
Abstract
The Mannich reaction is commonly used to introduce N atoms into compound molecules and is thus widely applied in drug synthesis. The Mannich reaction accounts for a certain proportion of structural modifications of natural products. The introduction of Mannich bases can significantly improve the activity, hydrophilicity, and medicinal properties of compounds; therefore, the Mannich reaction is widely used for the structural modification of natural products. In this paper, the application of the Mannich reaction to the structural modification of natural products is reviewed, providing a method for the structural modification of natural products.
Introduction
Natural products are the basis of the traditional medicinal system, and their use in the treatment of diseases can be traced back to approximately 2600 BC. Cedrus spp. (cedar), Cupressus sempevirens (cypress), Glycyrrhiza glabra (licorice), Commiphora spp. (myrrh), and Papaver somniferum (poppy juice) extracts and derivatives are still used today to treat diseases such as coughs, inflammations, and parasitic infections.
Natural products constitute a treasury for drug development and have always been valuable sources in drug design. Pharmaceutical researchers are often inspired by natural product molecules with specific reactive backbones, reactive groups, and excellent biological activities. For example, khellin from Ammi visnaga (L) Lamk led to the development of chromolyn (in the form of sodium chromoglycate) as a bronchodilator and galegine from Galega officinalis L, which is a model for the synthesis of metformin and other bisguanidine-type antidiabetic drugsCitation1. In addition, data analysis has shown that a considerable number of drugs approved by the Food and Drug Association (FDA) since 1939 contain natural product fragments. The natural product database contains 210,213 natural products. This demonstrates the importance of natural products for drug research. Therefore, the structural modifications of natural products are of great significance.
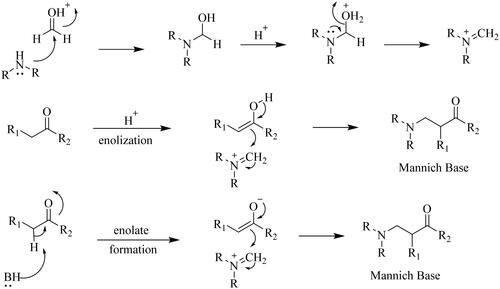
The Mannich reaction, also known as the amine methylation reaction, is an organic chemical reaction in which a compound (typically a carbonyl compound) containing reactive hydrogen is condensed with formaldehyde and secondary amine or ammonia to form α,β-amino (carbonyl) compound. It is also an important reaction type and a key reaction step in the synthesis of numerous drugs and natural products. The Mannich reaction mechanism is as follows (. The product of the reaction β-amino (carbonyl) compound is referred to as the Mannich base. It contains nitrogen atoms, which can increase the water solubility of the compound, increase its affinity for the receptor, and increase the bioavailability and pharmacological activity of the drug molecule. Investigation of the literature has shown that Mannich bases have a wide range of biological characteristics, including anti-cancerCitation2, anti-inflammatoryCitation3, analgesicCitation4, and antimicrobial activitiesCitation5. In this paper, we review the progress of the Mannich reaction for the modification of natural products according to the classification of their biological activities.
Figure 1. Mannich reaction under acidic conditions and basic conditions.
Applications in modification of natural products for antitumor activity
Chalcones
Reddy et al.Citation6 found that Mannich bases in chalcones may increase biopotency because the nitrogen-containing groups in Mannich bases increase the number of molecular sites for electrophilic attack in cells as well as increase the affinity of the compound for water. A series of Mannich bases of heterocyclic chalcones were synthesised using the one-step Claisen-Schmidt condensation of heterocyclic aldehydes with the Mannich bases of acetophenones, and the target compounds were tested against three human cancer cell lines (prostate, PC-3; breast, MCF-7; and nasopharyngeal, KB) and a multidrug-resistant subline (KB-VIN) (). Of the 39 synthesised chalcones, 31 showed potent activity against at least one cell line, with IC50 values ranging 0.03–3.80 µg/mL. The B ring is a benzene ring and the 1-position is a methoxy group of compound 1 (), which exhibited the strongest inhibitory activity on the MCF-7 cell line with an IC50 value of 0.03 µg/mL, significantly increasing the anti-proliferative activity of the compound.
Figure 2. Compounds 1–34 for antitumor activity.
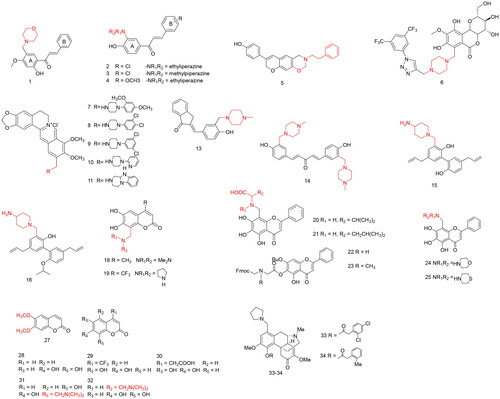
Table 1. Applications in modification of natural products for antitumor activity.
Bui et al.Citation7 found that the biological activity of Mannich bases can be attributed to the deamination of Mannich bases in chalcones and the corresponding cyclohexadienones, which may generate additional sites for the cellular sulphur nucleophilic attack of alcohols and the chemical structures of α,β-unsaturated ketones that can alkylate nucleophiles. Therefore, p-hydroxyacetophenone and different substituted benzaldehyde became α,β-unsaturated ketones after Claisen-Schmidt condensation under the condition of NaOH as the base and methanol as the solvent. The compound α,β-unsaturated ketones were synthesised as a series of Mannich base derivatives under formaldehyde, different secondary amines, and methanol reflux conditions, and the cytotoxic activity of the derivatives in vitro against human hepatocellular carcinoma (HepG2), human lung cancer (SK-LU-1), and MCF-7 were evaluated (). Compound 2 () was the most potent against the three cell lines, with IC50 values of 1.57, 1.16, and 1.21 µg/mL, respectively. In addition, compounds 3 and 4 () exhibited significant activity against MCF-7 cells, with IC50 values of 2.0 µg/mL. Interestingly, the 4-chloro group in the B ring was beneficial for enhancing cytotoxic activity against the three cell lines in almost all cases compared to the methoxy group.
Phenoxodiol
Phenoxodiol, an isoflavone analogue, is clinically used to treat drug-resistant ovarian and prostate cancers. Chen et al.Citation8 reported a series of Mannich base derivatives obtained through the Mannich reaction using phenoxodiol, primary amines, and formaldehyde as reactants and methanol as solvent at room temperature. All 25 compounds inhibited tumour cell proliferation in a dose-dependent manner (). Compared with the lead compound, phenoxodiol, the majority of compounds exhibited higher antiproliferative activity against the MDA MB-231 breast cancer cell line, among which compound 5 () exhibited the highest antiproliferative activity with an IC50 of 0.7 µM, which was 50-fold higher than that of phenoxodiol (IC50 = 35.6 µM). Compound 5 showed low toxicity to the MRC-5 cell line (IC50 = 197.0 µM), and its specificity value to the MDA-MB-231 cell line was 266. Therefore, compound 5 should be considered for further in-depth studies.
Bergenin
Bergenin is a C-glucoside of 4-O-methyl gallic acid, which has demonstrated a wide range of biological characteristics including anticancer, anti-inflammatory, antioxidant activitiesCitation9. In addition, it has few side effects, low toxicity, and no drug resistance. Kumar et al.Citation10 fused various 1,2,3-triazole and amine molecules through click and Mannich reactions to synthesise a series of bergnin-1,2,3-triazole manichaeine derivatives. Moreover, their anticancer activities against a panel of cancer cell lines was investigated. The results showed that the antiproliferative activity of all compounds (ranging 1.33–21.25 µM) against HeLa cell lines is stronger than that of the lead compound bergenin with an IC50 value of 22 µM (). Compound 6 () exhibited the strongest antiproliferative activity, with an IC50 value of 1.33 µM, and was equipotent to doxorubicin.
Berberine
Berberine, an isoquinoline alkaloid, is a primary metabolite isolated from the stems, skin, and roots of several Berberis species, and has anticancer and antioxidant properties. Piperazine rings can effectively inhibit the growth of cancer cells and prevent caspase-dependent apoptosis caused by various signalling pathways in tumours. In addition, the introduction of different electron-absorbing and electron-donating functional groups into the piperazine ring system enhances the anticancer effect. Therefore, based on the principle of molecular hybridisation, Mistry et al.Citation11 used berberine as the lead compound and synthesised a series of berberine Mannich base derivatives by introducing different substituted piperazine rings through the Mannich reaction. Their inhibitory effects on HeLa and CaSki cell lines and their cytotoxicity in Madin-Darby canine kidney (MDCK) cell lines were evaluated (). All the synthesised compounds showed better inhibition of the growth of HeLa and CaSki cell lines than berberine, with IC50 values of 7.33–11.12 µM and 5.76–10.70 µM, respectively. Compounds 8 and 9 () containing 3, 4-dichlorophenylpiperazine and 3,5-dichlorophenylpiperazine, respectively, showed the strongest inhibitory effects on HeLa cells, with IC50 values of 7.53 and 7.33 µM, respectively. The TI values are 42.53 and 42.39, respectively. Compound 7 (), which contains 2,4-dimethoxyphenylpiperazine, had the strongest inhibitory effect on CaSki cells, with and IC50 value of 5.76 µM, CC50 value of 248.9 µM, and TI value of 43.24. In summary, the introduction of a dichloropiperazine group into the berberine structure was beneficial for improving the antiproliferative ability of the HeLa cell line, and dioxypiperazine substitution was important for the antiproliferative ability of the CaSki cell line.
Kim et al.Citation12 also synthesised a series of berberine Mannich base derivatives by introducing a piperazine ring into the berberine structure through the Mannich reaction and evaluated their inhibitory effects on the HeLa and CaSki cell lines (). All synthesised compounds showed good anti-proliferative activity, with IC50 values of 7.31–11.35 µM and 6.11–12.33 µM for HeLa and CaSki cells, respectively. In addition, CC50 values of 193.1–490.5 µM towards the normal cell line (MDCK) and SI values of 23.22–57.47 and 23.03–56.39 were found for HeLa and CaSki cells, respectively. Among them, compound 10 () with pyridinyl piperazine and compound 11 () with carrazole showed the strongest inhibitory activity against HeLa and CaSki cell lines, which was equivalent to that of the lead compound berberine. The results showed that the piperazine ring could improve the activity of the compound through the Mannich reaction; however, further modification and research are required.
Chalcones and Mannich bases are a group of compounds known for their cytotoxicity. Mannich bases may induce more cytotoxicity than their chalcone analogues because they generate additional alkylation centres in the cells through thiols. Tugrak et al.Citation13 designed and synthesised a series of new Mannich base derivatives with compound 12 () as the lead compound. All compounds were tested against Ca9–22, HSC-2, HSC-3, and HSC-4 as tumour cell lines and HGF, HPC (pulp cells), and HPLF human normal oral cells as non-tumour cell lines (). The cytotoxicity, selectivity index (SI), and potency selectivity expression (PSE), expressed as percentages, were determined for the compounds.
