
Abstract
Autophagy is induced during differentiation of human monocytes into macrophages that is mediated by CSF1/CSF-1/M-CSF (colony stimulating factor 1 [macrophage]). However, little is known about the molecular mechanisms that link CSF1 receptor engagement to the induction of autophagy. Here we show that the CAMKK2-PRKAA1-ULK1 pathway is required for CSF1-induced autophagy and human monocyte differentiation. We reveal that this pathway links P2RY6 to the induction of autophagy, and we decipher the signaling network that links the CSF1 receptor to P2RY6-mediated autophagy and monocyte differentiation. In addition, we show that the physiological P2RY6 ligand UDP and the specific P2RY6 agonist MRS2693 can restore normal monocyte differentiation through reinduction of autophagy in primary myeloid cells from some but not all chronic myelomonocytic leukemia (CMML) patients. Collectively, our findings highlight an essential role for PRKAA1-mediated autophagy during differentiation of human monocytes and pave the way for future therapeutic interventions for CMML.
Abbreviations:
| ACTB | = | actin, β |
| CAMKK2 | = | calcium/calmodulin-dependent protein kinase kinase 2, β |
| CASP8 | = | caspase 8, apoptosis-related cysteine peptidase |
| CFLAR | = | CASP8 and FADD-like apoptosis regulator |
| CMML | = | chronic myelomonocytic leukemia |
| CSF1 | = | colony stimulating factor 1 (macrophage) |
| CSF1R | = | colony stimulating factor 1 receptor |
| DEFA1 | = | defensin, α 1 |
| DEFA3 | = | defensin, α 3, neutrophil-specific |
| DRS | = | dorsomorphin |
| EMR1 | = | EGF-like module-containing mucin-like hormone receptor-like 1 |
| FADD | = | Fas (TNFRSF6)-associated via death domain |
| ITGAM | = | integrin, α M |
| MAP1LC3B LC3B | = | microtubule-associated protein 1 light chain 3 β |
| P2RY6 | = | pyrimidinergic receptor P2Y, G-protein coupled, 6 |
| PLCB3 | = | phospholipase C, β 3 (phosphatidylinositol-specific) |
| PLC | = | phospholipase |
| PLCG2 | = | phospholipase C, gamma 2 (phosphatidylinositol-specific) |
| PRKAA | = | protein kinase, AMP-activated, α |
| PRKAA1 | = | protein kinase, AMP-activated, α 1 catalytic subunit |
| PRKAA2 | = | protein kinase, AMP-activated, α 2 catalytic subunit |
| PRKAG1 | = | protein kinase, AMP-activated, gamma 1 noncatalytic subunit |
| RIPK1 | = | receptor (TNFRSF)-interacting serine-threonine kinase 1 |
| STK11 | = | serine/threonine kinase 11 |
| TFRC | = | transferrin receptor |
| UDP | = | uridine diphosphate |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| WT | = | wild-type |
Introduction
Monocytes have the unique property among peripheral blood cells of migrating into tissues and further differentiating into morphologically and functionally heterogeneous cells that include macrophages, myeloid dendritic cells, and osteoclasts.Citation1,2 In the absence of differentiation, circulating monocytes are believed to undergo apoptotic cell death. Once stimulated by an inflammatory response, monocytes activate prosurvival pathways, migrate to tissues, and differentiate into macrophages.Citation3 The differentiation of human peripheral blood monocytes into macrophages can be recapitulated ex vivo by CSF1.Citation4 Previous studies have established that physiological monocyte differentiation triggered by CSF1R (colony stimulating factor 1 receptor) engagement is critically dependent on the formation of a multimolecular complex consisting of FADD (Fas [TNFRSF6]-associated via death domain), CFLAR/FLIP (CASP8 and FADD-apoptosis regulator), CASP8/caspase-8, RIPK1 (receptor [TNFRSF]-interacting serine-threonine kinase 1) and other protein partners; CASP8 acts as the initiatory caspase in this process.Citation5,6 More recently, we have established that autophagy also plays a crucial role during CSF1-induced differentiation of monocytes.Citation7,8
Autophagy (macroautophagy) is an evolutionarily conserved catabolic pathway that delivers cytoplasmic substrates, such as damaged organelles and cytoplasmic proteins, to lysosomes for degradation.Citation9,10 There is growing evidence that supports a role of autophagy in the rapid cellular changes that are necessary for proper differentiation of different cell types.Citation11–13 However, the molecular mechanisms that regulate autophagy in the context of cell differentiation have remained largely unexplored. The induction of autophagy critically requires ULK1 (unc-51 like autophagy activating kinase 1), which acts in a multimolecular complex to initiate autophagy.Citation14 ULK1 phosphorylation on Ser555 by PRKAA/AMPK (protein kinase, AMP-activated) is one of the main mechanisms leading to the induction of autophagy via the phosphorylation and activation of PIK3C3/VPS34.Citation15
Results from studies presented herein establish the complete signaling pathway that links CSF1R engagement to the induction of autophagy and differentiation of monocytes into macrophages. We found that CSF1 increases the expression of the purinergic receptor P2RY6 and activates the CAMKK2-PRKAA1-ULK1 pathway that is responsible for autophagy induction. Notably, inhibition of this pathway using pharmacological inhibitors, siRNA approaches, and prkag1/ampkγ1 knockout mice, abrogated CSF1-mediated induction of autophagy and differentiation.
