
Abstract
This research study describes the development of new small molecules based on 2,4-thiazolidinedione (2,4-TZD) and their aldose reductase (AR) inhibitory activities. The synthesis of 17 new derivatives of 2,4-TZDs hybrids was feasible by incorporating two known bioactive scaffolds, benzothiazole heterocycle, and nitro phenacyl moiety. The most active hybrid (8b) was found to inhibit AR in a non-competitive manner (0.16 µM), as confirmed by kinetic studies and molecular docking simulations. Furthermore, the in vivo experiments demonstrated that compound 8b had a significant hypoglycaemic effect in mice with hyperglycaemia induced by streptozotocin. Fifty milligrams per kilogram dose of 8b produced a marked decrease in blood glucose concentration, and a lower dose of 5 mg/kg demonstrated a noticeable antihyperglycaemic effect. These outcomes suggested that compound 8b may be used as a promising therapeutic agent for the treatment of diabetic complications.
Introduction
Diabetes mellitus is a major cause of several progressive and chronic diseases that adversely affect many organs, including vascular and nervous systems, with an approximated 200 million causalities of mortality and morbidityCitation1,Citation2. Type 2 diabetes (T2D) is a chronic life-threatening disorder exhibiting abnormal elevated blood glucose (BG) concentration resulting from diminished response of the target tissues to insulin action (insulin resistance) and progressive impaired function of β cells in pancreasCitation2,Citation3. Thus, diabetes is considered a major and growing public health burden, and has gained prime global health importance because of the several long-term complications, such as neuropathy, nephropathy, retinopathy, cataract, and cardiovascular disordersCitation3,Citation4. Most of the currently available drugs can cause problems including hypoglycaemia, compliance, and obesityCitation5,Citation6. Hence, there is a crucial necessity to develop new safe and potent antidiabetic drugs with improved compliance and reduced side effectsCitation3,Citation4.
Under hyperglycaemic state, higher than 30% of the BG is bio-transformed into sorbitol by aldose reductase (AR) enzyme resulting in the major diabetic secondary complicationsCitation7–11. Subsequently, sorbitol dehydrogenase converts sorbitol to fructose through polyol pathway, which is a necessary mechanism for regulation of glucose metabolism in mammalian cells. AR is a key enzyme that belongs to aldo-keto reductase super-family involved in the polyol pathway for glucose reduction to sorbitol (). It is believed that activation of this metabolic pathway is associated with the chronic diabetic complications like retinopathy, diabetic cataract, neuropathy, and nephropathy. Therefore, aldose reductase inhibitors (ARIs) emerged as a fruitful therapeutic tool to prevent the development of these metabolic complications via inhibition of the first step of polyol pathwayCitation1,Citation7,Citation8. ARIs have been found to supress and prevent sorbitol accumulation in specific tissues such as peripheral nerves, lens, and kidney. Accordingly, the decreased sorbitol flux by ARIs could be exploited as emerging approach for the management of major diabetes complications. Furthermore, the pathogenesis of sorbitol-induced diabetic complications may be result from interruption in cellular redox, sorbitol-osmotic effects, free radical defence, in addition to elevated oxidative and glycation stressCitation1.
Figure 1. Design of new 5-arylidene-2,4-TZDs-based hybrids as ARIs based on some reported AR inhibitors.
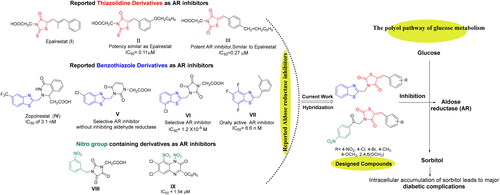
Orally active ARIs vary structurally and are classified into two major chemical groups: cyclic imides (mainly spirohydantoins) and carboxylic acid derivatives, such as Epalrestat (I)Citation4. The majority of carboxylic acid derivatives were evaluated as ARIs preclinically and clinically, nonetheless, their development is generally restrained by their diminished in vivo potency, several side effects, and pharmacokinetic obstaclesCitation12,Citation13. The carboxylic acid ARIs show potent in vitro activity as ARIs; however, their effectiveness decreases in vivo. Carboxylic acid derivatives could be completely ionised at physiological pH, and thus, their in vivo activity is generally lower than that of less ionised compounds. This effect is possibly due to the impaired penetration of physiological membranes of such ionised compoundsCitation14–16. Therefore, the development of a new generation of more selective non-carboxylic acid ARIs is prioritised to pursue the desired pharmacokinetic and therapeutic properties with reduced toxicity, fewer side effects and enhanced tissue permeability and drug uptake at the physiological pH. Currently, Epalrestat (I), which is a carboxylic acid derivative bearing 2-thioxo-4-thiazolidinone moiety (), is the only approved ARI commercially available in Japan, China, and India. Epalrestat (I) is easily absorbed into the neural tissue and inhibits AR with minimum side effectsCitation17–19.
Likewise, there is a great interest in 2,4-thiazolidinediones (2,4-TZDs) as a new class of antidiabetic drugs acting as ARIs with dual activity controlling both glucose and lipid metabolismCitation12. 2,4-TZDs, as antidiabetic agent, has been previously reported to act by activating peroxisome proliferator-activated receptors (PPARs), specifically PPAR-gamma (PPARγ). Upon activation of these receptors, increasing transcription of a number of specific genes and decreasing transcription of others would happen. As a result, these receptors play transcriptional regulation of some genes and in turn control glucose and lipid metabolism. Briefly, the main effect of expression and repression of these receptors is an increase in the storage of fatty acids in adipocytes, and thereby decreasing the amount of fatty acids existing in the circulation. Thus, cells become more dependent on the oxidation of glucose to provide energy for other cellular processesCitation12.
As a result, 2,4-TZDs are efficient in metabolic regulation of lipid and glucose associated with insulin resistance and therefore, they markedly differ from other antihyperglycaemic agents in having dual activity used for the treatment of both T2D and obesityCitation12,Citation20,Citation21. Interestingly, several 5-arylidene-2,4-TZDs such as compound II and III () have been reported as safer bioisosteres of the other AR inhibitors and have exerted appreciable AR inhibitory activitiesCitation22,Citation23.
Furthermore, benzothiazole-based carboxylic acids, such as Zopolrestat (IV), have shown potent and selective inhibition of AR (). Zopolrestat is a potent, orally active AR inhibitor used for the treatment of diabetic complications with IC50 value of 3.1 nMCitation24,Citation25. Thus, benzothiazole side chains featured in Zopolrestat were then incorporated into several other derivatives, such as compounds V, VI, and VII, which have been proved to show potent AR inhibition activitiesCitation12,Citation26–30.
Moreover, several molecules containing nitro group, for example, aromatic nitro compounds VIII and IX, have discovered with potential AR inhibitory activity (). The nitro group was anticipated to play an important role for AR active site binding interactions with Tyr48 and His110 residues, which are the residues essential for binding with carboxylic acids ARIs through their anionic formsCitation13,Citation31,Citation32.
In the present study and in the light of the aforesaid investigations, the structure features of our designed compounds were based on the reported AR inhibitory abilities of 5-arylidene-2,4-TZDs pharmacophore as well as benzothiazole nucleus and aromatic nitro compounds (). For this endeavour, 5-arylidene-2,4-TZDs-based derivatives as a privileged scaffold, have been designed, synthesised, and biologically evaluated for their AR inhibitory impact. As well, based on a hybrid pharmacophore design, the incorporation of benzothiazole or nitro phenyl moieties was hoped to generate new hybrid candidates with better AR inhibitionCitation4. The present study focuses on the synthesis of 5-arylidene-2,4-TZDs hybrids substituted at position 3 with either benzothiazole pharmacophore or 4-nitrophenyl-2-oxoethyl substituent in an attempt to investigate the role of these versatile bioactive functionalities in AR inhibition (). Finally, all the synthesised compounds were investigated for their potential to inhibit AR where the most active compound would be also evaluated for its antihyperglycaemic influence. Further, molecular docking studies were conducted to rationalise their possible binding interactions in AR binding site.