Based on the data, only compound 13 () (CC50 = 7.1 µmol/L, CC50 = 6.8 µmol/L) was more toxic to the HSC-2 and HSC-4 cell lines than melphalan (CC50 = 8.5 µmol/L, CC50 = 11.9 µmol/L) and 5-FU (CC50 = 13 µmol/L, CC50 = 13 µmol/L).
The SI value represents the selectivity of the compound against tumour and normal cells. Six of the seven compounds synthesised showed significant tumour specificity against HSC-4 cells, with SI values ranging 1.13–7.9, whereas compound 13 showed significant tumour specificity against HSC-2 and HSC-4 cells. The selectivity coefficients were 7.6 and 7.9 for HSC-2 and HSC-4 cells, respectively.
PSE can be used to evaluate the cytotoxicity and selective toxicity of compounds comprehensively. Most Mannich base derivatives exhibited higher PSE values than compound 12 (PSE = 0.0144). Compound 13, which contains an N-methylpiperazine moiety in its chemical structure, had the highest PSE value (PSE = 0.2262, a 15.7-fold increase compared with compound 12. Therefore, compound 13 with the highest PSE value is a good candidate for the development of new cytotoxic compounds.
Curcumin
Curcumin is a chemical substance isolated from the turmeric rhizome that has various biological characteristics, including anticancerCitation14, antiviralCitation15, chemopreventiveCitation16, anti-inflammatoryCitation17, antioxidantCitation18, anti-Alzheimer’s disease (AD)Citation19, and anti-HIV activitiesCitation20. Unsaturated ketones in curcumin are considered to be active groups; therefore Kadir Ozden Yerdelen et al.Citation21retained the unsaturated ketone structure, introduced various secondary amines at the methoxy position, synthesised five different compounds, and evaluated their antiproliferative activities against the HL-60, HSC-2, HSC-3, and HSC-4 cell lines (). Compared with the positive control drug, the CC50 of curcumin ranged 6.3–11 µM, the anti-proliferative activities of the synthetic compound were improved, and the CC50 values ranged 0.92–17 µM. The antiproliferative activity of compound 14 () (CC50 HSC-2 = 3.9 µM) containing N-methyl piperazine group was significantly stronger than that of the lead compound (CC50 HSC-2 = 7.8 µM), the positive control drug Melphalan (CC50 HSC-2 = 6.2 µM), and 5-FU (CC50 HSC-2 = 4.9 µM). Compound 14 also had the highest average SI value (3.96) and the highest PSE value (136.6), indicating that compound 14 has both antiproliferative activity and selective toxicity to tumour cells.
Magnolol and honokiol
Magnolol and honokiol are the main bioactive compounds of Magnolia officinalis and are multi-target compounds with various biological characteristics, such as anti-inflammatoryCitation22, antioxidantCitation23, antiarthritic propertiesCitation24, and anticancer activitiesCitation25. Tang et al.Citation26 isolated a series of honokiol derivatives from an ethanolic extract of the stem bark of M. officinalis and evaluated their effects on HCC827 (19del EGFR mutation), H1975 (L858 R/T790 M EGFR mutation), and H460 (KRAS mutation) cell lines (). Furthermore, isolated magnolol was used as starting material for the partial synthesis of further derivatives. Among them, piperitylmagnolol showed the best antiproliferative activity, with IC50 values of 15.85, 15.60, and 18.60 µM for the cell lines, respectively. Therefore, using piperitylmagnolol as the lead compound, 31 Mannich base derivatives were designed and synthesised, of which compound 15 () exhibited broad-spectrum anti-proliferative activity with IC50 values of 5.01, 5.61, and 5.98 µM for the three cell lines, respectively. This anti-proliferative activity was 8-fold higher than that of honokiol and magnolol. Moreover, the aqueous solubility of compound 15 was remarkably improved (105-fold) compared to that of honokiol and magnolol. Flow cytometry and western blot assays showed that compound 15 induced cell cycle arrest at the G0/G1 phase, causing efficient apoptosis in H1975 cells. The wound-healing migration assay revealed that it prevented the migration of HUVECsA in a dose-dependent manner through CDK2, CDK4, Cyclin E, and Cyclin D1 inhibition, as well as through upregulation of cleaved-PARP and cleaved-caspase 3 levels. Compound 15 (10, 30, and 100 mg/kg, p.o.) dose-dependently inhibited in vivo tumour growth in an H1975 xenograft model without significant body weight loss or behavioural disorders, with tumour inhibition rates of 46.3, 59.3, and 61.2, respectively, suggesting that compound 15 is a potential oral anticancer agent warranting further investigation.
As mentioned above, the introduction of a nitrogen-containing heterocycle at the C-3 position of honokiol and magnolol significantly improves the anti-proliferation activity and water solubility of the compound; the allyl group at the 3′/5′ position is essential for activity, and its oxidation reduces the antitumor activity of the compoundCitation26. Therefore, Zhao et al.Citation27 retained the allyl group on the basis of compound 15, changing the structure of the heterocycle and the extension of the carbon chain between the magnolol moiety and the 4-aminopiperidin-1-yl moiety. In this study, 51 new Mannich base derivatives were designed and synthesised, their antiproliferative activities were evaluated, and their mechanism of action was explored. Among them, compound 16 () exhibited the most potent antiproliferative activities on H460, HCC827, and H1975 cells with IC50 values of 0.86, 0.63, and 0.93 µM, which were approximately 10- and 100-fold higher potent than that of compound 15 and magnolol, respectively. Flow cytometry and western blotting revealed that compound 16 induced G0/G1 phase cell cycle arrest, apoptosis, and autophagy in cancer cells. Moreover, blocking autophagy enhanced the anticancer activity of compound 16 in vitro and in vivo, suggesting that autophagy plays a cytoprotective role in induced cancer cell death. In conclusion, compound 16 has the potential for use in cancer treatment and thus requires further study.
Esculetin
Esculetin (6,7-dihydroxycoumarin), a naturally occurring coumarin, is the main active ingredient in the traditional Chinese medicine Cortex FraxiniCitation28. Esculetin and its derivatives exhibit various bioactivities including anticancerCitation29, antioxidantCitation30, anti-diabetic propertiesCitation31. Studies have shown that the catechol group is easily hydrolysed by stage II metabolic enzymes and that the 7-OH of esuletin is the main site of UGT metabolism. Therefore, to improve both the metabolic stability and the anti-tumour activities, Wang et al.Citation32 synthesised a series of C-4 and C-8 substituted derivatives through Knoevenagel, Wittig, Perkin, and Mannich reactions and evaluated their antiproliferative activity against the A549 cell line and half-life in human liver S9 fractions in vitro ().
Metabolic stability experiments showed that among seven 4-substituted esculetin derivatives, most compounds demonstrated higher metabolic stability than the parent, compound 17 (), with an elimination t1/2 value of 44.7 min and a range of elimination t1/2 value of 47.4–91.4 min. In addition, all 8-substituted esculetin Mannich derivatives, with elimination t1/2 values ranging 102–176 min, demonstrated significantly higher metabolic stability than compound 17. Among them, compound 18 (), which contains a dimethylamino group, demonstrated optimal metabolic stability, with an elimination t1/2 value of 176.4 min, which is 3-fold that of compound 17. This suggests that amine-containing substitutes at the C-8 position of esculetin Mannich derivatives are likely to improve the metabolic stability of the compound. This may be because of the introduction of nitrogen-containing groups at the C-8 position, which increases the solubility of the compound, reduces the affinity of the substrate for metabolic enzymes, increases the molecular volume, increases the potential for hindrance of the reaction, and reduces the possibility of the substrate entering the enzyme active site.
Experiments indicate that the antiproliferative activity of synthesised compounds (24.2–1.9 µM for A549) were significantly increased compared with that of compound 17 (IC50 48.9 µM for A549). Among them, compound 19 (), which contains both trifluoromethyl and pyrrolidinyl groups, has the strongest inhibitory activity; its IC50 value is 1.9 µM, which is 20-fold that of the lead compound. Moreover, the elimination t1/2 value of compound 19 was 115.3, indicating that the stability of the compound was good and that it warrants further study.
Baicalein
Baicalein has a variety of pharmacological activities and clinical studies have shown that it has a significant inhibitory effect on liver cancer with high selectivityCitation33. Ikemoto et al.Citation34 showed that baicalein has a significant inhibitory effect on KU-1, EJ-1, and MBT-2 cell lines, with IC50 values ranging 0.9–4.4 µg/mL.
However, poor water solubility and low bioavailability of baicalein seriously affect its clinical application. Li et al.Citation35 introduced nitrogen-containing groups into the structures of lead compounds through the Mannich and condensation reactions to improve their solubility and bioavailability. Thirteen baicalein derivatives were synthesised, and the inhibitory activities of the target compounds against human hepatoma cells HepG2 were evaluated using the 3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (). The antiproliferative activities of compounds 20, 21, 22, and 23 () was significantly improved, and with IC50 values of 19.38, 11.70, 9.56, and 13.49 µg/mL, respectively. Among them, compound 22 showed the highest antiproliferative activity (4.2-fold that of the parent compound), with an IC50 value of 40.24 µg/mL. This has the potential for further study.
Flavopiridol
Flavopiridol is a semi-synthetic flavonoid derivative of rohitukine that has a strong inhibitory effect on a variety of tumour cells and is currently undergoing phase II clinical trials as an anti-tumour agent. Baicalein and quercetin are also flavonoids derivatives that can induce cell cycle arrest and apoptosis in cancer cells; however, flavopiridol and baicalein have poor inhibitory activity on CDKs. Zhang et al.Citation36 studied the chemical structure of flavopiridol and introduced a nitrogen-containing D ring in the baicalein and quercetin structures to increase their CDK inhibitory activity. Sixteen flavopiridol, baicalein, and quercetin analogues were synthesised through the Mannich reaction and evaluated for their inhibitory activity against CDK1/cyclin B (). Among them, compounds 24 and 25 () exhibited the strongest CDK1/Cyclin B inhibitory activity and had IC50 values of 0.27 and 0.28 µM, respectively, which is comparable to that of the positive control drug flavopiridol (IC50 = 0.33 µM). Therefore, compounds 24 and 25 warrants further study.
Coumarin
Studies have shown that natural coumarins exhibit antitumor activity by inhibiting the Mcl-1 pathway; however, there is currently a lack of systematic research on their structure and Mcl-1 inhibitory activityCitation37. Xia et al.Citation38 designed and synthesised a series of coumarin derivatives through acid-catalysed Pechmann condensation and Mannich reactions and evaluated their inhibitory activity against Mcl-1, determining the relationship between structure and inhibitory activity ().