A pathological situation in which the differentiation of monocytes is clearly altered is chronic myelomonocytic leukemia (CMML).Citation16 CMML is associated with monocytosis and characterized by defects in monocyte to macrophage differentiation.Citation17 This defect in differentiation can be attributed to the presence of immature dysplastic granulocytes that secrete high levels of α-defensins DEFA1/HNP1/2 and DEFA3/HNP3 that antagonize P2RY6, in CMML patients. We show that a contingent of immature granulocyte cells that is present in variable proportion in CMML patients may affect PRKAA activation in leukemic monocytes. In light of this observation, we showed that triggering P2RY6 with its physiological ligand (UDP) or the P2RY6 agonist could restore autophagy and normal differentiation of monocytes in some but not all CMML patients. Collectively our findings highlight an essential role for P2RY6-mediated autophagy through PRKAA activation during the differentiation of human monocytes and pave the way for future therapeutic interventions for CMML.
Results
CSF1-induced differentiation of monocytes is associated with the induction and activation of PRKAA1/AMPKα1
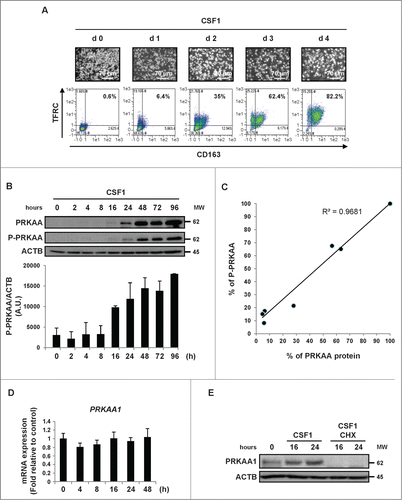
When stimulated with CSF1, human monocytes differentiate into macrophages as shown by an increase in cell adherence and the acquisition of specific markers such as TFRC/CD71 and CD163. After 4 d, more than 80% of myeloid cells were found to be positive for TFRC and CD163 expression (), and increased cell surface expression correlated with a rise in TFRC and CD163 mRNA expression (Figs. S1A and S3A). During differentiation, monocytes accumulated the protein kinase AMP-activated (PRKAA) (). Increased expression of PRKAA was detected one d after CSF1 stimulation and was maximal 2 d later. The CSF1-induced augmentation in PRKAA expression tightly correlated with increased phosphorylation of PRKAA on Thr172 (). Indeed, there was a strict correlation between CSF1-induced expression of PRKAA and its phosphorylation on Thr172 during human monocyte differentiation (). We next sought to identify the PRKA α catalytic subunits (PRKAA1/α1 and/or PRKAA2/α2) expressed during human monocyte differentiation. Of note, unstimulated monocytes exhibited undetectable levels of the PRKAA2/AMPKα2 mRNA and the corresponding protein and no induction of PRKAA2 mRNA was detected during CSF1-mediated monocyte differentiation, indicating that only PRKAA1/AMPKα1 was expressed in differentiating human monocytes (, S1B and C). Importantly, the increased expression of PRKAA1 was not accompanied by a concomitant rise in the PRKAA1 mRNA level, as shown by real-time qPCR analysis (). These findings suggest that increased expression of PRKAA1 is regulated at the post-transcriptional level during CSF1-mediated differentiation of human monocytes and accordingly, cycloheximide treatment of monocytes inhibited CSF1-mediated accumulation of PRKAA1 expression ().