Results and discussion
Chemistry
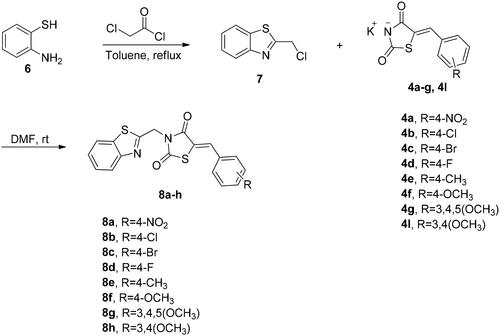
The synthetic approach exploited for preparation of the target compounds, (Z)-5-(4 or 3-substitutedbenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5a–k) and (Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(4-subtitutedbenzylidene)thiazolidine-2,4-dione (8a–h) is outlined in Schemes 1 and 2. The synthesis of N-unsubstituted 5-arylidene-thiazolidine-2,4-diones (3a–k) in acceptable yield has been accomplished employing Knoevenagel condensation, where the thiazolidine-2,4-dione was condensed with the corresponding arylaldehyde as reported previouslyCitation33–36. After that, compounds 3a–k were refluxed in ethanol with potassium hydroxide to obtain the subsequent potassium salts 4a–k. After that, the potassium salts 4a–k were stirred with 2-bromo-4′-nitroacetophenone in DMF to attain the corresponding derivatives 5a–k (Scheme 1). On the other hand, the benzothiazole counterparts 8a–h were synthesised through refluxing the potassium salts 4a–g and 4l with benzothiazole methyl chloride 7 in DMF (Scheme 2). The newly synthesised thiazolidine-2,4-dione hybrids 5a–k and 8a–h were elucidated utilising spectroscopic analyses (1H NMR, 13C NMR, and MS). All spectral and analytical results were compatible with the assigned compounds.
Scheme 1. Synthetic pathway for the target 4-nitro-phenacyl tethered thiazolidine-2,4-dione hybrids 5a–k.
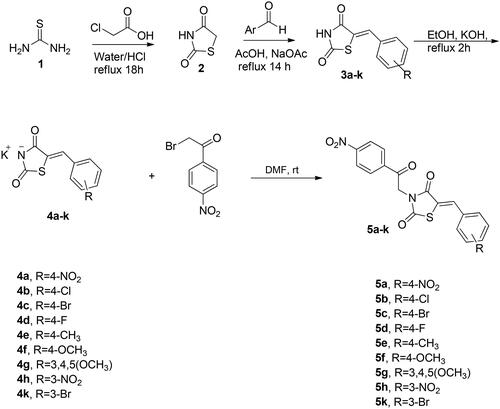
Scheme 2. Synthetic pathway for the target benzothiazole tethered thiazolidine-2,4-dione hybrids 8a–h.
Regarding IR spectral data for the target, thiazolidine-2,4-dione hybrids 5a–k and 8a–h showed two strong absorption bands at 1754–1737 cm−1, 1704–1687 cm−1 resulting from the starching of the two C═O in thiazolidine-2,4-dione scaffold. For 1H NMR spectra of thiazolidine-2,4-diones 5a–k and 8a–h, they demonstrated one singlet signal around 8 ppm for the benzylidene proton that supported the occurrence of Knoevenagel reaction between 2,4-diones 3a–k and the selected aromatic aldehydes. Also, the absence of a singlet signal corresponding to the NH from the thiazolidine-2,4-dione ring at 12.50–12.52 ppm confirmed the success of N-substitution of potassium salts 4a–k.Citation37 Moreover, 1H NMR spectral data of the hybrids 5a–k and 8a–h showed one signal for N-CH2 methyl group as singlet signal around 5.4 ppm. Concerning 13C NMR of the hybrids 5a–k and 8a–h, it was revealed that the appearance of either three or two signals at 170–157 ppm confirmed the presence of olefinic carbons. Moreover, the existence of one signal at about 45 ppm was assigned to the methyl carbon. Finally, the mass spectral data gave the fragmentation patterns of the target compounds and their corresponding mass revealing the molecular ion peaks (M+) as predicted by their molecular formulas.
Biological evaluation
Inhibitory activity against human aldose reductase
All the newly synthesised thiazolidine-2,4-dione hybrids 5a–k and 8a–h were screened for their in vitro inhibitory activity against human AR using Epalrestat as a positive control. The results of in vitro AR inhibition activity are presented as IC50 values in . The obtained results indicated that the tested compounds demonstrated variable inhibitory potencies against AR enzyme at sub-micromolar level, except for compound 8a. Regarding benzothiazole-tethered thiazolidine-2,4-dione 8b, it was found to be the most potent AR inhibitor with IC50 of 0.16 μM, falling closer to Epalrestat reference drug (IC50 of 0.10 μM). Moreover, the phenacyl-thiazolidine-2,4-dione hybrids 5a, 5f as well as the benzothiazole-based candidates 8c, 8e, and 8g exhibited a strong inhibitory impact with IC50 range spanning from 0.21 μM to 0.29 μM. Furthermore, the phenacyl-derived hybrids 5b–e, 5h, and 5k and the benzothiazole-based analogues 8d and 8h displayed moderate inhibitory effect with IC50 range from 0.33 to 0.57 μM. On contrary, the counterparts 5g, 8a, and 8f showed weak inhibitory influence with IC50 range of 0.72–1.98 μM.
Table 1. IC50 values of the tested compounds 5a–k and 8a–h against human aldose reductase.
The SAR analysis for the assessed compounds hinted out that the enzyme inhibitory action is influenced by the various substituents appended to the aromatic ring of benzylidene moiety either in phenacyl-thiazolidine-2,4-dione hybrids 5a–k or in benzothiazole-thiazolidine-2,4-dione hybrids 8a–h. Regarding the phenacyl-derived thiazolidine-2,4-dione hybrids 5a–k, the appending of NO2 functionality to aromatic ring had a potential impact on AR inhibition (5a, IC50 = 0.22 µM) affording the highest potent AR inhibitor within these series. Of special note, shifting of NO2 group to meta position to provide the regioisomer 5h (IC50 = 0.51 µM) declined the inhibitory action by more than two-fold. In a similar behaviour, the inclusion of 4-chloro 5b, 4-bromo 5c, 4-fluoro 5d, 4-methyl 5e, or 3,4,5-OCH3 5g to benzylidene moiety sharply reduced the AR inhibitory action (IC50 values equal 0.46, 0.49, 0.57, 0.55, and 0.72 µM, respectively). Moreover, the movement of bromo appendage from para position (5c, IC50 = 0.49 µM) to meta position (5k, IC50 = 0.41 µM) slightly enhanced the inhibitory action. On contrast, the para methoxy counterpart 5f revealed a strong inhibitory influence (IC50 value of 0.28 μM) coming at the second order after the 4-nitro containing analogue (5a, IC50 = 0.22 µM).
Concerning benzothiazole-based thiazolidine-2,4-dione hybrids 8a–h, it was obviously noted that replacement of the phenacyl moiety with benzothiazole motif improved the AR inhibitory effect by about twofold, except for the 4-nitro containing hybrid 8a (IC50 = 1.98 µM) and 4-methoxy-grafted analogue 8f (IC50 = 0.94 µM). It was detected that the incorporation of 4-chloro to the benzylidene moiety furnished the most efficient AR inhibitor within this study (IC50 value equals 0.16 µM) followed by the 4-bromo hybrid 8c (IC50 = 0.21 µM). Thereafter, the other substitution pattern provided potent inhibition in the following order 3,4,5-trimethoxy hybrid 8g, 4-methyl derivative 8e, 4-fluoro analogue 8d, and 3,4-dimethoxy derivative 8h displaying IC50 values of 0.27, 0.29, 0.33, and 0.45 µM, respectively. Strikingly, it was deduced that the presence of 4-nitro 8a or 4-methoxy 8e on benzylidene moiety exerted negative influence on the inhibitory potency providing the least potent AR inhibitors within this study (IC50 values of 1.98 and 0.94 µM, respectively). Notably, it was deduced that the presence of methoxy substitutions on benzylidene moiety had an observable influence on the inhibitory potencies, where number of methoxy groups was directly proportional to the inhibitory activity; compound 8g (IC50 of 0.27 μM) with three methoxy groups showed better activity than the two methoxy groups-appended derivative 8h (IC50 of 0.45 μM) and the one methoxy-affixed analogue 8f (IC50 of 0.94 μM), which possessed the least activity. Furthermore, the hybrid 8e possessing a para methyl instead of methoxy functionality established a remarkable increment in its activity with IC50 of 0.29 μM.