First, the effects of the position and number of hydroxyl groups on the coumarin scaffold towards the inhibitory activity against Mcl-1 were explored. It was found that compound 28 () exhibited Mcl-1 inhibitory activity, with a Ki value of 1.75 µM. Furthermore, compound 27 () was a methylated product of the catechol group of compound 26 () and its inhibitory activity against Mcl-1 was significantly reduced, with a Ki value of 9.54 µM. Therefore, the catechol group of coumarin derivatives is a key inhibitor of Mcl-1 activity. Compounds 29 and 30 () are products of C-4 of Compound 26 substituted with hydrophobic trifluoromethyl and hydrophilic ethyl carboxylate groups, respectively, with Ki values of 0.21 and 9.36 µM, respectively. This shows that the introduction of a hydrophobic group at the C-4 level of compound 26 was beneficial for enhancing the inhibitory activity of Mcl-1, whereas the hydrophilic group was not. Compounds 31 and 32 () are the products of compound 26 obtained by introducing N,N-2 methyl at C-8 and C-5 through Mannich reaction, and their Ki values are 11.55 µM, 4.79 µM, and IC50 values are 53.6 and 21.79 µM, respectively. This suggests that the introduction of hydrophilic groups at the C-8 and C-5 positions of compound 26 was not conducive to increasing the inhibition of Mcl-1, possibly because the nitrogen-containing groups and C-6 and C-7 hydroxyl groups formed hydrogen bonds. Among the 37 compounds synthesised, compound 29 exhibited the strongest inhibitory activity against Mcl-1, with Ki and IC50 values ofare 0.21 and 1.21 µM, respectively. This is comparable to the positive control drug gossypol, which exhibited Ki and IC50 values of 0.19 and 1.52 µM, respectively. These findings are of significant value for the design and development of more potent Mcl-1 inhibitors for biomedical applications.
Sinomenine
S. acutum, which carries a phenanthrene nucleus and an ethylamine bridge, has a variety of pharmacological activities, such as amelioration of immunosuppression and anti-inflammation, and anticancer propertiesCitation39. Nitrogenous groups, such as piperidine and tetrahydropyrrole, can increase the solubility and activity of compounds. Li et al.Citation40 used sinomenine as a lead compound and introduced piperidine methylene or tetrahydropyrrole methylene at the C-1 position through the Mannich reaction to obtain two series of 4-dissubstituted sinomenine derivatives. The inhibitory activities of the compounds against five cancer cell lines (MCF-7, HeLa, and SW480) were evaluated using the MTT assay ().
The results indicated that compound 33 () with a chlorine atom in the R group had significant anticancer activity compared to compound 34 () with a methyl group in the R group, indicating that the chlorine substitution of benzene can improve the antitumor activity of compounds. Moreover, compound 33 showed the highest inhibitory activity against MCF-7, HeLa, and SW480 cells, with IC50 values of 5.73, 8.20, and 6.08 µM, respectively. This indicates that the selective toxicity of compound 33 on cancer cells was higher than that on normal cells, and it warrants further study.
Application in modification of natural products for anti-AD
AD is a chronic age-related neurodegenerative disease that is mainly characterised by memory loss, behavioural abnormalities, and cognitive impairmentCitation41. Low levels of acetylcholine (ACh) can lead to memory and cognitive deficits in patients with AD, and inhibition of acetylcholinesterase (AChE) activity can reduce the hydrolysis of ACh and relieve the symptoms of ADCitation42. β-Amyloid (Aβ) deposition can lead to severe neurodegeneration or neuronal death, and it is also an important factor in the development of ADCitation43. Higher levels of biometal ions, such as Cu2+ (0.4 mM, 5.7-fold higher), Zn2+ (1.0 mM, 2.8-fold higher), and Fe2+/3+ (0.9 mM, 2.9-fold higher), were found in Aβ deposits from patients with AD than in healthy, normal cellsCitation44. These biomaterials accelerate Aβ aggregation and exacerbate the overproduction of reactive oxygen species (ROS), leading to oxidative damage to neuronal cellsCitation45. The pathogenesis of AD is closely related to monoamine oxidase B (MAO-B), and the overexpression of MAO-B directly enhances the processing of γ-secretase substrates to increase Aβ productionCitation46.
Chalcone
AD is a multi-mechanism neurological disease, and its specific pathogenesis is still unclear; therefore, there is an urgent need to discover new effective drugs that can alleviate AD symptoms. Based on a multitarget-directed ligand design strategy, Zhang et al.Citation47 designed and synthesised a series of chalcone Mannich base derivatives and evaluated their anti-AD activity. In vitro assays showed that most compounds exerted potent selective inhibition towards AChE, significant inhibition towards MAO-B, good antioxidant activities, and moderate inhibitory activities of self- and Cu2+-induced Aβ1 − 42 aggregation (). Among them, compound 35 () exhibited moderate inhibition of AChE (IC50 = 0.44 µM) and MAO-B (IC50 = 1.21 µM), good inhibitory effect on self-induced Aβ1 − 42 aggregation (55.0%, at 25 µM), biometal chelating properties, and moderate antioxidant activity, with a value 1.93-fold of Trolox. Therefore, compound 35 requires further investigation.
Figure 3. Compounds 35–47 for anti-Alzheimer’s disease.
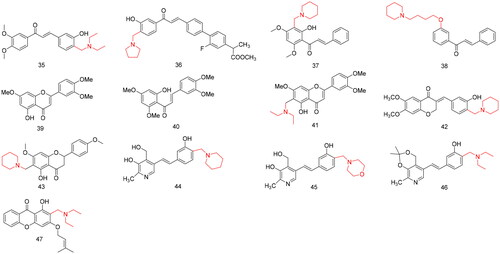
Table 2. Application in modification of natural products for anti-Alzheimer’s disease.
Flurbiprofen
Flurbiprofen is a non-steroidal anti-inflammatory drug (NSAID) with significant anti-neuroinflammatory activity; the α, β unsaturated ketone structure of chalcone is a flexible structure with Michael acceptor characteristics, and it is a special scaffold for molecular hybridisation. Studies have shown that multi-target-directed ligands (MTDLs), which can simultaneously modulate multiple biological targets, appear to be more effective in ADCitation48. Tian et al.Citation49 synthesised a series of low-molecular-weight (MW) flurbiprofen-chalcone hybrid Mannich base derivatives through Friedel-Crafts acylation, Michael addition, esterification, and Mannich reactions and evaluated their anti-AD activity. Biological screening indicated that most of the derivatives exhibited potent multitarget effects in AD (). Among them, compound 36 (), which bears a pyrrolidine group, exhibited the highest activities against itself and Cu2+-induced Aβ1–42 aggregation (70.65 and 54.89% at 25.0 µM, respectively), highly selective inhibition towards AChE and MAO-B (IC50 = 7.15 µM and 0.43 µM respectively), and good antioxidant abilities and metalchelating properties. Therefore, compound 36 requires further study.
Flavokawain B
Flavokawain B, a 20-hydroxy-4, 6-dimethoxy chalcone isolated from the roots of kava, showed remarkable anti-proliferative activity against different cancer cell lines. However, the content of flavokawain B in plants is low; therefore, Liu et al.Citation50 first synthesised the lead compound flavokawain B through Houben-Hoesch acetylization, regioselective O-methylation, and Claisen–Schmidt condensation with benzaldehyde, and seven flavokawain B Mannich base derivatives were synthesised through the Mannich reaction. Their inhibitory activity on AChE was evaluated (). Pharmacological data showed that four compounds displayed potent activities against AChE with IC50 values <20 µM. Among them, the inhibitory activity of AChE of compound 37 () was the strongest (IC50 = 4.15 µM), at twice that of the positive control drug rivastigmine (IC50 = 10.54 µM). The log p values of compound 37 was 1.75, indicating that it was sufficiently lipophilic to cross the blood-brain barrier in vivo.
Liu et al.Citation51 designed and synthesised 20 new nitrogen-containing chalcone derivatives using flavokawain B as the lead compound, and evaluated their inhibitory effects on AChE and BuChE (). In vitro activity experiments showed that all the synthesised compounds had stronger inhibitory activity on AChE than the lead compound flavokawain B, among which compound 38 () had the strongest inhibitory activity on AChE (IC50 = 0.85 µmol/L). Its selectivity to AChE was 35.79-fold higher than that of BuChE (IC50 = 30.42 µmol/L). An enzyme kinetic study showed that the inhibition mechanism of compound 38 against AChE was mixed-type inhibition. Therefore, Flavokawain B/compound 38 requires further study.
Hesperidin
Hesperidin is a bioflavonoid and plant pigment with antioxidant and anti-inflammatory effects that is found primarily in citrus fruits. Duan et al.Citation52 used hesperidin as a lead compound and obtained compounds 39 and 40 () through hydrolysis, dehydrogenation, regioselective O-methylation, and base-catalysed ring-opening reactions (). Eleven new hesperidin Mannich base derivatives were synthesised by introducing secondary amines at the C-3′ position of compound 39 and the C-6 position of compound 40 through the Mannich reaction. The AChE inhibitory activities of the synthesised compounds were also evaluated. Among them, compound 41 () with the introduction of a diethylamine group had the strongest inhibitory activity on AChE with an IC50 of 0.54 µmol·L−1, which was 2.5-fold that the positive control drug neoeserine methyl sulphate (IC50 = 1.38 µM). Therefore, compound 41 has the potential for further study.
Homoisoflavonoids
Homoisoflavonoids, 3-benzylidene-4-chromanones, are flavonoid natural products with MAO-B inhibitory activityCitation53 and high affinity towards Aβ aggregatesCitation54. In addition, phenolic Mannich base derivatives have good antioxidantCitation55, anti-inflammatoryCitation56, and AChE inhibitory activitiesCitation50. Based on the MTDLs strategy, Li et al.Citation57 designed and synthesised a series of homoisoflavonoid Mannich base derivatives with homoisoflavonoids as the lead compounds, combined with the structural features of phenolic Mannich bases and donepezil, and evaluated their inhibitory activities on AChE and BuChE (). In vitro experiments showed that most of the compounds exhibited good selective inhibitory activity against AChE and MAO-B, among which compound 42 () exhibited comprehensive inhibitory activity with IC50 values of 2.49, 19.8, and 1.74 µM for AChE, BuChE, and MAO-B, respectively. The selectivity for the AChE receptor was 7662-fold higher than that for the BuChE receptor; therefore, it is a good selective AChE receptor inhibitor. Compound 42 inhibited self- and Cu2+-induced Aβ1-42 aggregation by 39.3 and 47.0%, respectively. Therefore, compound 42 has the potential for further study.
As a universal ingredient in the peels of some fruits and medicinal plants, naringin is typically used as a raw material for the synthesis of biologically active derivatives because of its easy availability and low cost. Liu et al.Citation58 synthesised eight new acacetin-7-O-methyl ether Mannich base derivatives through dehydrogenation, regioselective methylation, glycosidic bond hydrolysis, and Mannich reactions using naringin as the lead compound. In addition, their AChE inhibitory activities were evaluated (). Bioactivity evaluation revealed that most compounds exhibited moderate or potent AChE inhibitory activity. Among them, compound 43 () showed a potent activity and high selectivity with IC50 values of 0.82 and 46.30 µM for AChE and BuChE, respectively. The inhibitory effects of rivastigmine (IC50 (AChE) = 10.54 µM, IC50 (BuChE) = 0.26 µM), the inhibitory effect on AChE was 12.85-fold higher. A kinetic study suggested that compound 43 bound to AChE with a mixed-type inhibitory profile. Therefore, it is a promising compound for the treatment of AD as a novel AChE inhibitor derived from natural products, and thus requires further study.
