung cancer (NSCLC). Bioorg Chem. 2021;115:105234.
- Chen L, Zhang Y, Tian L, Wang C, Deng T, Zheng X, Wang T, Li Z, Tang Z, Meng Q, et al. Noncovalent EGFR T790M/L858R inhibitors based on diphenylpyrimidine scaffold: design, synthesis, and bioactivity evaluation for the treatment of NSCLC. Eur J Med Chem. 2021;223:113626.
- Shaheen MA, El-Emam AA, El-Gohary NS. Design, synthesis and biological evaluation of new series of hexahydroquinoline and fused quinoline derivatives as potent inhibitors of wild-type EGFR and mutant EGFR (L858R and T790M). Bioorg Chem. 2020;105:104274.
- Chen L, Zhang Y, Wang C, Tang Z, Meng Q, Sun H, Qi Y, Ma X, Li L, Li Y, et al. Design, synthesis, and biological evaluation of hydroxamic acid-substituted 2, 4-diaryl aminopyrimidines as potent EGFRT790M/L858R inhibitors for the treatment of NSCLC. Bioorg Chem. 2021;114:105045.
- Elzahhar PA, Abd El Wahab SM, Elagawany M, Daabees H, Belal ASF, El-Yazbi AF, Eid AH, Alaaeddine R, Hegazy RR, Allam RM, et al. Expanding the anticancer potential of 1,2,3-triazoles via simultaneously targeting cyclooxygenase-2, 15-lipoxygenase and tumor-associated carbonic anhydrases. Eur J Med Chem. 2020;200:112439.
- Gouda AM, Beshr EA, Almalki FA, Halawah HH, Taj BF, Alnafaei AF, Alharazi RS, Kazi WM, AlMatrafi MM. Arylpropionic acid-derived NSAIDS: new insights on derivatization, anticancer activity and potential mechanism of action. Bioorg Chem. 2019;92:103224.
- Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94(4):252–266.
- Afifi OS, Shaaban OG, Abd El Razik HA, Shams El-Dine SE-DA, Ashour FA, El-Tombary AA, Abu-Serie MM. Synthesis and biological evaluation of purine-pyrazole hybrids incorporating thiazole, thiazolidinone or rhodanine moiety as 15-LOX inhibitors endowed with anticancer and antioxidant potential. Bioorg Chem. 2019;87:821–837.
- Omar YM, Abdel-Moty SG, Abdu-Allah HH. Further insight into the dual COX-2 and 15-LOX anti-inflammatory activity of 1,3,4-thiadiazole-thiazolidinone hybrids: the contribution of the substituents at 5th positions is size dependent. Bioorg Chem. 2020;97:103657.
- Ghanim AM, Rezq S, Ibrahim TS, Romero DG, Kothayer H. Novel 1,2,4-triazine-quinoline hybrids: the privileged scaffolds as potent multi-target inhibitors of LPS-induced inflammatory response via dual COX-2 and 15-LOX inhibition. Eur J Med Chem. 2021;219:113457.
- Rao CV, Janakiram NB, Madka V, Devarkonda V, Brewer M, Biddick L, Lightfoot S, Steele VE, Mohammed A. Simultaneous targeting of 5-LOX-COX and EGFR blocks progression of pancreatic ductal adenocarcinoma. Oncotarget. 2015;6(32):33290–33305.
- Mohammed A, Janakiram NB, Pant S, Rao CV. Molecular targeted intervention for pancreatic cancer. Cancers. 2015;7(3):1499–1542.
- Qin X, Yang L, Liu P, Yang L, Chen L, Hu L, Jiang M. Design, synthesis and biological evaluation of 2,3-dihydro-[1,4] dioxino [2,3-f] quinazoline derivatives as EGFR inhibitors. Bioorg Chem. 2021;110:104743.
- Elmetwally SA, Saied KF, Eissa IH, Elkaeed EB. Design, synthesis and anticancer evaluation of thieno [2,3-d] pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers. Bioorg Chem. 2019;88:102944.
- Marzaro G, Castagliuolo I, Schirato G, Palu’ G, Dalla Via M, Chilin A, Brun P. Substituted quinazolinones as kinase inhibitors endowed with anti-fibrotic properties. Eur J Med Chem. 2016;115:416–425.
- Ewes WA, Elmorsy MA, El-Messery SM, Nasr MN. Synthesis, biological evaluation and molecular modeling study of [1,2,4]-triazolo [4,3-c] quinazolines: new class of EGFR-TK inhibitors. Bioorg Med Chem. 2020;28(7):115373.
- Emam AM, Dahal A, Singh SS, Tosso RD, Ibrahim SM, El‐Sadek M, Jois SD, Enriz RD, Kothayer H. Quinazoline‐tethered hydrazone: a versatile scaffold toward dual anti‐TB and EGFR inhibition activities in NSCLC. Arch Pharm. 2021;354(12):2100281.
- Aziz MW, Kamal AM, Mohamed KO, Elgendy AA. Design, synthesis and assessment of new series of quinazolinone derivatives as EGFR inhibitors along with their cytotoxic evaluation against MCF7 and A549 cancer cell lines. Bioorg Med Chem Lett. 2021;41:127987.