Figure 1. CSF1-induced differentiation of human monocytes is associated with the induction of PRKAA expression and activation. Human peripheral blood monocytes from healthy donors were exposed to 100 ng/mL CSF1 for the indicated times. (A) Macrophage differentiation was examined morphologically (fibroblastic shape) and by 2-color flow cytometric analysis. The percentage indicates cells that express both TFRC/CD71 and CD163. (B) Immunoblot analysis of PRKAA and phospho-PRKAA (Thr172) in monocytes following CSF1 stimulation. The ratio between phospho-PRKAA protein and ACTB was determined from 3 independent experiments using the ImageJ software. (C) The ratio of the PRKAA to phospho-PRKAA protein level was determined from the results of using the ImageJ software. (D) Real-time qPCR analysis of PRKAA1 gene expression in monocytes exposed to 100 ng/mL CSF1 for the indicated times (mean ± SD of 3 independent experiments). (E) Immunoblot analysis of PRKAA1 and phospho-PRKAA1 (Thr172) in monocytes exposed to 100 ng/mL CSF1 alone or in association with 10 μg/mL cycloheximide (CHX), which was added 45 min before CSF1 treatment. ACTB was detected as the loading control. Each panel is representative of at least 3 independent experiments.
The CAMKK2-PRKAA1 axis is required for monocyte differentiation
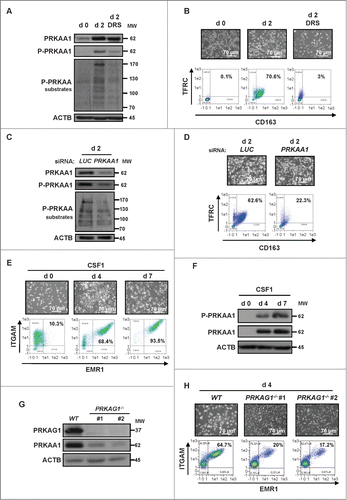
The increased expression of PRKAA1 during monocyte differentiation prompted us to investigate whether it could play a role in this process. Pharmacological inhibition of PRKAA1 by dorsomorphin (DRS, 2 μM) dampened CSF1-induced PRKAA1 phosphorylation on Thr172 () and inhibited CSF1-mediated monocyte differentiation, as illustrated by the drastic decrease in the double-positive TFRC+ and CD163+ cell population at d 2 (). Accordingly, inhibition of PRKAA1 expression by a specific siRNA resulted in a concomitant decrease of PRKAA1 phosphorylation on Thr172 and a reduction of the phosphorylation of PRKAA1 substrates (). In light of the effect of DRS on monocyte differentiation, PRKAA1 knockdown also led to a robust decrease in the double-positive TFRC+ and CD163+ cell population (). It has been previously reported that STK11/LKB1 and CAMKK2/CaMKKβ can phosphorylate PRKAA1 on Thr172 resulting in its activation.Citation18,19 Of note, we found that human monocytes failed to express the STK11 mRNA (Fig. S2A). To investigate if low or technically undetectable levels of STK11 were nevertheless expressed by human monocytes, we analyzed whether STK11 silencing could affect CSF1-induced monocyte differentiation. Figure S2B shows that STK11 silencing failed to strongly affect CSF1-induced monocyte differentiation, suggesting that STK11 is neither involved in PRKAA1 phosphorylation nor in the differentiation of human monocytes induced by CSF1. The efficiency of the siRNA targeting STK11 was verified in the K562 cell line (Fig. S2C). To investigate monocyte differentiation in another context, we treated murine bone marrow monocytes with CSF1. After 4 and 7 d, differentiation of monocytes into macrophages was assessed by flow cytometry. The ITGAM+/CD11b+ and EMR1+/F4/80+ cell population corresponding to macrophages increased tremendously at d 4 and d 7 (). As it is the case with purified human monocytes, PRKAA1 expression and phosphorylation on Thr172 also increased in murine macrophages (). Finally, to specifically assess the role of PRKAA1 in the differentiation process, we stimulated bone marrow monocytes from wild-type and prkag1−/- mice for 4 d with CSF1. As already reported, PRKAG1 deficiency led to a drastic reduction of PRKAA1 expressionCitation20 in 2 different transgenic mice () and to a concomitant inhibition of monocyte differentiation (). All together, these findings highlight the key role of PRKAA1 in monocyte differentiation in 2 different contexts.