Resveratrol
Resveratrol, a natural product with a stilbene structure, is an anti-atopic dermatitis agent with antioxidant, anti-inflammatory, and neuroprotective propertiesCitation59. Pyridoxine (vitamin B6) is a potent antioxidantCitation60 and inhibits free radical productionCitation61 suggesting that it may help treat AD. Yang et al.Citation62 integrated the pyridoxine moiety with the stilbene structure into one molecular entity and introduced the Mannich base moiety into their hybrids with to make them work synergistically and improve the anti-AD potential and BBB permeability of the compound. A series of novel pyridoxine-resveratrol hybrid Mannich base derivatives were synthesised and evaluated for their biological activity. Most of these compounds selectively inhibited AChE and MAO-B (). Among them, compounds 44 and 46 () exhibited the highest potency for AChE inhibition with IC50 values of 2.11 and 1.56 µM, respectively. and compound 45 () exhibited the highest MAO-B inhibition with an IC50 value of 2.68 µM. In addition, all target compounds displayed good antioxidant and metal-chelating properties. These preliminary findings provide a new starting point for the further development of multifunctional agents for AD.
Xanthone
Xanthone (dibenzo-γ-pyrone) is an active ingredient in natural products with a wide range of biological activities, such as anti-bacterial, anti-inflammatory, antitumourCitation63, and α-glucosidase inhibitory propertiesCitation64. Xanthone derivatives have been reported to inhibit AChE and block acetylcholinesterase-induced Aβ aggregationCitation65. In addition, Mannich bases can increase the relative biological potency and solubility of compoundsCitation66. Qin et al.Citation67 designed and synthesised a series of 1, 3-dihydroxyxanthone Mannich base derivatives with alkoxy and alkenoxy substituents at position 3 of xanthone and dialkylamine methyl substituted at position 2 using xanthone as the lead compound, and evaluated their inhibitory effects on AChE and BuChE (). Most of the target compounds exhibited moderate to good inhibitory activities, with IC50 values at the micromolar level against both AChE and BuChE. Among them, compound 47 () exhibited the strongest inhibitory activity against AChE and BuChE, with IC50 values of 2.61 ± 0.13 µM and 0.51 µM, respectively. The inhibitory activity of compound 47 against BuChE was 59-fold that of the positive control drug, galanthamine·HB (IC50 = 29.99 µM). Therefore, compound 47 requires further study.
Application in modification of natural products for antimicrobial activity
Magnolol
Magnolol, a natural product of M. officinalis, exhibits a wide range of biological activities. Yan et al.Citation68 found that magnolol (EC50 = 3.07 µg/mL, p ≤ .001) exhibited better antifungal activity than tebuconazole against Rhizoctonia solani, with an EC50 value of 3.48 µg/mL. Therefore, Li et al.Citation69 used magnolol as the lead compound and performed structural modifications at the C-3 and C-3′ positions and the hydroxyl and C-5 and C-5′ allyl positions to synthesise a series of magnolol derivatives. The in vitro biological activities of the compounds against phytopathogenic fungi (R. solani, Fusarium graminearum, Botrytis cinerea, and Sclerotinia sclerotiorum) were systematically evaluated ().
Table 3. Application in modification of natural products for antimicrobial activity.
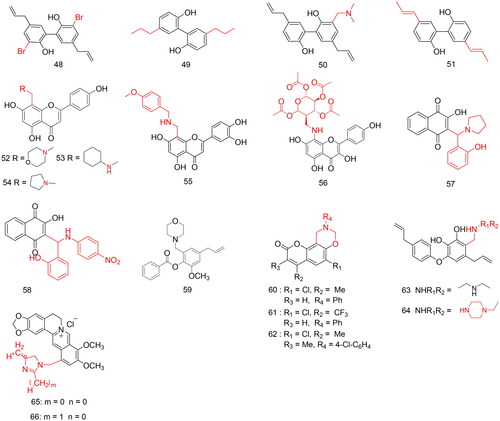
The results showed that 11 compounds were active against the four phytopathogenic fungi with EC50 values ranging 1.40 − 20.00 µg/mL. Compound 48 (), with bromine atoms at the C-3 and C-3′ positions, exhibited excellent antifungal properties against B. cinerea with an EC50 value of 2.86 µg/mL, making it approximately 2.8-fold more potent than magnolol (EC50 = 8.13 µg/mL). Moreover, compound 49 (), with saturated propyl groups at the C-5 and C-5′ positions, exhibited the highest antifungal activity against F. graminearum and R. solani with EC50 values of 4.39 and 1.40 µg/mL, respectively. Compound 51 (), with a propenyl group at the C-5 and C-5′ positions, showed good antifungal activity against S. sclerotiorum, with an EC50 value of 4.64 µg/mL. Physiological and biochemical studies have shown that compound 50 () inhibits the growth of B. cinerea by changing the shape of the mycelium, increasing cell membrane permeability, and destroying mitochondrial function. Furthermore, structure-activity relationship (SAR) analysis showed that the hydroxyl group was key to maintaining the activity and the reduction and transfer of the C-5 and C-5′ double bonds. In addition, the bromine substitution significantly increased the antibacterial activity, which was more effective than alkylamines. These modification results are helpful for researchers to perform structural modifications, improve the activity of the compound, and reduce its toxicity.
Figure 4. Compounds 48–66 for antimicrobial activity.
Apigenin
Liu et al.Citation70 synthesised a series of Mannich base derivatives with apigenin as the lead compound, introduced different primary or secondary amines at the C-8 position through the Mannich reaction, and evaluated their antibacterial activities against Staphylococcus aureus, Bacillus subtilis, Escherichia coli, and Pseudomonas aeruginosa (). All compounds exhibited stronger antibacterial activity than the lead compound ampicillin, with minimum inhibitory concentration (MIC) values ranging 1.95–62.5 µg/mL. Among them, compounds 52 and 53 () exhibited significant inhibitory activity against S. aureus and B. subtilis, with MIC values of 1.95 and 3.91 µg/mL, respectively, which were comparable to that of the positive control tetracycline (MIC = 1.95 and 3.91 µg/mL), but lower than that of ampicillin (MIC = 31.25 µg/mL). Moreover, SAR analysis showed that the six-membered cyclic compound 52, containing two heteroatoms, was better at improving the antibacterial activity of apigenin.
Flavonoids
Orientins, isoorientins, and luteolins are flavonoids. Lv et al.Citation71 evaluated antibacterial activity against Gibberella sanbinetti. Orientin (inhibition rate of 77.23%) and isoorientin (inhibition rate of 82.2%) at 20 mg/L showed excellent antifungal activity against G. sanbinetti (). However, luteolin (inhibition rate of 0.00%) was barely active. Their structural differences are reflected in the 8th or 6th position of the A ring. It has been reported that the 8th position substitution of flavonoid derivatives is better than the 6th position substitution; therefore, drawing on the structure of orientin and isoorientin, flavonoids can be substituted at the 8th position. A series of 19 compounds was synthesised and evaluated for their antifungal activity against G. sanbinetti and Gaeumannomyces graminis. However, in vitro, antifungal activity experiments showed that the antifungal activity of these compounds did not reach the expected levels. The inhibition rate range was 0.00–47.5%. They were then evaluated for their antibacterial activity against two gram-positive bacteria (S. aureus and Listeria monocytogenes) and two gram-negative bacteria (E. coli and Salmonella gallinarum). Compound 56 () exhibited broad-spectrum antibacterial activity against all four bacteria (MIC = 1, 0.5, 2, and 0.05 mg/L). Compound 55 () (MIC = 2, 0.125 mg/L) showed significant inhibitory activity against S. aureus and S. gallinarum compared to novobiocin (MIC = 2, 0.25 mg/L).
To determine the relationship between the introduced 4-methoxybenzylamine, tetrahydro-2H-pyran structure, and antibacterial activity, the inhibitory activities of compounds 55 and 56 against topoisomerases II and IV, isolated from E. coli, were tested. Compounds 55 and 56 exhibited inhibitory effects on both topoisomerases, and the IC50 values of compound 54 were 0.5 and 16 mg/L, respectively, and the IC50 values of compound 56 were 0.25 and 8 mg/L, respectively. Compound 56 inhibited both enzymes more strongly than compound 55. This suggests that the inhibition of topoisomerases II and IV by 4-methoxybenzylamine and tetrahydro-2H-pyran inhibited bacterial cell growth.
Lawsone
Lawsone (2-hydroxy-1,4-naphthoquinone) is a naphthoquinone with an antifungal activity. Allochio et al.Citation72 used lawsone as a lead compound, designed and synthesised a series of lawsone Mannich base derivatives using the Mannich reaction, and evaluated their inhibitory activities against Candida spp (). Among these, compounds 57 and 58 () showed the highest inhibitory activity against Candida albicans American Tissue Culture Collection (ATCC) 10231, with MIC values ranging 20–330 µg mL−1.
Compared with the positive control drug ketoconazole (MIC = 62.5 µg·L−1), compound 57 (MIC = 20 µg·L−1) had a higher inhibitory effect on the growth of C. albicans. Moreover, C. glabrata ATCC 2001 showed sensitivity to compounds 57 and 58 with MIC values of 170 and 330 µg·L−1, respectively. The effect of the compounds on membrane ergosterol was determined using an exogenous ergosterol assay, and the results showed that in the presence of exogenous ergosterol (MIC = 80 µg mL−1); the MIC value of compound 57 against C. albicans ATCC 10231 was enhanced 4-fold, indicating that the antifungal activity of compound 57 against C. albicans ATCC 10231 can be attributed to its effect on membrane ergosterol.
3.5. Eugenol
Eugenol is a natural allyl phenol that has a simple structure, is easy to modify, and has a wide range of biological activities. Mannich base derivatives contain nitrogen-containing groups, which improve the water solubility of compounds and also protonate nitrogen atoms under physiological conditions to strengthen the interaction between ligands and receptorsCitation2. The structures of linezolid, eprazolid, and itraconazole antibacterial drugs suggest that the bioisosteric ring of piperazine or morpholine plays a vital role in their antibacterial activity. Pedro Henrique Oliveira Abrão et al.Citation73 introduced a morpholino group into the lead compound eugenol through the Mannich reaction, synthesised a series of eugenol Mannich base derivatives, and evaluated their inhibitory activities against C. albicans ATCC10231 and Candida krusei ATCC 6258 (). In addition, they explored the effect of the phenolic hydroxyl groups of these compounds, being freed or masked as esters of different polarities and volumes, on their anticandidal activity. The results showed that compound 59 () exhibited the strongest antifungal activity, with IC50 values ranging 0.63–10.2 µg/mL. C. albicans and C. krusei were the most sensitive to compound 59 with an IC50 value of 0.63 µg/mL, which was much higher than that of the positive control drug fluconazole, of which the IC50 values were 3.27 and 104.5 µg/mL, respectively.
Osthole
Osthole, a natural O-methylated coumarin, exhibits antifungal activity against R. solani and a broad spectrum of other phytopathogenic fungiCitation74. Studies have shown that compounds containing coumarin scaffolds exhibit highly effective inhibitory effects against phytopathogenic fungi. Therefore, Zhang et al.Citation75 introduced an oxazine ring into the coumarin structure through a microwave-assisted three-pot Mannich reaction because the oxazine ring contains nitrogen atoms that can significantly increase the solubility and balance the lipophilicity of the compound. A series of coumarin–Mannich base derivatives were synthesised. The antifungal activities of the synthesised compounds were evaluated against B. cinerea, Colletotrichum capsici, and R. solani (). The preliminary bioassays showed that the inhibitory effects of compounds 60, 61, 62, and osthole () were 88.2, 98.7, 78.6, and 86.8%, respectively. Among these, compound 61 was the most potent, with broad-spectrum antifungal activity. The EC50 values of compound 61 against B. cinerea, C. capsici, and R. solani were 2.1, 19, and 5.8 nM, respectively, which were superior to those of osthole. Therefore, compound 61 warrants further investigation as a potential antifungal drug.