- do Amaral DN, Lategahn J, Fokoue HH, da Silva EMB, Sant’Anna CMR, Rauh D, Barreiro EJ, Laufer S, Lima LM. A novel scaffold for EGFR inhibition: introducing n-(3-(3-phenylureido) quinoxalin-6-yl) acrylamide derivatives. Sci Rep. 2019;9(1):1–12.
- Jiang N, Bu Y, Wang Y, Nie M, Zhang D, Zhai X. Design, synthesis and structure-activity relationships of novel diaryl urea derivatives as potential EGFR inhibitors. Molecules. 2016;21(11):1572.
- Sakr A, Rezq S, Ibrahim SM, Soliman E, Baraka MM, Romero DG, Kothayer H. Design and synthesis of novel quinazolinones conjugated ibuprofen, indole acetamide, or thioacetohydrazide as selective COX-2 inhibitors: Anti-inflammatory, analgesic and anticancer activities. J Enzyme Inhib Med Chem. 2021;36(1):1810–1828.
- Wang C, Li Y, Liu Z, Wang Z, Liu Z, Man S, Zhang Y, Bao K, Wu Y, Guan Q, et al. Design, synthesis and biological evaluation of 1-aryl-5-(4-arylpiperazine-1-carbonyl)-1 h-tetrazols as novel microtubule destabilizers. J Enzyme Inhib Med Chem. 2021;36(1):549–560.
- Aboul-Enein MN, El-Azzouny AA, Attia MI, Maklad YA, Amin KM, Abdel-Rehim M, El-Behairy MF. Design and synthesis of novel stiripentol analogues as potential anticonvulsants. Eur J Med Chem. 2012;47(1):360–369.
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6(10):813–823.
- RCSB protein data bank (pdb). Available from: http://www.rcsb.org.
- Molecular operating environment (MOE 2019.0102). Montreal: Chemical Computing Group Inc. Available from: http://www.chemcomp.com.2019.0102
- Lee SY, Kim HJ, Han JS. Anti-inflammatory effect of oyster shell extract in LPS-stimulated raw 264.7 cells. Prev Nutr Food Sci. 2013;18(1):23–29.
- Perkins DJ, Kniss DA. Blockade of nitric oxide formation down-regulates cyclooxygenase-2 and decreases PGE2 biosynthesis in macrophages. J Leukoc Biol. 1999;65(6):792–799.
- Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, Squadrito F, Altavilla D, Bitto A. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longevity. 2017;2017:1–13.
- Qi S, Feng Z, Li Q, Qi Z, Zhang Y. Myricitrin modulates NADPH oxidase-dependent ROS production to inhibit endotoxin-mediated inflammation by blocking the JAK/STAT1 and NOX2/P47(PHOX) pathways. Oxid Med Cell Longev. 2017;2017:9738745.
- Chan A, Murin S. Up in smoke: the fallacy of the harmless hookah. Chest. 2011;139(4):737–738.
- Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60(1):6–20.
- Fujita M, Somasundaram V, Basudhar D, Cheng RYS, Ridnour LA, Higuchi H, Imadome K, No JH, Bharadwaj G, Wink DA. Role of nitric oxide in pancreatic cancer cells exhibiting the invasive phenotype. Redox Biol. 2019;22:101158.
- Wang W, Xu R-L, He P, Chen R. Mar1 suppresses inflammatory response in LPS-induced raw 264.7 macrophages and human primary peripheral blood mononuclear cells via the SIRT1/PGC-1Α/PPAR-γ pathway. J Inflamm. 2021;18(1):8.
- Soromou LW, Zhang Z, Li R, Chen N, Guo W, Huo M, Guan S, Lu J, Deng X. Regulation of inflammatory cytokines in lipopolysaccharide-stimulated raw 264.7 murine macrophage by 7-o-methyl-naringenin. Molecules. 2012;17(3):3574–3585.
- Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Structural analysis of NSAID binding by prostaglandin H2 synthase: Time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry. 2001;40(17):5172–5180.
- Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384(6610):644–648.
- Kobe MJ, Neau DB, Mitchell CE, Bartlett SG, Newcomer ME. The structure of human 15-lipoxygenase-2 with a substrate mimic. J Biol Chem. 2014;289(12):8562–8569.
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277(48):46265–46272.
- Hanan EJ, Baumgardner M, Bryan MC, Chen Y, Eigenbrot C, Fan P, Gu X-H, La H, Malek S, Purkey HE, et al. 4-Aminoindazolyl-dihydrofuro [3,4-d] pyrimidines as non-covalent inhibitors of mutant epidermal growth factor receptor tyrosine kinase. Bioorg Med Chem Lett. 2016;26(2):534–539.
- Heald R, Bowman KK, Bryan MC, Burdick D, Chan B, Chan E, Chen Y, Clausen S, Dominguez-Fernandez B, Eigenbrot C, et al. Noncovalent mutant selective epidermal growth factor receptor inhibitors: a lead optimization case study. J Med Chem. 2015;58(22):8877–8895.
- Hanan EJ, Eigenbrot C, Bryan MC, Burdick DJ, Chan BK, Chen Y, Dotson J, Heald RA, Jackson PS, La H, et al. Discovery of selective and noncovalent diaminopyrimidine-based inhibitors of epidermal growth factor receptor containing the T790M resistance mutation. J Med Chem. 2014;57(23):10176–10191.