Collectively, the introduction of 4-chloro, 4-bromo, or 3,4,5-trimethoxy substituents to the benzylidene moiety along with benzothiazole scaffold resulted in potent AR inhibitors. Besides, applying 4-nitro or 4-methoxy functionalities to the benzylidene moiety parallel with 4-nitro phenacyl group led to efficient AR inhibitors. Generally, the inferred SAR insights for the tested thiazolidine-2,4-dione hybrids can be employed for additional manipulations to develop more potent and safe AR inhibitors for the management of long-term diabetic complications.
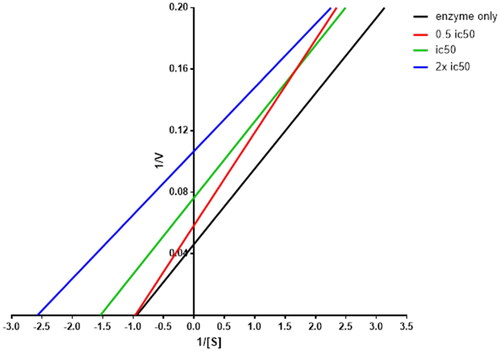
Analysis of kinetic parameters for determination of 8b inhibition mode
The most potent AR inhibitor 8b was selected for this study, the analysis of KM and Vmax of AR enzyme alone and with different concentrations of 8b showed that at 0.5 IC50, the compound led to significant reduction in Vmax and slight reduction in Km. In contrary, the Km and Vmax have been decreased significantly at concentrations equal to IC50 and double of IC50 with approximately fixed slope values, which is a unique pattern of non-competitive inhibitors, where KM and Vmax were reduced when the concentration of the inhibitor increased as shown in . These findings indicate that the rationale design of 8b was successful to produce similar compounds as the reported inhibitors.
Figure 2. Lineweaver–Burk plot showing kinetics of aldose reductase in the absence and presence of different concentrations of 8b.
Investigation of in vivo hypoglycaemic impact of the hybrid 8b
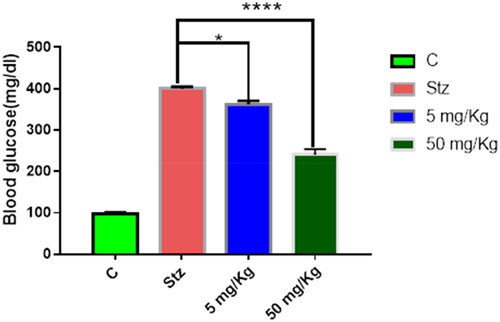
Streptozotocin (STZ)-induced diabetes is a widely used model to assess the hypoglycaemic effect of compounds. Since compound 8b showed very comparable AR inhibitory activity to Epalrestat, its potential to decrease BG level in vivo was assessed using two doses (5 mg/kg and 50 mg/kg). In case of the lower dose, the BG level was reduced from 399.8 mg/dL (STZ group) to 362.333 mg/dL, while in the higher dose, it was decreased to 240 mg/dL after six weeks of treatment as depicted in . This result highlighted that beside the observed in vitro AR inhibitory activity of the hybrid 8b, it also has pronounced hypoglycaemic effect in dose-dependent manner as demonstrated in the experimental animals. This could be attributed due to the ability of thiazolidinedione to regulate other molecular targets associated with diabetes such as peroxisome proliferator-activated receptor gamma (PPAR-γ).Citation38
Figure 3. In vivo hypoglycaemic effect of compound 8b in STZ-induced diabetes model after 6 weeks of treatment. Data shown are averages of six independent experiments *p > 0.05 and ****p > 0.0001.
Target fishing and molecular docking study of the most efficient hybrid 8b
Molecular docking tools have been used routinely in assessing the binding mode of bioactive compounds to explain their ability to inhibit several molecular targetsCitation39–47. So, taking into consideration the results of the kinetic study of compound 8b and its observed hypoglycaemic effect in vivo, target fishing was used to identify potential pharmacological targets using Pharmmaper server. This server used preprepared pharmacophore model to identify molecular features in the compound of interest, then fit score is calculated and converted to z-score, which is modified fit score that takes statistic factor in consideration where the positive value indicates high significance of the target. After pharmacophore-based screening, AR, PPAR-γ, and glycogen synthase kinase-3 (GSK-3) emerged as the most relevant targets to diabetes with z-scores, 1.15, 0.54, and 0.6, respectively. Benzothiazole ring and thiazolidinedione moiety were highlighted as important pharmacophore in case of all the three enzymes. This could be explained in the light of the alignment of molecular features in compound 8b with the pharmacophore model generated for aldose, reductase PPAR-γ and GSK-3 inhibitors as shown in (S) and Table 1S. This finding agrees with previous studies reporting the ability of this class of compounds to inhibit these targets and could support the observed hypoglycaemic effect of compound 8bCitation48–50.
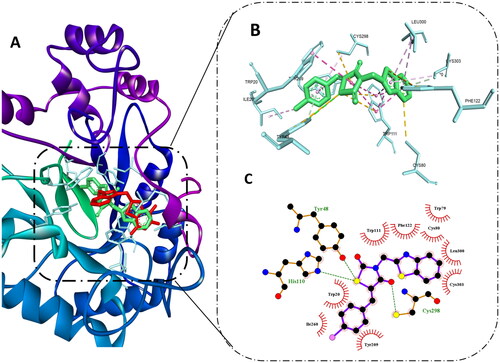
Molecular docking of compound 8b at AR active site showed that it was able to bind to the same site of the reported non-competitive AR inhibitors. Also, it exerted remarkable binding energy −11.3 kcal/mol in comparison to −12 kcal/mol of the co-crystallised ligand. Since the co-crystallised ligand is one of the AR carboxylic acid inhibitors, it was able to exert several hydrogen bonds with Tyr48, HIS110, and TRP111 through its carboxylic acid moiety, also several hydrophobic interactions were observed with TRP20, TRP111; moreover, the trifluoro-benzothiazole ring was able to access the secondary hydrophobic pocket and formed hydrophobic interaction with Leu303A and PHE122.
Interestingly, compound 8b best pose was aligned to the co-crystallised ligand, hence, it was able to reproduce most of these interaction to some extent, which explains its excellent binding affinity. Still, instead of the carboxylic group in the co-crystallised ligand, the thiazolidinedione ring formed hydrogen bonds with TYR48, HIS110, and CYS298 maintaining the ability to halt the catalytic activity of the enzyme.
In addition, it formed hydrophobic interactions with amino acid residues in the backbone such as TRP20, and TRP111, while the benzothiazole ring demonstrated hydrophobic interactions with PHE122, LEU300, and CYS303 in the second selectivity hydrophobic site, which is in agreement to previous reports indicating that planar aromatic ring could improve the selectivity by accessing such hydrophobic pocketCitation51. Finally, the benzylidene moiety interacted with TRP20, TYR209, ILE260, and CYS298 as shown in . These binding interactions infer the importance of thiazolidinedione and benzothiazole moiety as assumed previously in the predicted pharmacophore model and previous reports showing their important role in achieving good AR inhibitory activityCitation52,Citation53.
Figure 4. Molecular docking of 8b in the active site of aldose reductase PDB: 3g5e. (A) Compound 8b (green) aligned with the co-crystallised ligand (red). (B) 3D binding interaction of the hybrid 8b with active site residues of aldose reductase. (C) 2D interaction of compound 8b with active site residues of aldose reductase.