Figure 2. PRKAA1 is required for macrophagic differentiation of monocytes. (A) Human monocytes were exposed for 2 d to 100 ng/mL CSF1 alone or in combination with 2 μM DRS, which was added 45 min before CSF1 treatment. The expression of PRKAA1, Phospho-PRKAA1 and Phospho-PRKAA substrates was analyzed by immunoblotting. (B) Human monocytes were exposed for 2 d to 100 ng/mL CSF1 alone or in combination with 2 μM DRS, which was added 45 min before CSF1 treatment. Differentiation was measured as described in . (C) Monocytes were transfected with siRNAs targeting LUCIFERASE (LUC) or PRKAA1 and exposed 2 d to 100 ng/mL CSF1. The expression of PRKAA1, Phospho-PRKAA1 and Phospho-PRKAA substrates was analyzed by immunoblotting. (D) Monocytes were transfected with siRNA targeting LUCIFERASE (LUC) or PRKAA1 and exposed for 2 d to CSF1. Differentiation was assessed as described in . (E) Enriched bone marrow murine monocytes were exposed for the indicated time to 100 ng/mL CSF1. Differentiation was studied by morphological examination (fibroblastic shape) and by 2-color flow cytometry analysis at indicated day. Percentages indicate cells that express both high ITGAM/CD11b and EMR1/F4/80 staining. (F) Immunoblot analysis of PRKAA1 and phospho-PRKAA1 (Thr172) in monocytes following CSF1 stimulation. (G) Immunoblot analysis of PRKAG1/AMPKγ1 and PRKAA1 in monocytes obtained from WT or prkag1−/- mice (n = 2). ACTB was detected as a loading control. (H) Monocytes obtained from WT or prkag1−/- mice were exposed for the indicated times to 100 ng/mL CSF1. Differentiation was assessed as described in . For each experiment, ACTB was detected as the loading control. Each panel is representative of at least 3 independent experiments.
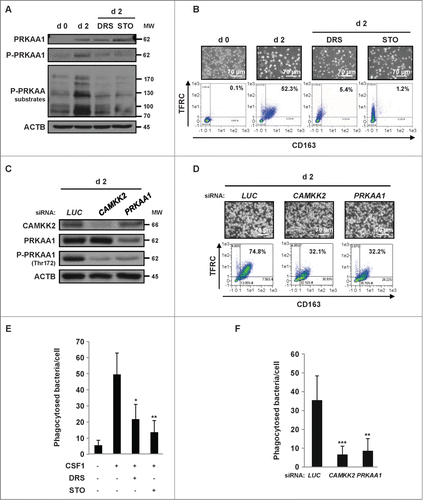
Next, we hypothesized that CAMKK2 might act as the upstream kinase responsible for PRKAA1 phosphorylation and activation in human monocytes. Accordingly, the specific CAMKK2 inhibitor ST0–609 was as efficient as DRS in inhibiting the phosphorylation of PRKAA1 on Thr172 and PRKAA substrates () and the CSF1-induced differentiation of human monocytes (). The importance of CAMKK2 in this process was further highlighted using a siRNA that specifically targeted CAMKK2 (). CAMKK2 silencing resulted in decreased phosphorylation of PRKAA1 on Thr172 and the inhibition of monocyte differentiation, similar to knockdown of PRKAA1 (). Furthermore, pharmacological inhibition of CAMKK2 by STO-609 or PRKAA by DRS or knockdown of CAMKK2 or PRKAA1 furthermore induced a significant decrease in the phagocytic function of macrophages (). Taken together our data clearly illustrate a key role of the CAMKK2-PRKAA1 axis in the differentiation of monocytes into macrophages and the acquisition of phagocytic function.