Obovatol
Obovatol, a novel lignan bearing a diphenyl ether skeleton, exhibits a wide range of biological activities, including antifungalCitation76, anti-tumorCitation77, anti-inflammatoryCitation78, anxiolyticCitation79, anti-plateletCitation80, and anti-AD activitiesCitation81. The biological activity of obovatol can be significantly improved by introducing active groups at the ortho position of the phenolic hydroxyl groupCitation82. Yang et al.Citation83 designed and synthesised a series of C-4-aminomethylated derivatives of obovatol as lead compounds through the Mannich reaction. Inhibition of spore germination and mycelial growth by these derivatives has been investigated against several phytopathogenic fungi.
The fungicidal activities of 21 synthesised obovatol derivatives against four phytopathogenic fungal strains were examined using spore germination assays (). The results of the study show that compounds 63 and 64 () exhibited broad-spectrum and strong inhibitory effects on A. solani, F. solani, and B. cinerea fungal spores. Compound 64 exhibited greater inhibitory activity against A. solani, F. solani, and B. cinerea than the other two positive controls, with IC50 values of 63.19, 59.13, and 16.9 µg/mL, respectively. In addition, compounds 63 and 64 have the strongest inhibitory effect on B. cinerea, with IC50 values of 28.68 and 16.9 µg/mL, respectively, which are higher than that of the positive controls hymexazol (IC50 = 66.80 µg/mL) and difenoconazole (IC50 = 96.76 µg/mL), and that of the lead compound obovatol (IC50 = 143.13 µg/mL).
The antifungal activities of the 21 compounds against six strains of phytopathogenic fungi were preliminarily investigated in vitro. The average inhibition rates (AIRs) was determined to be 100 g/mL. The results showed that seven compounds had potent inhibitory effects on one or more fungi, with AIRs > 60%. Among them, compound 64 had the strongest inhibitory effect on B. cinerea, with an AIR value of 92.18%, which was higher than that of the positive controls hymexazole (AIR = 88.44%), difenoconazole (AIR = 16.33%), and the lead compound obovatol (AIR = 51.70%). Additionally, the SARs of the compounds suggested that the introduction of 1-methyl-piperazinyl and diethylamine groups significantly improved their antibacterial activity. Collectively, compounds 63 and 64 can be used to synthesise lead candidates as environmentally friendly fungicides to be tested in future studies.
Berberine
Berberine, a derivative of 5,6-dihydrodibenzo[a,g]quinolizinium, exerts antimicrobial effects not only by DNA breakage but also through the accumulation of NorA substrate in bacterial cellsCitation84. The imidazole nucleus contains nitrogen atoms, which, to a certain extent, improve the antibacterial activity of the compound and help improve water solubility. Considering the potential of berberine in the field of anti-infection and the extension of research on the development of azole compounds, Wen et al.Citation85 introduced imidazole and its alkyl-substituted derivatives at the C-12 position of the berberine ring and synthesised a series of new antibacterial drugs. The antimicrobial activities of the synthesised compounds in vitro against four gram-positive bacteria, four gram-negative bacteria, and five fungi were evaluated (). The results showed that all synthesised compounds exhibited effective antibacterial activity against all the tested bacteria and fungi, with MIC values of 1–256 µg/mL, which were better than those of their precursor berberine. Among them, compound 65 () showed excellent antimicrobial activity with a broad spectrum in comparison with other compounds. Compound 65 exhibited the most potent antibacterial activity against E. typhosa, B. subtilis, and MRSA, with MIC values of 1–32 µg/mL, which were higher than those of chloromycin (MIC = 32–64 µg/mL) and berberine (MIC = 128–512 µg/mL). Compound 66 () exhibited better activity against B. yeast (MIC = 16 g/mL) and A. flavus (MIC = 32 g/mL) than fluconazole, which was used a sthe reference compound.
The lipid-water partitioning of drugs plays an important role in exerting bioactivities by influencing transportation, distribution, metabolism, and secretion in organisms. ClogP values have been widely used to predict the transportation and bioactivities of drugs, and it has been revealed that compounds with lower ClogP values exhibit more efficient antimicrobial activities. The berberine-based imidazoles showed lower ClogP values than their precursor berberine, indicating that the newly synthesised compounds possessed more reasonable water solubility, which was favourable for permeation through biological membranes and be delivered to the binding sites.
The cytotoxic properties of compound 65 against HepG2 cells were examined using the colorimetric cell proliferation MTT assay. cytotoxicity results showed that the cell viability of compound 65 against HepG2 cells was more than 87% at a concentration of 100 µg/mL.
The relationship between antimicrobial mechanisms and cell toxicity has not been fully elucidated but is thought to involve ROS. Therefore, DCFH-DA was used as a fluorescent probe to investigate the ROS levels. The results showed that compound 65 generated less ROS than berberine did. Thus, compound 65 exhibited a better safety profile than berberine through the downregulation of ROS generation.
Interactive studies of compound 65 with calf thymus DNA at the molecular level show that compound 65 effectively intercalated into calf thymus DNA to form a 3a-DNA complex which may further block DNA replication and exert powerful antimicrobial activities. Therefore, compound 65 warrants further investigation as a potential antibacterial agent.
Application in modification of natural products for antidiabetic activity
Flavonoids
Flavonoid compounds exhibit potential glucosidase inhibitory activity, providing protection against diabetes in vitro and in vivoCitation86. However, natural products suffer from low water solubility and a lack of selectivity, which severely limits their clinical use. The introduction of nitrogenous bases not only increases the solubility of the parent flavonoid compound but also increases the binding affinity with the target enzyme, leading to higher therapeutic efficacy.
Oroxylin A, a flavonoid from the Indian herb Oroxylum indicum, is a potent inhibitor. Babu et al.Citation87 designed and synthesised a series of 8-aminomethylated derivatives based on oroxylin A with a methoxy group at the 6th position through the Mannich reaction and evaluated their effects on rat intestinal and yeast α-glucosidase inhibitory activities. The synthesised compounds significantly enhanced the a-glucosidase-inhibitory activity of the lead compounds (). Among them, compound 67 () with N-methyl piperazine at the C8 position showed the strongest inhibitory activity, with IC50 values of 35.43 and 98.84 µg/mL for rat intestinal a-glucosidase and yeast a-glucosidase, respectively. Regarding the structural features of the active Mannich bases, the results indicate that the 5,7-dihydroxy-6-methoxy groups on the A-ring of the flavonoid are the basis of glucosidase inhibition activity. Alicyclic amine substituents significantly improved the intestinal a-glucosidase inhibitory potential of the parent compound. Among them, the N-methylpiperazinyl substitution had the strongest inhibitory effect. This experiment showed that the introduction of alicyclic amines into natural compounds can significantly improve their activity, providing a direction for the results of natural products.
Figure 5. Compounds 67–70 for antidiabetic activity.
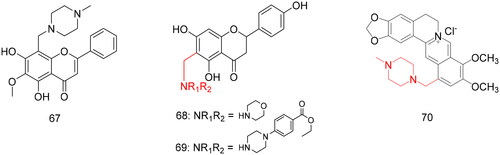
Table 4. Application in modification of natural products for antidiabetic activity.
Zhen et al.Citation88 designed and synthesised a series of novel flavonoid alkaloids using different flavonoids and nitrogen-containing groups as raw materials. These new compounds were screened for their inhibitory activity against yeast (). The concentration of compounds measured against a-glucosidase ranged 4.13–806 µM, among which compounds 68 and 69 () exhibited the strongest inhibitory activity with IC50 values of 23.4 and 4.1 µM, respectively. Enzyme kinetic assays showed that compounds 68 and 69 were close to the non-competitive model inhibitor against a-glucosidase. Their Ki value were 37.8 and 13.2 µM, respectively.
The SAR indicated that the carbonyl group in the parent structure was essential for activity. For the A ring, the introduction of tertiary amines into its structure results in better activity than secondary amines, and the hydrogen bond acceptor increases the affinity; however, the increase in hydrophobic groups inhibited the activity. In addition, the hydroxyl group at the 4′ position increased inhibitory activity.
Berberine
Berberine (BBR) is a quaternary ammonium salt derived from isoquinoline alkaloids. Research has shown that BBR has strong effects on hypoglycaemic and insulin-sensitizing effectsCitation89.
Using berberine as the lead compound, Li et al.Citation90 synthesised a series of berberine Mannich base derivatives by introducing different nitrogenous bases at the C-12 position through the Mannich reaction and evaluated their anti-diabetic activities against type 2 diabetes mellitus in 3T3-L1 adipocytes and L6 myotubes16 using rosiglitazone and insulin as positive controls, respectively (). The results showed that the insulin resistance reversal activities of the synthesised compounds were better than those of the positive control, rosiglitazone, and were concentration-dependent. Among them, compound 70 () exhibited the best activity, and its sensitisation was 1.26-fold that of rosiglitazone. Therefore, introducing various aminomethyl groups at the 12-position of berberrubine can markedly improve insulin resistance reversal activity and stimulate glucose transport in type 2 diabetes mellitus. Moreover, the six-membered nitrogen-containing heterocyclic group plays an important role in the reversal of insulin resistance. This structural modification provided useful information for the subsequent modification of berberine derivatives with enhanced activity.
Application in modification of natural products for anti-inflammatory activity
Flavonoids
Hesperidin (hesperetin-7-O-rutinoside) and hesperetin (hesperetin-7-O-glycoside) are the main active components of orange plants of the Rutaceae family, with various pharmacological activities, such as anti-inflammatoryCitation91, antiox-idantCitation92, antibacterialCitation93, anticancerCitation94, anti-hepatic fibrosisCitation95, and immunomodula-tion propertiesCitation96. The bioavailability of hesperetin is lower than that of hesp-eridin, likely because the rutinoside moiety attached to flavonoids enhances their water solubility.