In silico prediction of physicochemical properties and pharmacokinetic profile
Lipinski’s rule and Veber’s parameters calculation
The oral bioavailability is one of the most important criteria in design and discovery of therapeutically active moleculesCitation54. Therefore, to predict the oral bioavailability and drug-likeness features for a candidate, Lipinski formulated “Rule of Five” using some main molecular descriptors like molecular weight, partition coefficient (logP), counts of hydrogen bond donors and acceptorsCitation55. Later, Veber added additional descriptors affecting drug oral bioavailability such as topological polar surface area (TPSA) and rotatable bonds numbers (nRB)Citation56. The assessment of compliance of the most potent hybrids 5a, 5f, 8b, 8c, 8e, and 8g to Lipinski’s rule and Veber’s standardCitation57 revealed that all the tested compounds can serve as successful drug candidates where hybrids 5a, 5f, and 8g are completely in agreement with Lipinski’s rule with no violations, while compounds 8b, 8c, and 8e displayed one violation (Log P > 5) as shown in . Concerning Veber’s measures, the number of rotatable bonds of the tested hybrids was ≤10, exhibiting accepted molecular flexibility with subsequent good permeability and efficient oral bioavailability. Moreover, TPSA value of all the tested hybrids was in accordance with Veber’s standards (TPSA < 140 Å2) except for compound 5a (TPSA (146.09) >140 Å2). These TPSA assessments are used to estimate of oral absorption % (%ABS) theoretically applying the equation: %ABS = 109 – (0.345 TPSA)Citation58. The hybrids 8b, 8c, and 8e had % ABS of 91.65%, as well compound 5g displayed % ABS of 82.10%, which may potentially allow better passive oral absorption relative to Epalrestat reference drug (78.05%). Based on these findings, it could be proposed that the hybrids 8b, 8c, 8e, and 8g had reasonable drug-likeness with good physicochemical characteristics and may be served as good orally active antidiabetic candidates.
Table 2. Calculated parameters for Lipinski’s rule and Veber’s standards for the hybrids 5a, 5f, 8b, 8c, 8e, and 8g.
ADMET analysis
Calculation of the pharmacokinetic parameters (ADMET) is a significant step in early stage drug discovery for improvement of both drug efficacy and safety profile, in addition to avoidance of therapeutic agent failure as an effective clinical candidateCitation57. Thus, ADMET profile of the most active hybrids 5a, 5f, 8b, 8c, 8e, and 8g was calculated theoretically using online Pre-ADMET server.
As shown in , the obtained calculations stated that all tested hybrids are predicted to have negative carcinogenic activities like reference drug except compound 8c, which is predicted to be positive carcinogen. All compounds exerted excellent intestinal absorption with HIA values ranging 77.0–100%, displaying their potency as orally active drugs. They also showed medium CNS penetration (BBB values ranged 0.1–1.86) and low cellular permeability in MDCK cell model (MDCK < 25 nm/s). Besides, compound 5a and 5f showed low cellular permeability in CaCo2 cell model (CaCo2 values <4 nm/s); however, the hybrids 8b, 8c, 8e, and 8g displayed medium permeability with CaCo2 values ranging from 23.56 to 41.54 nm/s. Moreover, hybrids 5a, 8b, 8c, and 8e are not predicted to be involved in drug–drug interactions as they cannot inhibit CYP3A4 enzyme, in contrast to the positive control and the analogues 5f and 8g, which have CYP3A4 inhibitory action. Similarly, as the reference drug, all the assessed hybrids are inhibitors to P-glycoprotein (PgP); hence, it was anticipated to enhance bioavailability of the co-administered drugs.
Table 3. ADMET profile for the active hybrids 5a, 5f, 8b, 8c, 8e, and 8g.
Conclusions
Molecular hybridisation approach was exploited to design two series of 2,4-TZD hybrids 5a–k and 8a–h appending to either benzothiazole scaffold or para nitro phenacyl moiety, followed by their synthesis utilising different synthetic methodologies and spectral analyses for all analogues. After that, all the prepared analogues were assessed for their in vitro AR inhibitory effect. Interestingly, the majority of all derivatives displayed potential inhibitory impact at sub-micromolar level comparable to the positive control (Epalrestat). Noteworthy, the benzothiazole-tethered 2,4-TZD hybrid 8b stood out as the most potent AR inhibitor within this study (8b, IC50 = 0.16 µM; Epalrestat, IC50 = 0.10 µM). Moreover, it was able to reduce BG level in STZ-induced diabetic animal model at 50 mg/kg dose from 399.8 mg/dL to 240 mg/dL. Furthermore, diverse in silico calculations suggested that these candidates possess drug-likeness characters and acceptable pharmacokinetics as non-competitive AR inhibitors. Also, target fishing shows that compound 8b has several pharmacophoric features similar to the inhibitors of AR, PPAR-γ, and GSK-3 Als. Molecular docking study displayed that hybrid 8b can bind to AR active site in a similar pattern as the reported AR inhibitors. Collectively, the new 2,4-TZD hybrids could be used as candidates for development of more efficient and selective AR inhibitors for management of serious diabetic complications.
Experimental
Chemistry
General
Melting points (°C) were determined using Stuart apparatus (SMP 30) and are uncorrected. IR spectra (KBr) were recorded on FT-IR 200 spectrophotometer (ύ cm−1), Faculty of Pharmacy, Mansoura University, Mansoura, Egypt. 1H NMR and 13C NMR spectra were measured in DMSO-d6 at 1H NMR (400 MHz), 13C NMR (100 MHz) using TMS as an internal standard, at NMR Unit, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt. The following abbreviations are used as follows: s, singlet; d, doublet; t, triplet; m, multiplet; br, broad, chemical shift (δ ppm). Mass spectra were carried out on direct inlet part to mass analyser utilising Thermo Scientific GCMS model ISQ at the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Assiut, Egypt. All the chemicals and reagents used were purchased from Aldrich Chemicals Co. (Milwaukee, WI) and commercial sources. Reaction times were determined using TLC on silica gel plates 60F245 E. Merck, using hexane/EtOAc (1:1) as eluting system and the spots were visualised by UV (366–245 nm).
The key precursors, thiazolidinedione (2), 1,3-thiazolidine-2,4-dione derivative (4a–l) and benzothiazole chloride 7 could be easily prepared according to the previously described literature proceduresCitation33,Citation59.
General procedure for synthesis of (Z)-5-(4 or 3-substitutedbenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5a–k)
The potassium salts 4a–k (0.30 mmol, 1 equiv.) and 2-bromo-4′-nitroacetophenone 95% (0.30 mmol, 1 equiv.) were mixed together in a round-bottomed flask along with 10 mL dry DMF. Thereafter, the reaction mixture was stirred at room temperature and the reaction progress was monitored by TLC. Twelve hours later, the reaction was stopped, and the reaction mixture was quenched with water. The separated solid was filtrated off and crystallised from ethanol to produce the corresponding nitro-phenacyl-based thiazolidine-2,4-dione hybrids in pure form 5a–k.
(Z)-5-(4-nitrobenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5a)
Pale yellow solid; (0.095 g, 77%). M.p. 204–206 °C. IR (νmax/cm−1): 3075, 2982 (CH), 1748, 1691 (C═O), 1609, 1525, 1220. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.8 Hz, 2H), 8.39 (d, J = 8.8 Hz, 2H), 8.35 (d, J = 8.6 Hz, 2H), 8.16 (s, 1H), 7.96 (d, J = 8.6 Hz, 2H), 5.49 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 191.4, 167.0, 165.3, 151.1, 148.3, 139.5, 138.7, 132.0, 131.7, 130.4, 125.5, 124.8, 124.5, 48.9. MS m/z (%): 413.9 (M+, 48.61).
(Z)-5-(4-chlorobenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5b)
Yellow solid; (0.105 g, 88%). M.p. 260–262 °C. IR (νmax/cm−1): 3111, 2975 (CH), 1738, 1684 (C═O), 1601, 1526, 1221. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.8 Hz, 2H), 8.34 (d, J = 8.8 Hz, 2H), 8.04 (s, 1H), 7.72 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 8.5 Hz, 2H), 5.46 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 192.1, 167.3, 165.5, 151.5, 139.0, 136.6, 134.5, 133.3, 132.4, 130.4, 130.0, 124.5, 122.0, 48.7. MS m/z (%): 402.7 (M+, 17.58).