Figure 3. The CAMKK2-PRKAA1 axis is required for human monocyte differentiation. (A) Human monocytes were exposed for 2 d to 100 ng/mL CSF1 alone or in combination with 2 μM DRS or 10 μM STO-609, which were added 45 min before CSF1 treatment. The expression of PRKAA1, Phospho-PRKAA1 and Phospho-PRKAA substrates was analyzed by immunoblotting. (B) Human monocytes were exposed for 2 d to 100 ng/mL CSF1 alone or in combination with either 2 μM DRS or 10 μM STO-609, which were added 45 min before CSF1 treatment. Differentiation was examined as described in . (C) Monocytes were transfected with siRNA targeting LUCIFERASE (LUC), CAMKK2 or PRKAA1 and exposed for 2 d to 100 ng/mL CSF1. Expression of CAMKK2, PRKAA1, and Phospho-PRKAA1 was analyzed by immunoblotting. (D) Monocytes were transfected with siRNA and treated as in . Differentiation was examined as previously described. (E) Functional assay of monocytes exposed for 2 d to 100 ng/mL CSF1 alone or in combination with 2 μM DRS or 10 μM STO-609. The results are expressed as the number of phagocytosed bacteria per cell and represent the mean ± SD of 4 independent experiments performed in triplicate. *P < 0.05 and **P < 0.01 (vs. untreated cells) according to a Student paired t test. (F) Functional assay of monocytes transfected with LUCIFERASE (LUC), CAMKK2, or PRKAA1 siRNA and treated for 2 d with CSF1. The results are expressed as in **P < 0.01 and **P < 0.001 (vs d2 siLUC) according to a paired Student t test. Each panel is representative of at least 3 independent experiments.
PLCG2 downstream of the CSF1 receptor is required for monocyte differentiation but not PRKAA1 activation
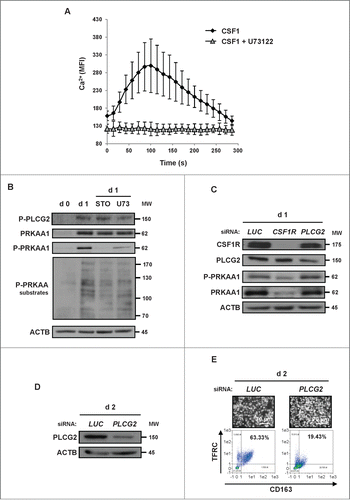
To further decipher the signaling events that link the CSF1 receptor to the induction of monocyte differentiation, we next studied the implication of calcium release that is a prerequisite for CAMKK2 activation.Citation21 It is well established that PLCG2 interacts with the CSF1 receptor to induce Ca2+ release.Citation22 Accordingly, CSF1 triggered a rapid release of Ca2+ in human monocytes that was abrogated by the pan-phospholipase C (PLC) inhibitor U73122 (). U73122 also inhibited CSF1-mediated PLCG2 and PRKAA1 phosphorylation on Thr172, suggesting that the effect of CSF1 on PRKAA1 phosphorylation is dependent on PLC (). Of note, knockdown of the CSF1R decreased PRKAA1 expression and phosphorylation in identical conditions (). Finally, knockdown of PLCG2 failed to affect CSF1-induced PRKAA1 phosphorylation at d one () but efficiently inhibited CSF1-induced monocyte differentiation at d 2 (). Collectively, these findings show that PLCG2 which acts downstream of CSF1 receptor activation is required for CSF1-induced differentiation of human monocytes but is dispensable for PRKAA1 phosphorylation and activation.
Figure 4. PLCG2 downstream of the CSF1 receptor is involved in monocyte differentiation but not in PRKAA1 activation. (A) Monocytes were loaded using the Fluo-4 Direct Calcium Assay kit and stimulated with 100 ng/mL CSF1 alone or in combination with one μM U73122. Calcium fluxes are expressed as the median of the fluorescence Intensity (MFI, arbitrary unit) and represent the mean ± SD of 3 independent experiments performed in duplicate. (B) Human monocytes were exposed for one d to 100 ng/mL CSF1 alone or in combination with either 10 μM STO-609 or 1 μM U73122, which were added 45 min before CSF1 treatment. The expression of Phospho-PLCG2, PRKAA1, Phospho-PRKAA1 and Phospho-PRKAA substrates was analyzed by immunoblotting. (C) Monocytes were transfected with siRNAs targeting LUCIFERASE (LUC), CSF1R or PLCG2 and exposed for one d to 100 ng/ml CSF1. The expression of the different proteins was analyzed by immunoblotting. (D and E) Monocytes were transfected with siRNAs targeting LUCIFERASE (LUC), or PLCG2 and exposed for 2 d to 100 ng/ml CSF1. The expression of PLCG2 was analyzed by immunoblotting (D), and differentiation was evaluated as described previously (E). Each panel is representative of at least 3 independent experiments.