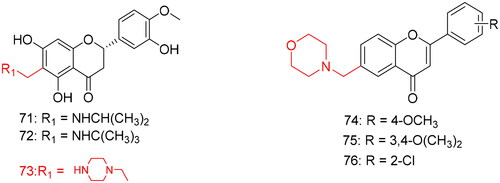
To improve the water solubility and bioavailability of hesperetin, Wang et al.Citation97 synthesised 16 novel hesperetin derivatives containing Mannich base moieties by introducing different primary and secondary amines at the C6 position of the hesperetin A ring through the Mannich reaction. Its anti-inflammatory activity was assessed by inhibiting lipopolysaccharide (LPS)-induced tumour necrosis factor-α (TNF-α) and interleukin-6 (IL-6) in mouse RAW264.7 macrophages (). The MTT assay showed that the stimulator LPS (0.5–1 µg/mL) and all compounds (1–10 µm) exhibited no toxic effects on RAW 264.7 cells. In addition, compounds with aliphatic and alicyclic amines had better inhibitory effects on LPS-induced TNF-α and IL-6 than hesperetin; however, aromatic amine-substituted compounds had weaker effects on TNF-α and IL-6 than hesperetin, which may be due to their lower solubility. Compounds 71, 72, and 73 () exhibited the strongest inhibitory effects. To explore the relationship between the water solubility of the compounds and their anti-inflammatory activities, the logP of the compounds was measured using the shake-flask method. The results showed that the logP value was negatively correlated with the anti-inflammatory activity. The smaller the logP value, the better the water solubility and anti-inflammatory activity. For example, compounds 71, 72, and 73 had the smallest logP values of −0.26, −0.76, and −0.75, respectively, and exhibited the strongest inhibitory effects on LPS-induced TNF-α and IL-6. In conclusion, this study demonstrated the relationship between water solubility and the anti-inflammatory activity of compounds, providing a direction for future structural modification of natural products.
Figure 6. Compounds 71–76 for anti-inflammatory activity.
Nitrogen-containing groups play an important role in the structure of drugs by promoting binding to receptors and enhancing water solubility. Therefore, to improve the anti-inflammatory activity of flavonoids, Hasan et al.Citation98 introduced a secondary amine structure at the C6 position of flavonoids, designed, and synthesised a series of flavonoid Mannich base derivatives, and evaluated their anti-inflammatory, antimicrobial, and analgesic properties action. Gastrointestinal tract (GIT) protection from ulcerogenesis and lipid peroxidation (LPO) was considered.
The anti-inflammatory activities of the 18 synthesised compounds were evaluated using by the carrageenan-induced rat paw oedema method, and the results showed that six compounds showed anti-inflammatory activities similar to that of the positive control drug ibuprofen (89.50%) (). Among them, compounds 75 and 76 () showed the strongest inhibitory effects, with inhibition rates of 80.86 and 82.51%, respectively.
The acetic acid-induced writing method was used to evaluate the analgesic activity of six compounds with obvious anti-inflammatory activity, and the results showed that compounds 75 and 76 had 53.61 and 54.81% protective effects on acetic acid-induced writhing, which were comparable to that of the positive control drug ibuprofen (65.06%).
Acute ulcerogenic activity testing of six compounds showed lower ulcerogenic activity for these compounds, ranging 0.1–0.4, compared with a high severity index of 0.9 for the standard drug ibuprofen. Compounds 74 (), 75, and 76 with a morpholino group at the C6 position exhibited the lowest ulcer-causing activities, with severity indices of 0.2 and 0.1, respectively, indicating that the introduction of a morpholine group into the lead compound can significantly improve the anti-ulcer activity of the compounds.
To explore the relationship between the ulcerogenic activity and lipid peroxidation activity, the lipid peroxidation activity of compounds was evaluated, and the results showed that the lipid peroxidation activity of the synthesised compounds ranged 0.331–0.526 nM MDA/mg of protein, whereas that of ibuprofen was 0.608. This shows that compounds with low ulcerogenic activity also have low lipid peroxidation activity and that the synthesised compounds can inhibit the induction of gastric mucosal injury, further indicating that their protective effect may be related to the inhibition of gastric mucosal lipid peroxidation.
Application in modification of natural products for other activities
As one of the leading causes of morbidity and mortality worldwide, ischaemic cerebrovascular disease not only causes physical and emotional pain in patients but also imposes a great financial burden on their families and societyCitation99. Increasing evidence suggests that thrombin plays an important role in ischaemic cerebrovascular diseasesCitation100.
Oxidative stress plays a key role in ischaemic cerebrovascular diseaseCitation101. Research shows that Scutellarin can improve microcirculation, dilate blood vessels, inhibit platelet aggregation, and increase cerebral blood flowCitation102. Furthermore, scutellarin is clinically used in China to treat acute cerebral infarction and paralysis caused by cerebrovascular diseases such as hypertension, cerebral haemorrhage, and cerebral thrombosisCitation103. Scutellarein is a hydrolysate of scutellarin that has a strong protective effect against cerebral ischaemia. Docking studies of thrombin (2R2M) with scutellarein indicated that the B- and C-rings of the ligand interacted well with the S1 and S2 pockets, respectively. However, ring A could only partially interact with the S3 pocket of thrombin. Zhong et al.Citation104 synthesised the compound LR3d through the Mannich reaction and evaluated its biological activity. The results showed that morpholinyl aminomethylene substituent derivative LR3d exhibited stronger anticoagulant ability and better antioxidant activity than scutellarein.
Considering that the fixed structure may be beneficial for the compound to interact with the S3 pocket of thrombin (2R2M), Zhong et al.Citation104 used a scaffold-hopping strategy to design a series of novel compounds fused to 1,3-oxazine rings at the 7th and 8th positions, and the thrombin inhibitory activity of these compounds was studied using thrombin time (TT), activated partial thromboplastin time (APTT), prothrombin time (PT), and fibrinogen (FIB). The antioxidant abilities of these analogues were evaluated using the MTT assay with the 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay.
Biological activity evaluation showed that compound 77 () prolonged TT and APTT, increased PT, and decreased FIB content significantly compared to scutellarein, and it exhibited strong antioxidant activity with an IC50 value of 24.57 µM. Therefore, compound 94 has the potential to treat ischaemic cerebrovascular disease.
Figure 7. Compounds 77–79 for other activities.
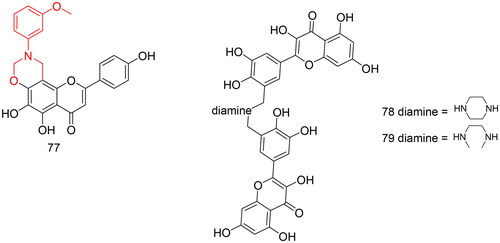
A2E, a pyridinium bisretinoid, is a lipofuscin pigment found in retinal pigment cells. In addition, A2E is thought to play an important role in retinal diseases, such as macular degeneration, and antioxidants could serve as therapeutic agents for these diseases. The Mannich reaction can combine amine molecules into organic compounds. Amine molecules, as water-soluble groups, can improve the physicochemical properties (such as solubility) of compounds, thereby increasing their bioavailability. Chalcone, quercetin, and sesamol have antioxidant activities and can undergo regioselective Mannich reactions under certain conditions. Therefore, Joshi et al.Citation105 used chalcone, quercetin, and sesamol as lead compounds, introduced different primary and secondary amines, synthesised a series of Mannich base compounds, and evaluated their inhibitory activity against the oxidant A2E. Single concentration determinations at 100 µM showed that compounds 78 and 79 () were superior to quercetin in inhibiting A2E photooxidation, indicating that the diamine (piperazine or N1,N2-dimethylethane-1,2-diamine, respectively) groups contained in compounds 78 and 79 can enhance the antioxidant capacity; the closer the diamine group is to the phenolic hydroxyl group, the more likely it is to increase the antioxidant effect.
Conclusion remarks and future perspectives
As prodrugs or as molecules eliciting a response from biological targets, numerous novel Mannich bases have been synthesised and evaluated as potential treatments for a multitude of diseases and medical conditions. Significant progress has been made in the field of anticancer, antimicrobial, and anti-AD agents, and numerous biologically active natural products have been produced through the Mannich reaction to increase their biological activity. Therefore, the Mannich reaction has earned a place as a powerful tool in medicinal chemistry, both for the synthesis of novel chemical entities endowed with various interesting biological properties and for the modification of the physicochemical properties of a candidate, which ultimately influences the bioavailability, performance, and pharmacological activity of the drug candidate.
Acknowledgement
We would like to thank Editage (www.editage.cn) for English language editing.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Fabricant DS, Farnsworth NR. The value of plants used in traditional medicine for drug discovery. Environ Health Perspect. 2001;109(Suppl 1):69–75.
- Shivarama Holla B, Veerendra B, Shivananda MK, Poojary B. Synthesis characterization and anticancer activity studies on some Mannich bases derived from 1,2,4-triazoles. Eur J Med Chem. 2003;38(7–8):759–767.
- Sujith KV, Rao JN, Shetty P, Kalluraya B. Regioselective reaction: synthesis and pharmacological study of Mannich bases containing ibuprofen moiety. Eur J Med Chem. 2009;44(9):3697–3702.
- Malinka W, Swiatek P, Filipek B, Sapa J, Jezierska A, Koll A. Synthesis, analgesic activity and computational study of new isothiazolopyridines of Mannich base type. Farmaco. 2005;60(11–12):961–968.
- Ashok M, Holla BS, Poojary B. Convenient one pot synthesis and antimicrobial evaluation of some new Mannich bases carrying 4-methylthiobenzyl moiety. Eur J Med Chem. 2007;42(8):1095–1101.
- Reddy MV, Su CR, Chiou WF, Liu YN, Chen RY, Bastow KF, Lee KH, Wu TS. Design, synthesis, and biological evaluation of Mannich bases of heterocyclic chalcone analogs as cytotoxic agents. Bioorg Med Chem. 2008;16(15):7358–7370.
- Hieu BT, Thuy LT, Thuy VT, Tien HX, Van LV, Hoang VD, Vu TK. Design, synthesis and in vitro cytotoxic activity evaluation of new Mannich bases. B Korean Chem Soc. 2012;33(5):1586–1592.
- Chen Y, Cass SL, Kutty SK, Yee EM, Chan DS, Gardner CR, Vittorio O, Pasquier E, Black DS, Kumar N. Synthesis, biological evaluation and structure-activity relationship studies of isoflavene based Mannich bases with potent anti-cancer activity. Bioorg Med Chem Lett. 2015;25(22):5377–5383.
- Bajracharya GB. Diversity, pharmacology and synthesis of Bergenin and its derivatives: potential materials for therapeutic usages. Fitoterapia. 2015;101:133–152.
- Pavan Kumar P, Siva B, Venkateswara Rao B, Dileep Kumar G, Lakshma Nayak V, Nishant Jain S, Tiwari AK, Purushotham U, Venkata Rao C, Suresh Babu K. Synthesis and biological evaluation of Bergenin-1,2,3-triazole hybrids as novel class of anti-mitotic agents. Bioorg Chem. 2019;91:103161.
- Mistry B, Keum Y-S, Noorzai R, Gansukh E, Kim DH. Synthesis of piperazine based N-Mannich bases of berberine and their antioxidant and anticancer evaluations. J Iran Chem Soc. 2016;13(3):531–539.
- Mistry B, Patel RV, Keum YS, Noorzai R, Gansukh E, Kim DH. Synthesis of Mannich base derivatives of berberine and evaluation of their anticancer and antioxidant effects. J Chem Res. 2016;40(2):73–77.
- Tugrak M, Yamali C, Sakagami H, Gul HI. Synthesis of mono Mannich bases of 2-(4-hydroxybenzylidene)-2,3-dihydroinden-1-one and evaluation of their cytotoxicities. J Enzyme Inhib Med Chem. 2016;31(5):818–823.
- Manohar S, Thakur A, I. Khan S, Sun G, Ni N, Wang B, S. Rawat D. Synthesis of unsymmetrical C5-curcuminoids as potential anticancer agents. LDDD. 2013;11(2):138–149.