(Z)-5-(4-bromobenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5c)
Yellow solid; (0.100 g, 75%). M.p. 241–243 °C. IR (νmax/cm−1): 3076, 2969 (CH), 1748, 1696 (C═O), 1605, 1527, 1216. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.7 Hz, 2H), 8.34 (d, J = 8.7 Hz, 2H), 8.02 (s, 1H), 7.80 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 5.46 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 191.5, 167.2, 165.5, 151.1, 138.8, 136.0, 133.3, 132.4, 132.2, 130.4, 130.0, 124.5, 122.0, 48.8. MS m/z (%): 447.1 (M+, 22.32).
(Z)-5-(4-fluorobenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5d)
White solid; (0.098 g, 85%). M.p. 201–203 °C. IR (νmax/cm−1): 3111, 2982 (CH), 1748, 1691 (C═O), 1596, 1520, 1151. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.4 Hz, 2H), 8.34 (d, J = 8.4 Hz, 2H), 8.05 (s, 1H), 7.78 (d, J = 7.0 Hz, 2H), 7.44 (d, J = 7.0 Hz, 2H), 5.46 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 191.5, 167.3, 165.6, 151.1, 138.8, 133.5, 133.3, 130.4, 129.9, 124.5, 120.9, 117.2, 117.0, 48.7. MS m/z (%): 386.2 (M+, 24.88).
(Z)-5-(4-methylbenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5e)
Yellow solid; (0.08 g, 70%). M.p. 239–241 °C. IR (νmax/cm−1): 3076, 2969 (CH), 1732, 1678 (C═O), 1596, 1535, 1222. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.8 Hz, 2H), 8.35 (d, J = 8.8 Hz, 2H), 8.00 (s, 1H), 7.60 (d, J = 8.2 Hz, 2H), 7.41 (d, J = 8.2 Hz, 2H), 5.45 (s, 2H), 2.40 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 191.6, 167.8, 165.6, 162.3, 151.7, 136.8, 132.3, 130.9, 130.3, 125.2, 124.5, 118.4, 114.6, 48.7, 21.6. MS m/z (%): 382.0 (M+, 40.09).
(Z)-5-(4-methoxybenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5f)
Pale yellow solid; (0.107 g, 90%). M.p. IR (νmax/cm−1): 3068, 2968 (CH), 1742, 1680 (C═O), 1598, 1520, 1325. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.8 Hz, 2H), 8.34 (d, J = 8.8 Hz, 2H), 7.99 (s, 1H), 7.67 (d, J = 8.8 Hz, 2H), 7.16 (d, J = 8.8 Hz, 2H), 5.43 (s, 2H), 3.87 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 191.6, 167.5, 165.7, 161.9, 151.1, 138.8, 134.6, 132.96, 130.4, 125.7, 124.5, 117.9, 115.6, 56.1, 48.6. MS m/z (%): 398.05 (M+, 25.10).
(Z)-3-(2-(4-nitrophenyl)-2-oxoethyl)-5-(3,4,5-trimethoxybenzylidene)thiazolidine-2,4-dione (5g)
Yellow solid; (0.110 g, 80%). M.p. 184–186 °C. IR (νmax/cm−1): 3070, 2969 (CH), 1740, 1680 (C═O), 1596, 1535, 1220. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.6 Hz, 2H), 8.34 (d, J = 8.6 Hz, 2H), 7.98 (s, 1H), 7.01 (s, 2H), 5.46 (s, 2H), 3.87 (s, 6H), 3.76 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 191.6, 167.4, 165.6, 153.8, 151.1, 140.3, 138.8, 134.8, 130.4, 128.8, 124.5, 120.1, 108.3, 60.7, 56.5, 48.7. MS m/z (%): 458.13 (M+, 79.45).
(Z)-5-(3-nitrobenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5h)
Yellow solid; (0.085 g, 69%). M.p. 196–198 °C. IR (νmax/cm−1): 3080, 2980 (CH), 1751, 1693 (C═O), 1609, 1521, 1224. 1H NMR (400 MHz, DMSO-d6) δ 8.47–8.31 (m, 6H), 8.15 (s, 1H), 7.96 (d, J = 7.2 Hz, 2H), 5.48 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 191.7, 167.5, 165.6, 149.4, 147.8, 138.9, 136.2, 133.3, 133.1, 129.9, 129.4, 128.7, 124.5, 124.2, 123.0, 48.7. MS m/z (%): 413.3 (M+, 35.75).
(Z)-5-(3-bromobenzylidene)-3-(2-(4-nitrophenyl)-2-oxoethyl)thiazolidine-2,4-dione (5k)
Yellow solid; (0.101, 75%). M.p. 179–181 °C. IR (νmax/cm−1): 3108, 2985 (CH), 1752, 1692 (C═O), 1601, 1520, 1151. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.8 Hz, 2H), 8.35 (d, J = 8.8 Hz, 2H), 8.04 (s, 1H), 7.93 (s, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.55 (t, J = 7.9 Hz, 1H), 5.47 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 191.5, 167.1, 165.4, 151.1, 138.7, 135.7, 133.8, 133.7, 132.9, 131.9, 130.4, 128.7, 124.5, 123.0, 123.0, 48.8. MS m/z (%): 447.1 (M+, 13.52).
General procedure for the preparation of (Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(4-subtitutedbenzylidene)thiazolidine-2,4-dione (8a–h)
The potassium salts 4a–g, 4l (0.32 mmol, 1 equiv.) and 2-chloromethyl-1,3-benzothiazole 7 (0.32 mmol, 1 equiv.) were added together in a round-bottomed flask along with 10 mL dry DMF. After that, the reaction mixture was heated under reflux and the reaction progress was monitored by TLC. After 12 h, the reaction was stopped, and the reaction mixture was quenched with water. The separated solid was filtrated off and crystallised from ethanol to obtain the corresponding benzothiazole-based thiazolidine-2,4-dione hybrids in pure form 8a–h.
(Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(4-nitrobenzylidene)thiazolidine-2,4-dione (8a)
Yellow solid; (0.095 g, 74%). M.p. 190–192 °C. IR (νmax/cm−1): 3026, 2937 (CH), 1745, 1680 (C═O), 1608, 1580, 1145. 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 7.9 Hz, 2H), 8.17 (s, 1H), 8.13 (d, J = 7.8 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.95 (d, J = 7.9 Hz, 2H), 7.60–7.50 (m, 2H), 5.36 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.9, 165.2, 164.9, 152.4, 148.3, 139.5, 135.4, 132.0, 131.6, 131.1, 126.9, 126.1, 124.8, 123.2, 122.9, 43.4. MS m/z (%): 397.4 (M+, 22.5).
(Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(4-chlorobenzylidene)thiazolidine-2,4-dione (8b)
White solid; (0.082 g, 70%). M.p. 159–161 °C. IR (νmax/cm−1): 3022, 2930 (CH), 1738, 1678 (C═O), 1605, 1585, 1130.1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J = 7.8 Hz, 1H), 8.05 (s, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H), 7.60–7.50 (m, 2H), 5.34 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.2, 165.4, 165.1, 152.5, 136.0, 135.3, 133.3, 132.4, 132.2, 130.0, 126.9, 126.0, 123.2, 122.9, 122.0, 43.3. MS m/z (%): 386.6 (M+, 14.20).
(Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(4-bromobenzylidene)thiazolidine-2,4-dione (8c)
Pale yellow solid; (0.085 g, 66%). M.p. 174–176 °C. IR (νmax/cm−1): 3027, 2930 (CH), 1740, 1683 (C═O), 1607, 1580, 1132. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.2 Hz, 1H), 7.93 (s, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.51 (t, J = 7.7 Hz, 1H), 7.46–7.39 (m, 3H), 5.38 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.4, 165.4, 165.2, 152.4, 135.4, 133.5, 133.4, 130.0, 126.9, 126.0, 123.2, 122.9, 120.9, 117.3, 117.1, 43.1. MS m/z (%): 431.1 (M+, 30.1).
(Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(4-fluorobenzylidene)thiazolidine-2,4-dione (8d)
Pale yellow solid; (0.079 g, 71%). M.p. 190–192 °C. IR (νmax/cm−1): 3020, 2930 (CH), 1738, 1685 (C═O), 1607, 1580, 1130.1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J = 7.9 Hz, 1H), 8.07 (s, 1H), 7.99 (d, J = 7.9 Hz, 1H), 7.81–7.73 (m, 2H), 7.55–7.42 (m, 4H), 5.34 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 167.3, 165.5, 165.2, 152.5, 135.3, 133.5, 133.3, 133.2, 126.9, 126.1, 123.2, 122.9, 120.9, 117.2, 117.0, 43.3. MS m/z (%): 370.93 (M+, 45.49).
(Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(4-methylbenzylidene)thiazolidine-2,4-dione (8e)
Off-white solid; (0.090 g, 82%). M.p. 163–165 °C. IR (νmax/cm−1): 3025, 2940 (CH), 1740, 1691 (C═O), 1607, 1575, 1145. 1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J = 7.4 Hz, 1H), 8.00 (s, 1H), 7.97 (d, J = 7.4 Hz, 1H), 7.58 (d, J = 7.3 Hz, 2H), 7.56–7.44 (m, 2H), 7.40 (d, J = 7.3 Hz, 2H), 5.33 (s, 2H), 2.51 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.5, 165.5, 165.3, 152.5, 141.8, 135.3, 134.7, 130.8, 130.7, 130.6, 126.9, 126.0, 123.2, 122.9, 119.9, 43.3, 21.6. MS m/z (%): 366.4 (M+, 18.2).
(Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(4-methoxybenzylidene)thiazolidine-2,4-dione (8f)
Pale yellow solid; (0.085 g, 70%). M.p. IR (νmax/cm−1): 3024, 2939 (CH), 1740, 1680 (C═O), 1610, 1580. 1H NMR (400 MHz, DMSO) δ 8.10 (d, J = 7.6 Hz, 1H), 7.99 (s, 1H), 7.98 (d, J = 7.6 Hz, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.55–7.44 (m, 2H), 7.14 (d, J = 8.5 Hz, 2H), 5.32 (s, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.5, 166.0, 165.4, 152.5, 135.3, 134.6, 133.0, 132.6, 132.3, 126.9, 126.0, 123.2, 122.9, 117.8, 115.5, 56.0, 43.2. MS m/z (%): 382.2 (M+, 20.4).
(Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(3,4,5-trimethoxybenzylidene)thiazolidine-2,4-dione (8g)
Pale yellow solid; (0.066 g, 50%). M.p. 140–142 °C. IR (νmax/cm−1): 3020, 2950 (CH), 1738, 1685 (C═O), 1607, 1580, 1200. 1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J = 7.9 Hz, 1H), 8.00 (s, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.60–7.50 (m, 2H), 7.01 (s, 2H), 5.34 (s, 2H), 3.86 (s, 6H), 3.76 (s, 3H). MS m/z (%): 442.3 (M+, 22.92).
(Z)-3-(benzo[d]thiazol-2-ylmethyl)-5-(3,4-dimethoxybenzylidene)thiazolidine-2,4-dione (8h)
Yellow solid; (0.080 g, 60%). M.p. 180–182 °C. IR (νmax/cm−1): 3020, 2935 (CH), 1738, 1680 (C═O), 1609, 1585, 1140. 1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J = 7.8 Hz, 1H), 8.00 (s, 1H), 7.98 (d, J = 7.8 Hz, 1H), 7.60–7.50 (m, 2H), 7.28 (s, 2H), 7.17 (d, J = 8.8 Hz, 1H), 5.33 (s, 2H), 3.86 (s, 3H), 3.84 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.5, 165.9, 165.5, 155.2, 152.5, 149.1, 136.3, 134.8, 132.9, 132.6, 129.4, 126.5, 126.0, 123.4, 122.2, 113.9, 113.4, 60.1, 56.5, 43.6. MS m/z (%): 412.3 (M+, 25.2).
Biological evaluation of the synthesised compounds
In vitro enzyme inhibitory assay against human aldose reductase
Enzyme inhibitory assays for the synthesised compounds were carried out using AR activity assay kit (ab273276) according to the manufacturer instructions. From the obtained dose response curve, the concentration of the compounds inhibiting 50% of enzyme (IC50) was calculated.
Kinetic study of inhibitory effect of 8b against aldose reductase
Since compound 8b showed the best activity, it was subjected for kinetic study to determine its binding mode by determination of its effect on Km and Vmax of AR upon incubation with half IC50, IC50, and double IC50 of 8b. The Michaelis–Menten equation was used to calculate both KM and Vmax using non-linear regression by the aid of GraphPad Prism 8.0 (La Jolla, CA) and data were presented using Lineweaver–Burk plot.
Effect of compound 8b in blood glucose level in diabetic mice
Twenty-four male mice with an average weight of 25.41 ± 2.13 g, were used in this study. Six mice/cage were housed at room temperature with 12 h dark/light cycle under standardised environmental conditions with free access to water and food till initiation of the experimental protocol. All animal experiments were done in agreement with ethical guidelines for animal experimentation. Animals in specific groups were injected I.P. with solution of STZ (Sigma, Carlsbad, CA), at final dosage of 40 mg/kg BW, freshly prepared in 0.1 mol/L citrate–phosphate buffer, pH 4.5 for five successive days, then BG was determined, and the mice with established hyperglycaemia (>300 mg/dL) were involved in the study. Mice were randomly assigned as six mice per group as follows: vehicle control group (group I), received the STZ injection only (group II), received 5 mg/kg of compound 8b after STZ-induction (group III), and final group received 50 mg/kg after STZ-induction (group IV). After 6 weeks, fasting glucose levels were monitored and compared with initial glucose level. Statistical analysis was performed using one-way analysis of variance (ANOVA) using GraphPad Prism 8.0 (La Jolla, CA). A p value of less than 0.05 was considered significant. The mean and standard error of the mean were used to describe all of the obtained results. The research ethics committee at Kafrelsheikh University, Kafrelsheikh, Egypt examined and authorised the experiments (code number: KFS-IACUC/116/2023).
Molecular docking and ADME prediction studies
In silico tools were used to gain insights on potential mechanism of action of compound 8b and to assess its ability 8b to bind to AR. First, the 3D chemical structures of 8b were submitted to Pharmmaper server (http://www.lilab-ecust.cn/pharmmapper/) and the top targets related to diabetes were chosen based on their z-scoreCitation60.
For molecular docking studies, the crystal structure of AR was obtained from PDB using the code: 3g5e, which was subjected to protein repair and analysisCitation61,Citation62. Protein preparation module integrated in PyRx software was used to determine the active site coordinate, which was defined as X: 22, Y: −7, and Z: 23 and the grid box size was 22 × 22 × 32Citation63.
Compound 8b 2D structure was prepared using Marvin sketch version 21.17.0, ChemAxon (https://www.chemaxon.com) and saved as mol files and converted to PDBQT using AutoDock tools embedded in PyRX. Autodock vina was chosen to perform molecular docking, using default parameters. The validation of PyRx was confirmed by redocking the co-crystallised ligand and the RMSD was shown to be no more than 1.5 Å (Figure 1S). The best ranked pose in terms of binding free energy (ΔG) was analysed by visualisation of the interaction with the active site using discovery studio visualiser 20.0Citation64. The most active compounds in the biological assay were also submitted to PreADMET online server (https://preadmet.qsarhub.com/) to predict their Pharmacokinetic properties.
Supplemental Material
Download PDF (5 MB)Acknowledgements
We are thankful to the Research supporting project number (RSPD2023R740), King Saud University, Riyadh, Saudi Arabia.
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Pasala VK, Gudipudi G, Sankeshi V, Basude M, Gundla R, Singh Jadav S, Srinivas B, Goud EY, Nareshkumar D. Design, synthesis and biological evaluation of selective hybrid coumarin-thiazolidinedione aldose reductase-II inhibitors as potential antidiabetics. Bioorg Chem. 2021;114:104970.
- Imran M, Yar MS, Khan SA. Synthesis and antihyperglycemic activity of 2-(substituted phenyl)-3-{[4-(1-naphthyl)-1, 3-thiazol-2-yl] amino}-4-oxo-1, 3-thiazolidin-5-ylacetic acid derivatives. Acta Pol Pharm. 2009;66(1):51–56.