- Goel A, Aggarwal BB. Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr Cancer. 2010;62(7):919–930.
- Kawamori T, Lubet R, Steele VE, Kelloff GJ, Kaskey RB, Rao CV, Reddy BS. Chemopreventive effect of curcumin, a naturally occurring anti-inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res. 1999;59(3):597–601.
- Chen XJ, Ren LQ, Zhang XH, Guo L, Zhou JM, Liang G, Wang Y. Improved Pharmacokinetic Profile and Anti-Inflammatory Property of a Novel Curcumin Derivative, A50. LDDD. 2013;10 (6):535–542.
- Huang S-W, Frankel EN. Antioxidant activity of tea catechins in different lipid systems. J Agric Food Chem. 1997;45(8):3033–3038.
- Aggarwal BB, Sung B. Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci. 2009;30(2):85–94.
- Jordan WC, Drew CR. Curcumin – a natural herb with anti-HIV activity. J Natl Med Assoc. 1996;88(6):333.
- Yerdelen KO, Gul HI, Sakagami H, Umemura N, Sukuroglu M. Synthesis and cytotoxic activities of a curcumin analogue and its bis-Mannich derivatives. LDDD. 2015;12(8):643–649.
- Fu Y, Liu B, Zhang N, Liu Z, Liang D, Li F, Cao Y, Feng X, Zhang X, Yang Z. Magnolol inhibits lipopolysaccharide-induced inflammatory response by interfering with TLR4 mediated NF-kappaB and MAPKs signaling pathways. J Ethnopharmacol. 2013;145(1):193–199.
- Shen JL, Man KM, Huang PH, Chen WC, Chen DC, Cheng YW, Liu PL, Chou MC, Chen YH. Honokiol and magnolol as multifunctional antioxidative molecules for dermatologic disorders. Molecules. 2010;15(9):6452–6465.
- Kim KR, Park KK, Chun KS, Chung WY. Honokiol inhibits the progression of collagen-induced arthritis by reducing levels of pro-inflammatory cytokines and matrix metalloproteinases and blocking oxidative tissue damage. J Pharmacol Sci. 2010;114(1):69–78.
- Singh T, Prasad R, Katiyar SK. Inhibition of class I histone deacetylases in non-small cell lung cancer by honokiol leads to suppression of cancer cell growth and induction of cell death in vitro and in vivo. Epigenetics. 2013;8(1):54–65.
- Tang H, Zhang Y, Li D, Fu S, Tang M, Wan L, Chen K, Liu Z, Xue L, Peng A, et al. Discovery and synthesis of novel magnolol derivatives with potent anticancer activity in non-small cell lung cancer. Eur J Med Chem. 2018;156:190–205.
- Zhao M, Zheng YH, Zhao QY, Zheng W, Yang JH, Pei HY, Liu L, Liu KJ, Xue LL, Deng DX, Wang L, Ma X, et al. Synthesis and evaluation of new compounds bearing 3-(4-aminopiperidin-1-yl)methyl magnolol scaffold as anticancer agents for the treatment of non-small cell lung cancer via targeting autophagy. Eur J Med Chem. 2021;209:112922.
- Liang C, Ju W, Pei S, Tang Y, Xiao Y. Pharmacological activities and synthesis of esculetin and its derivatives: a mini-review. Molecules. 2017;22(3):387.
- Park SS, Park SK, Lim JH, Choi YH, Kim WJ, Moon SK. Esculetin inhibits cell proliferation through the Ras/ERK1/2 pathway in human colon cancer cells. Oncol Rep. 2010;25(1):223–230.
- Kim SH, Kang KA, Zhang R, Piao MJ, Ko DO, Wang ZH, Chae SW, Kang SS, Lee KH, Kang HK, et al. Protective effect of esculetin against oxidative stress-induced cell damage via scavenging reactive oxygen species. Acta Pharmacol Sin. 2008;29(11):1319–1326.
- Prabakaran D, Ashokkumar N. Protective effect of esculetin on hyperglycemia-mediated oxidative damage in the hepatic and renal tissues of experimental diabetic rats. Biochimie. 2013;95(2):366–373.
- Wang P, Xia YL, Yu Y, Lu JX, Zou LW, Feng L, Ge GB, Yang L. Design, synthesis and biological evaluation of esculetin derivatives as anti-tumour agents. RSC Adv. 2015;5(66):53477–53483.
- Okita K, Li Q, Murakamio T, Takahashi M. Anti-growth effects with components of Sho-saiko-to (TJ-9) on cultured human hepatoma cells. Eur J Cancer Prev. 1993;2(2):169–175.
- Ikemoto S, Sugimura K, Yoshida N, Yasumoto R, Wada S, Yamamoto K, Kishimoto T. Antitumor effects of Scutellariae radix and its components baicalein, baicalin, and wogonin on bladder cancer cell lines. Urology. 2000;55(6):951–955.
- Li L, Liu WY, Feng F, Wu CY, Xie N. Synthesis and in vitro cytotoxicity evaluation of baicalein amino acid derivatives. Chin J Nat Med. 2014;11(3):284–288.
- Zhang S, Ma J, Bao Y, Yang P, Zou L, Li K, Sun X. Nitrogen-containing flavonoid analogues as CDK1/cyclin B inhibitors: synthesis, SAR analysis, and biological activity. Bioorg Med Chem. 2008;16(15):7128–7133.
- Oh BS, Shin EA, Jung JH, Jung DB, Kim B, Shim BS, Yazdi MC, Iranshahi M, Kim SH. Apoptotic effect of galbanic acid via activation of caspases and inhibition of Mcl-1 in H460 non-small lung carcinoma cells. Phytother Res. 2015;29(6):844–849.
- Xia YL, Wang JJ, Li SY, Liu Y, Gonzalez FJ, Wang P, Ge GB. Synthesis and structure-activity relationship of coumarins as potent Mcl-1 inhibitors for cancer treatment. Bioorg Med Chem. 2021;29:115851.
- Zhao XX, Peng C, Zhang H, Qin LP. Sinomenium acutum: a review of chemistry, pharmacology, pharmacokinetics, and clinical use. Pharm Biol. 2012;50(8):1053–1061.
- Li S, Gao M, Nian X, Zhang L, Li J, Cui D, Zhang C, Zhao C. Design, synthesis, biological evaluation and silico prediction of novel sinomenine derivatives. Molecules. 2021;26(11):3466.
- Palmer AM. Neuroprotective therapeutics for Alzheimer’s disease: progress and prospects. Trends Pharmacol Sci. 2011;32(3):141–147.
- Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res. 2013;36(4):375–399.
- Jan A, Gokce O, Luthi-Carter R, Lashuel HA. The ratio of monomeric to aggregated forms of Abeta40 and Abeta42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J Biol Chem. 2008;283(42):28176–28189.
- Barnham KJ, Bush AI. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem Soc Rev. 2014;43(19):6727–6749.
- Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38(24):7609–7616.
- Schedin-Weiss S, Inoue M, Hromadkova L, Teranishi Y, Yamamoto NG, Wiehager B, Bogdanovic N, Winblad B, Sandebring-Matton A, Frykman S, et al. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with gamma-secretase and regulates neuronal amyloid beta-peptide levels. Alzheimers Res Ther. 2017;9(1):57.
- Zhang X, Song Q, Cao Z, Li Y, Tian C, Yang Z, Zhang H, Deng Y. Design, synthesis and evaluation of Chalcone Mannich base derivatives as multifunctional agents for the potential treatment of Alzheimer’s disease. Bioorg Chem. 2019;87:395–408.
- Rosini M, Simoni E, Caporaso R, Minarini A. Multitarget strategies in Alzheimer’s disease: benefits and challenges on the road to therapeutics. Future Med Chem. 2016;8(6):697–711.
- Tian C, Qiang X, Song Q, Cao Z, Ye C, He Y, Deng Y, Zhang L. Flurbiprofen-chalcone hybrid Mannich base derivatives as balanced multifunctional agents against Alzheimer’s disease: Design, synthesis and biological evaluation. Bioorg Chem. 2020;94:103477.
- Liu HR, Huang XQ, Lou DH, Liu XJ, Liu WK, Wang QA. Synthesis and acetylcholinesterase inhibitory activity of Mannich base derivatives flavokawain B. Bioorg Med Chem Lett. 2014;24(19):4749–4753.
- Liu H, Fan H, Gao X, Huang X, Liu X, Liu L, Zhou C, Tang J, Wang Q, Liu W. Design, synthesis and preliminary structure-activity relationship investigation of nitrogen-containing chalcone derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors: a further study based on Flavokawain B Mannich base derivatives. J Enzyme Inhib Med Chem. 2016;31(4):580–589.
- Duan KK, Liu HR, Fan HQ, Zhang J, Wang Q. Synthesis and anticholinesterase inhibitory activity of Mannich base derivatives of flavonoids. J Chem Res. 2014;38(7):443–446.
- Desideri N, Bolasco A, Fioravanti R, Monaco LP, Orallo F, Yanez M, Ortuso F, Alcaro S. Homoisoflavonoids: natural scaffolds with potent and selective monoamine oxidase-B inhibition properties. J Med Chem. 2011;54(7):2155–2164.
- Gan C, Zhao Z, Nan DD, Yin B, Hu J. Homoisoflavonoids as potential imaging agents for beta-amyloid plaques in Alzheimer’s disease. Eur J Med Chem. 2014;76:125–131.
- Park DH, Venkatesan J, Kim SK, Ramkumar V, Parthiban P. Antioxidant properties of Mannich bases. Bioorg Med Chem Lett. 2012;22(20):6362–6367.
- Kontogiorgis CA, Hadjipavlou-Litina DJ. Synthesis and antiinflammatory activity of coumarin derivatives. J Med Chem. 2005;48(20):6400–6408.
- Li Y, Qiang X, Luo L, Yang X, Xiao G, Zheng Y, Cao Z, Sang Z, Su F, Deng Y. Multitarget drug design strategy against Alzheimer’s disease: Homoisoflavonoid Mannich base derivatives serve as acetylcholinesterase and monoamine oxidase B dual inhibitors with multifunctional properties. Bioorg Med Chem. 2017;25(2):714–726.
- Liu HR, Men X, Gao XH, Liu LB, Fan HQ, Xia XH, Wang QA. Discovery of potent and selective acetylcholinesterase (AChE) inhibitors: acacetin 7-O-methyl ether Mannich base derivatives synthesised from easy access natural product naringin. Nat Prod Res. 2018;32(6):743–747.
- Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5(6):493–506.
- Mooney S, Leuendorf JE, Hendrickson C, Hellmann H. Vitamin B6: a long known compound of surprising complexity. Molecules. 2009;14(1):329–351.
- Hashim A, Wang L, Juneja K, Ye Y, Zhao Y, Ming LJ. Vitamin B6s inhibit oxidative stress caused by Alzheimer’s disease-related Cu(II)-beta-amyloid complexes-cooperative action of phospho-moiety. Bioorg Med Chem Lett. 2011;21(21):6430–6432.
- Yang X, Qiang X, Li Y, Luo L, Xu R, Zheng Y, Cao Z, Tan Z, Deng Y. Pyridoxine-resveratrol hybrids Mannich base derivatives as novel dual inhibitors of AChE and MAO-B with antioxidant and metal-chelating properties for the treatment of Alzheimer’s disease. Bioorg Chem. 2017;71:305–314.