- Sever B, Altıntop MD, Demir Y, Türkeş C, Özbaş K, Çiftçi GA, Beydemir Ş, Özdemir A. A new series of 2,4-thiazolidinediones endowed with potent aldose reductase inhibitory activity. Open Chem. 2021;19(1):347–357.
- Singh Grewal A, Bhardwaj S, Pandita D, Lather V, Singh Sekhon B. Updates on aldose reductase inhibitors for management of diabetic complications and non-diabetic diseases. Mini Rev Med Chem. 2016;16(2):120–162.
- Seltzer HS. Drug-induced hypoglycemia: a review of 1418 cases. Endocrinol Metab Clin N Am. 1989;18(1):163–183.
- O Moore-Sullivan TM, Prins JB. Thiazolidinediones and type 2 diabetes: new drugs for an old disease. Med J Aust. 2002;176(8):381–386.
- Lee AY, Chung SS. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999;13(1):23–30.
- Bozdağ-Dündar O, Evcimen ND, Ceylan-Ünlüsoy M, Ertan R, Sarıkaya M. Some new thiazolyl thiazolidinedione derivatives as aldose reductase inhibitors. Med Chem Res. 2008;16(1):39–47.
- Alexiou P, Pegklidou K, Chatzopoulou M, Nicolaou I, Demopoulos VJ. Aldose reductase enzyme and its implication to major health problems of the 21st century. Curr Med Chem. 2009;16(6):734–752.
- Pfeifer MA, Schumer MP, Gelber DA. Aldose reductase inhibitors: the end of an era or the need for different trial designs? Diabetes. 1997;46(Suppl. 2):S82–S89.
- Kador PF, Robison WG Jr, Kinoshita JH. The pharmacology of aldose reductase inhibitors. Annu Rev Pharmacol Toxicol. 1985;25:691–714.
- Costantino L, Rastelli G, Gamberini MC, Barlocco D. Pharmacological approaches to the treatment of diabetic complications. Expert Opin Ther Pat. 2000;10(8):1245–1262.
- Costantino L, Ferrari AM, Gamberini MC, Rastelli G. Nitrophenyl derivatives as aldose reductase inhibitors. Bioorg Med Chem. 2002;10(12):3923–3931.
- Kousaxidis A, Kovacikova L, Nicolaou I, Stefek M, Geronikaki A. Non-acidic bifunctional benzothiazole-based thiazolidinones with antimicrobial and aldose reductase inhibitory activity as a promising therapeutic strategy for sepsis. Med Chem Res. 2021;30(10):1837–1848.
- Kratky M, Sramel P, Bodo P, Prnova MS, Kovacikova L, Majekova M, Vinsova J, Stefek M. Novel rhodanine based inhibitors of aldose reductase of non-acidic nature with p-hydroxybenzylidene functional group. Eur J Med Chem. 2023;246:114922.
- Bacha MM, Nadeem H, Zaib S, Sarwar S, Imran A, Rahman SU, Ali HS, Arif M, Iqbal J. Rhodanine-3-acetamide derivatives as aldose and aldehyde reductase inhibitors to treat diabetic complications: synthesis, biological evaluation, molecular docking and simulation studies. BMC Chem. 2021;15(1):1–15.
- Hotta N, Sakamoto N, Shigeta Y, Kikkawa R, Goto Y, Diabetic Neuropathy Study Group in Japan. Clinical investigation of Epalrestat, an aldose reductase inhibitor, on diabetic neuropathy in Japan: multicenter study. J Diabetes Complications. 1996;10(3):168–172.
- Hotta N, Akanuma Y, Kawamori R, Matsuoka K, Oka Y, Shichiri M, Toyota T, Nakashima M, Yoshimura I, Sakamoto N, et al. Long-term clinical effects of Epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3-year, multicenter, comparative aldose reductase inhibitor-diabetes complications trial. Diabetes Care. 2006;29(7):1538–1544.
- Terashima H, Hama K, Yamamoto R, Tsuboshima M, Kikkawa R, Hatanaka I, Shigeta Y. Effects of a new aldose reductase inhibitor on various tissues in vitro. J Pharmacol Exp Ther. 1984;229(1):226–230.
- Das-Evcimen N, Sarıkaya M, Selen Gurkan-Alp A, Bozdag-Dundar O. Aldose reductase inhibitory potential of several thiazolyl-thiazolidine-2,4-diones. Lett Drug Des Discov. 2013;10(5):415–419.
- Bozdağ-Dündar O, Evranos B, Daş-Evcimen N, Sarıkaya M, Ertan R. Synthesis and aldose reductase inhibitory activity of some new chromonyl-2,4-thiazolidinediones. Eur J Med Chem. 2008;43(11):2412–2417.
- Maccari R, Del Corso A, Giglio M, Moschini R, Mura U, Ottanà R. In vitro evaluation of 5-arylidene-2-thioxo-4-thiazolidinones active as aldose reductase inhibitors. Bioorg Med Chem Lett. 2011;21(1):200–203.
- Ottanà R, Maccari R, Giglio M, Del Corso A, Cappiello M, Mura U, Cosconati S, Marinelli L, Novellino E, Sartini S, et al. Identification of 5-arylidene-4-thiazolidinone derivatives endowed with dual activity as aldose reductase inhibitors and antioxidant agents for the treatment of diabetic complications. Eur J Med Chem. 2011;46(7):2797–2806.
- Mylari BL, Larson ER, Beyer TA, Zembrowski WJ, Aldinger CE, Dee MF, Siegel TW, Singleton DH. Novel, potent aldose reductase inhibitors: 3,4-dihydro-4-oxo-3-[[5-(trifluoromethyl)-2-benzothiazolyl] methyl]-1-phthalazineacetic acid (Zopolrestat) and congeners. J Med Chem. 1991;34(1):108–122.
- Imran A, Shehzad MT, Shah SJA, Al Adhami T, Laws M, Rahman KM, Alharthy RD, Khan IA, Shafiq Z, Iqbal J. Development and exploration of novel substituted thiosemicarbazones as inhibitors of aldose reductase via in vitro analysis and computational study. Sci Rep. 2022;12(1):5734.
- Aotsuka T, Abe N, Fukushima K, Ashizawa N, Yoshida M. Benzothiazol-2-ylcarboxylic acids with diverse spacers: a novel class of potent, orally active aldose reductase inhibitors. Bioorg Med Chem Lett. 1997;7(13):1677–1682.
- Kotani T, Nagaki Y, Ishii A, Konishi Y, Yago H, Suehiro S, Okukado N, Okamoto K. Highly selective aldose reductase inhibitors. 3. Structural diversity of 3-(arylmethyl)-2,4,5-trioxoimidazolidine-1-acetic acids. J Med Chem. 1997;40(5):684–694.
- Kotani T, Ishii A, Nagaki Y, Toyomaki Y, Yago H, Suehiro S, Okukado N, Okamoto K. Highly selective aldose reductase inhibitors. II. Optimization of the aryl part of 3-(arylmethyl)-2,4,5-trioxoimidazolidine-1-acetic acids. Chem Pharm Bull. 1997;45(2):297–304.
- Zaher N, Nicolaou I, Demopoulos VJ. Pyrrolylbenzothiazole derivatives as aldose reductase inhibitors. J Enzyme Inhib Med Chem. 2002;17(2):131–135.
- Kousaxidis A, Petrou A, Rouvim P, Bodo P, Stefek M, Nicolaou I, Geronikaki A. A molecular hybridization approach for the design of selective aldose reductase (ALR2) inhibitors and exploration of their activities against protein tyrosine phosphatase 1B (PTP1B). J Mol Struct. 2023;1271:134116.
- Hussain S, Parveen S, Qin X, Hao X, Zhang S, Chen X, Zhu C, Ma B. Novel synthesis of nitro-quinoxalinone derivatives as aldose reductase inhibitors. Bioorg Med Chem Lett. 2014;24(9):2086–2089.
- Ishii A, Kotani T, Nagaki Y, Shibayama Y, Toyomaki Y, Okukado N, Ienaga K, Okamoto K. Highly selective aldose reductase inhibitors. 1. 3-(Arylalkyl)-2,4,5-trioxoimidazolidine-1-acetic acids. J Med Chem. 1996;39(9):1924–1927.