- Na Y. Recent cancer drug development with xanthone structures. J Pharm Pharmacol. 2009;61(6):707–712.
- Li GL, He JY, Zhang A, Wan Y, Wang B, Chen WH. Toward potent alpha-glucosidase inhibitors based on xanthones: a closer look into the structure-activity correlations. Eur J Med Chem. 2011;46(9):4050–4055.
- Belluti F, Rampa A, Piazzi L, Bisi A, Gobbi S, Bartolini M, Andrisano V, Cavalli A, Recanatini M, Valenti P. Cholinesterase inhibitors: xanthostigmine derivatives blocking the acetylcholinesterase-induced beta-amyloid aggregation. J Med Chem. 2005;48(13):4444–4456.
- Li NG, Song SL, Shen MZ, Tang YP, Shi ZH, Tang H, Shi QP, Fu YF, Duan JA. Mannich bases of scutellarein as thrombin-inhibitors: design, synthesis, biological activity and solubility. Bioorg Med Chem. 2012;20(24):6919–6923.
- Qin J, Lan W, Liu Z, Huang J, Tang H, Wang H. Synthesis and biological evaluation of 1, 3-dihydroxyxanthone Mannich base derivatives as anticholinesterase agents. Chem Cent J. 2013;7(1):78.
- Yan YF, Yang CJ, Shang XF, Zhao ZM, Liu YQ, Zhou R, Liu H, Wu TL, Zhao WB, Wang YL, et al. Bioassay-guided isolation of two antifungal compounds from Magnolia officinalis, and the mechanism of action of honokiol. Pestic Biochem Physiol. 2020;170:104705.
- Li H, He YH, Hu YM, Chu QR, Chen YJ, Wu ZR, Zhang ZJ, Liu YQ, Yang CJ, Liang HJ, et al. Design, synthesis, and structure-activity relationship studies of Magnolol derivatives as antifungal agents. J Agric Food Chem. 2021;69(40):11781–11793.
- Liu R, Zhao B, Wang DE, Yao T, Pang L, Tu Q, Ahmed SM, Liu JJ, Wang J. Nitrogen-containing apigenin analogs: preparation and biological activity. Molecules. 2012;17(12):14748–14764.
- Lv XH, Liu H, Ren ZL, Wang W, Tang F, Cao HQ. Design, synthesis and biological evaluation of novel flavone Mannich base derivatives as potential antibacterial agents. Mol Divers. 2019;23(2):299–306.
- Allochio Filho JF, Roldi LL, Delarmelina M, Fiorot RG, Andrade JT, Aleixo ÁA, Carvalho RS, Araújo MGF, Ferreira JMS, Taranto AG, et al. Synthesis, in vitroantifungal activity and molecular modeling studies of new Mannich bases derived from lawsone. J Brazil Chem Soc. 2016;27(11):2127–2140.
- Abrao PH, Pizi RB, De Souza TB, Silva NC, Fregnan AM, Silva FN, Coelho LF, Malaquias LC, Dias AL, Dias DF, et al. Synthesis and biological evaluation of new eugenol Mannich bases as promising antifungal agents. Chem Biol Drug Des. 2015;86(4):459–465.
- Vogl S, Zehl M, Picker P, Urban E, Wawrosch C, Reznicek G, Saukel J, Kopp B. Identification and quantification of coumarins in Peucedanum ostruthium (L.) Koch by HPLC-DAD and HPLC-DAD-MS. J Agric Food Chem. 2011;59(9):4371–4377.
- Zhang MZ, Zhang RR, Yin WZ, Yu X, Zhang YL, Liu P, Gu YC, Zhang WH. Microwave-assisted synthesis and antifungal activity of coumarin[8,7-e][1,3]oxazine derivatives. Mol Divers. 2016;20(3):611–618.
- Choi NH, Choi GJ, Min BS, Jang KS, Choi YH, Kang MS, Park MS, Choi JE, Bae BK, Kim JC. Effects of neolignans from the stem bark of Magnolia obovata on plant pathogenic fungi. J Appl Microbiol. 2009;106(6):2057–2063.
- Lee SK, Kim HN, Kang YR, Lee CW, Kim HM, Han DC, Shin J, Bae K, Kwon BM. Obovatol inhibits colorectal cancer growth by inhibiting tumor cell proliferation and inducing apoptosis. Bioorg Med Chem. 2008;16(18):8397–8402.
- Lee YJ, Lee YM, Lee CK, Jung JK, Han SB, Hong JT. Therapeutic applications of compounds in the Magnolia family. Pharmacol Ther. 2011;130(2):157–176.
- Seo JJ, Lee SH, Lee YS, Kwon BM, Ma Y, Hwang BY, Hong JT, Oh KW. Anxiolytic-like effects of obovatol isolated from Magnolia Obovata: involvement of GABA/benzodiazepine receptors complex. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1363–1369.
- Park ES, Lim Y, Lee SH, Kwon BM, Yoo HS, Hong JT, Yun YP. Antiplatelet activity of obovatol, a biphenolic component of Magnolia Obovata, in rat arterial thrombosis and rabbit platelet aggregation. J Atheroscler Thromb. 2011;18(8):659–669.
- Choi DY, Lee JW, Peng J, Lee YJ, Han JY, Lee YH, Choi IS, Han SB, Jung JK, Lee WS, Lee SH, et al. Obovatol improves cognitive functions in animal models for Alzheimer’s disease. J Neurochem. 2012;120(6):1048–1059.
- Yang C, Zhi X, Xu H. Semisynthesis and insecticidal activity of arylmethylamine derivatives of the neolignan honokiol against Mythimna separata Walker. Z Naturforsch C J Biosci. 2015;70(3–4):65–69.
- Yang C, Li T, Jiang L, Zhi X, Cao H. Semisynthesis and biological evaluation of some novel Mannich base derivatives derived from a natural lignan obovatol as potential antifungal agents. Bioorg Chem. 2020;94:103469.
- Tillhon M, Guaman Ortiz LM, Lombardi P, Scovassi AI. Berberine: new perspectives for old remedies. Biochem Pharmacol. 2012;84(10):1260–1267.
- Wen SQ, Jeyakkumar P, Avula SR, Zhang L, Zhou CH. Discovery of novel berberine imidazoles as safe antimicrobial agents by down regulating ROS generation. Bioorg Med Chem Lett. 2016;26(12):2768–2773.
- Wang GG, Lu XH, Li W, Zhao X, Zhang C. Protective effects of luteolin on diabetic nephropathy in STZ-induced diabetic rats. Evid Based Complement Alternat Med. 2011;2011:323171.
- Hari Babu T, Rama Subba Rao V, Tiwari AK, Suresh Babu K, Srinivas PV, Ali AZ, Madhusudana Rao J. Synthesis and biological evaluation of novel 8-aminomethylated oroxylin A analogues as alpha-glucosidase inhibitors. Bioorg Med Chem Lett. 2008;18(5):1659–1662.
- Zhen J, Dai Y, Villani T, Giurleo D, Simon JE, Wu Q. Synthesis of novel flavonoid alkaloids as alpha-glucosidase inhibitors. Bioorg Med Chem. 2017;25(20):5355–5364.
- Wang Y, Campbell T, Perry B, Beaurepaire C, Qin L. Hypoglycemic and insulin-sensitizing effects of berberine in high-fat diet- and streptozotocin-induced diabetic rats. Metabolism. 2011;60(2):298–305.
- Li R, Wu J, He Y, Hai L, Wu Y. Synthesis and in vitro evaluation of 12-(substituted aminomethyl) berberrubine derivatives as anti-diabetics. Bioorg Med Chem Lett. 2014;24(7):1762–1765.
- Crespo ME, Galvez J, Cruz T, Ocete MA, Zarzuelo A. Anti-inflammatory activity of diosmin and hesperidin in rat colitis induced by TNBS. Planta Med. 1999;65(7):651–653.
- Wilmsen PK, Spada DS, Salvador M. Antioxidant activity of the flavonoid hesperidin in chemical and biological systems. J Agric Food Chem. 2005;53(12):4757–4761.
- Huang YC, Liu KC, Chiou YL. Melanogenesis of murine melanoma cells induced by hesperetin, a citrus hydrolysate-derived flavonoid. Food Chem Toxicol. 2012;50(3–4):653–659.
- Park HJ, Kim MJ, Ha E, Chung JH. Apoptotic effect of hesperidin through caspase3 activation in human colon cancer cells, SNU-C4. Phytomedicine. 2008;15(1–2):147–151.
- Elshazly SM, Mahmoud AA. Antifibrotic activity of hesperidin against dimethylnitrosamine-induced liver fibrosis in rats. Naunyn Schmiedebergs Arch Pharmacol. 2014;387(6):559–567.
- Yeh CC, Kao SJ, Lin CC, Wang SD, Liu CJ, Kao ST. The immunomodulation of endotoxin-induced acute lung injury by hesperidin in vivo and in vitro. Life Sci. 2007;80(20):1821–1831.
- Wang QQ, Shi JB, Chen C, Huang C, Tang WJ, Li J. Hesperetin derivatives: synthesis and anti-inflammatory activity. Bioorg Med Chem Lett. 2016;26(5):1460–1465.
- Hasan SM, Alam MM, Husain A, Khanna S, Akhtar M, Zaman MS. Synthesis of 6-aminomethyl derivatives of benzopyran-4-one with dual biological properties: anti-inflammatory-analgesic and antimicrobial. Eur J Med Chem. 2009;44(12):4896–4903.
- Lu J, Wang D. Advances in endovascular therapy for ischemic cerebrovascular diseases. Chronic Dis Transl Med. 2016;2(3):135–139.
- Lapikova ES, Drozd NN, Tolstenkov AS, Makarov VA, Zvyagintseva TN, Shevchenko NM, Bakunina IU, Besednova NN, Kuznetsova TA. Inhibition of thrombin and factor Xa by Fucus evanescens fucoidan and its modified analogs. Bull Exp Biol Med. 2008;146(3):328–333.
- Cuzzocrea S, Riley DP, Caputi AP, Salvemini D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001;53(1):135–159.
- Liu YM, Lin AH, Chen H, Zeng FD. Study on pharmacokinetics of scutellarin in rabbits. Yao Xue Xue Bao. 2003;38(10):775–778.
- Pan Z, Feng T, Shan L, Cai B, Chu W, Niu H, Lu Y, Yang B. Scutellarin-induced endothelium-independent relaxation in rat aorta. Phytother Res. 2008;22(11):1428–1433.
- Zhong Y, Lu YT, Sun Y, Li NG, Gu T, Wu WY, Yu SP, Shi ZH. Scaffold hopping strategy for the design, synthesis and biological activity evaluation of novel hexacyclic scutellarein derivatives with a 1,3-oxazine ring fused at A-ring. Med Chem. 2018;14(5):478–484.
- Joshi D, Field J, Murphy J, Abdelrahim M, Schonherr H, Sparrow JR, Ellestad G, Nakanishi K, Zask A. Synthesis of antioxidants for prevention of age-related macular degeneration. J Nat Prod. 2013;76(3):450–454.