- Tilekar K, Upadhyay N, Hess JD, Macias LH, Mrowka P, Aguilera RJ, Meyer-Almes F-J, Iancu CV, Choe J-y, Ramaa C. Structure guided design and synthesis of furyl thiazolidinedione derivatives as inhibitors of GLUT 1 and GLUT 4, and evaluation of their anti-leukemic potential. Eur J Med Chem. 2020;202:112603.
- Bruno G, Costantino L, Curinga C, Maccari R, Monforte F, Nicolo F, Ottana R, Vigorita M. Synthesis and aldose reductase inhibitory activity of 5-arylidene-2,4-thiazolidinediones. Bioorg Med Chem. 2002;10(4):1077–1084.
- Kaminskyy D, Kryshchyshyn A, Lesyk R. 5-Ene-4-thiazolidinones – an efficient tool in medicinal chemistry. Eur J Med Chem. 2017;140:542–594.
- Xia Z, Knaak C, Ma J, Beharry ZM, McInnes C, Wang W, Kraft AS, Smith CD. Synthesis and evaluation of novel inhibitors of Pim-1 and Pim-2 protein kinases. J Med Chem. 2009;52(1):74–86.
- Stana A, Tiperciuc B, Duma M, Vlase L, Crişan O, Pîrnău A, Oniga O. Synthesis and antimicrobial activity of some new N‐substituted‐5‐arylidene‐thiazolidine‐2,4‐diones. J Heterocycl Chem. 2014;51(2):411–417.
- Hurren KM, Dunham MW. Are thiazolidinediones a preferred drug treatment for type 2 diabetes? Expert Opin Pharmacother. 2021;22(2):131–133.
- Othman DIA, Hamdi A, Tawfik SS, Elgazar AA, Mostafa AS. Identification of new benzimidazole-triazole hybrids as anticancer agents: multi-target recognition, in vitro and in silico studies. J Enzyme Inhib Med Chem. 2023;38(1):2166037.
- Al-Sanea MM, Hamdi A, Mohamed AAB, El-Shafey HW, Moustafa M, Elgazar AA, Eldehna WM, Ur Rahman H, Parambi DGT, Elbargisy RM, et al. New benzothiazole hybrids as potential VEGFR-2 inhibitors: design, synthesis, anticancer evaluation, and in silico study. J Enzyme Inhib Med Chem. 2023;38:2166036.
- El-Senduny FF, Elgazar A, Alwasify HA, Abed A, Foda M, Abouzeid S, Lewerenz L, Selmar D, Badria F. Bio-evaluation of untapped alkaloids from Vinca minor enriched by methyl jasmonate induced stress: an integrated approach. Planta Med. 2023.
- Hamdi A, El-Shafey HW, Othman DIA, El-Azab AS, AlSaif NA, Abdel-Aziz AAM. Design, synthesis, antitumor, and VEGFR-2 inhibition activities of novel 4-anilino-2-vinyl-quinazolines: molecular modeling studies. Bioorg Chem. 2022;122:105710.
- Hamdi A, Elhusseiny WM, Othman DI, Haikal A, Bakheit AH, El-Azab AS, Al-Agamy MH, Alaa A-M. Synthesis, antitumor, and apoptosis-inducing activities of novel 5-arylidenethiazolidine-2,4-dione derivatives: histone deacetylases inhibitory activity and molecular docking study. Eur J Med Chem. 2022;244:114827.
- Othman DI, Hamdi A, Abdel-Aziz MM, Elfeky SM. Novel 2-arylthiazolidin-4-one-thiazole hybrids with potent activity against Mycobacterium tuberculosis. Bioorg Chem. 2022;124:105809.
- Islam MS, Al-Majid AM, Azam M, Verma VP, Barakat A, Haukka M, Elgazar AA, Mira A, Badria FA. Construction of spirooxindole analogues engrafted with indole and pyrazole scaffolds as acetylcholinesterase inhibitors. ACS Omega. 2021;6(47):31539–31556.
- Al-Sanea MM, Hamdi A, Brogi S, Tawfik SS, Othman DIA, Elshal M, Ur Rahman H, Parambi DGT, Elbargisy RM, Selim S, et al. Design, synthesis, and biological investigation of oxadiazolyl, thiadiazolyl, and pyrimidinyl linked antipyrine derivatives as potential non-acidic anti-inflammatory agents. J Enzyme Inhib Med Chem. 2023;38(1):2162511.
- Al-Sanea MM, Chilingaryan G, Abelyan N, Mamikonyan M, Gasparyan H, Hovhannisyan S, Hamdi A, Ali AR, Selim S, Mohamed AA. Combination of ligand and structure based virtual screening approaches for the discovery of potential PARP1 inhibitors. PLOS One. 2022;17(9):e0272065.
- Martinez A, Alonso M, Castro A, Dorronsoro I, Gelpí JL, Luque FJ, Pérez C, Moreno FJ. SAR and 3D-QSAR studies on thiadiazolidinone derivatives: exploration of structural requirements for glycogen synthase kinase 3 inhibitors. J Med Chem. 2005;48(23):7103–7112.
- Kumar H, Aggarwal N, Marwaha MG, Deep A, Chopra H, Matin MM, Roy A, Emran TB, Mohanta YK, Ahmed R, et al. Thiazolidin-2,4-dione scaffold: an insight into recent advances as antimicrobial, antioxidant, and hypoglycemic agents. Molecules. 2022;27(19):6763.
- Long N, Le Gresley A, Wren SP. Thiazolidinediones: an in-depth study of their synthesis and application to medicinal chemistry in the treatment of diabetes mellitus. ChemMedChem. 2021;16(11):1717–1736.
- Kousaxidis A, Petrou A, Lavrentaki V, Fesatidou M, Nicolaou I, Geronikaki A. Aldose reductase and protein tyrosine phosphatase 1B inhibitors as a promising therapeutic approach for diabetes mellitus. Eur J Med Chem. 2020;207:112742.
- Lee YS, Chen Z, Kador PF. Molecular modeling studies of the binding modes of aldose reductase inhibitors at the active site of human aldose reductase. Bioorg Med Chem. 1998;6(10):1811–1819.
- Wilson DK, Tarle I, Petrash JM, Quiocho FA. Refined 1.8 A structure of human aldose reductase complexed with the potent inhibitor Zopolrestat. Proc Natl Acad Sci USA. 1993;90(21):9847–9851.
- Aungst BJ. Optimizing oral bioavailability in drug discovery: an overview of design and testing strategies and formulation options. J Pharm Sci. 2017;106(4):921–929.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25.
- Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623.
- El-Shafey HW, Gomaa RM, El-Messery SM, Goda FE. Quinazoline based HSP90 inhibitors: synthesis, modeling study and ADME calculations towards breast cancer targeting. Bioorg Med Chem Lett. 2020;30(15):127281.
- Wang R, Fu Y, Lai L. A new atom-additive method for calculating partition coefficients. J Chem Inf Comput Sci. 1997;37(3):615–621.
- Peprah K, Zhu XY, Eyunni SVK, Etukala JR, Setola V, Roth BL, Ablordeppey SY. Structure–activity relationship studies of SYA 013, a homopiperazine analog of haloperidol. Bioorg Med Chem. 2012;20(5):1671–1678.
- Wang X, Shen Y, Wang S, Li S, Zhang W, Liu X, Lai L, Pei J, Li H. PharmMapper 2017 update: a web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017;45(W1):W356–W360.
- Nnyigide OS, Nnyigide TO, Lee S-G, Hyun K. Modeling, protein repair and analysis server: a web server to repair PDB structures, add missing heavy atoms and hydrogen atoms, and assign secondary structures by amide interactions. J Chem Inf Model. 2022;62(17):4232–4246.
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786.
- Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243–250.
- Elimam DM, Elgazar AA, El-Senduny FF, El-Domany RA, Badria FA, Eldehna WM. Natural inspired piperine-based ureas and amides as novel antitumor agents towards breast cancer. J Enzyme Inhib Med Chem. 2022;37(1):39–50.