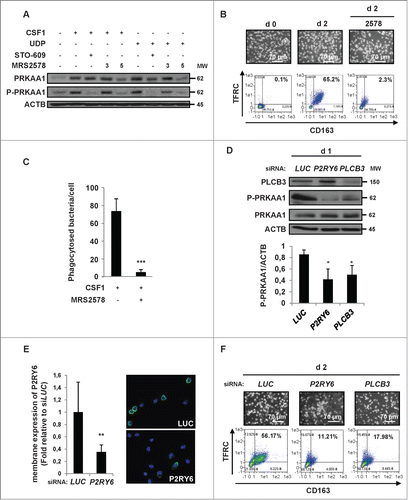
P2RY6 engagement activates the PLCB3-CAMKK2-PRKAA1 pathway that promotes human monocyte differentiation
We previously reported that P2RY6 is required for CSF1-induced human monocyte differentiation.Citation17 In line with this observation, we postulated that the CAMKK2-PRKAA1 pathway was triggered following an engagement of P2RY6. CSF1 was observed to significantly increase P2RY6 mRNA levels, while mRNA expression of other P2Y receptors was reduced in identical conditions (Fig. S3A). Increased expression of P2RY6 mRNA by CSF1 was associated with an increased cell surface expression of P2RY6 (Fig. S3B). In addition, confocal microscopy images, showed numerous puncta of CSF1 and P2Y6 receptor colocalization at the cell surface of differentiated monocytes (Fig. S4A). Moreover, specific silencing of the CSF1R decreased P2RY6 expression at the cell surface (Fig. S4B). Together, these results strongly suggest an interrelation of both receptors during monocyte differentiation.
Importantly, stimulation of monocytes with UDP, a physiological ligand of P2RY6, induced an increase of PRKAA1 expression and phosphorylation similar to that observed with CSF1. In both situations, the signal was abrogated by the P2RY6 antagonist MRS2578 (5 μM) (). As expected, CAMKK2 inhibition by STO-609 also abrogated CSF1 or UDP-induced PRKAA1 phosphorylation. In addition, inhibition of P2RY6 by MRS2578 resulted in a significant inhibition of CSF1-mediated monocyte differentiation () and phagocytic function (). Of note, the specific P2RY1 antagonist, MRS2179 failed to affect CSF1-induced monocyte differentiation in identical conditions (Fig. S5A). In addition and contrary to MRS2578, MRS2179 did not affect PRKAA1 phosphorylation on Thr172 (Fig. S5B). All together these data highlight the specific role of P2RY6 in the regulation of PRKAA1 activation and monocyte differentiation. To gain insights into PRKAA1 regulation during monocyte differentiation, we took advantage of a siRNA approach aimed at individually inhibiting the expression of P2RY6 or PLCB3. PLCB3 has been reported as the specific PLC isoform that is activated downstream of P2RY6.Citation23 Individual silencing of both proteins induced a significant inhibition of PRKAA1 expression and phosphorylation on Thr172 in monocytes stimulated for one d with CSF1 (). Because it has been recently demonstrated that the P2RY6 antibody commonly used for western blot detection of P2RY6 was poorly specific,Citation24 we verified P2RY6 expression using a specific monoclonal antibody that recognizes the native form of P2RY6 using flow cytometry and immunofluorescence. As shown in , P2RY6 silencing could be efficiently validated with this antibody by flow cytometry analysis and immunofluorescence. Of note, P2RY6 and PLCB3 knockdown potently inhibited CSF1-mediated monocyte differentiation at d 2 (). Taken together, these findings show that P2RY6 signaling activates the PLCB3-CAMKK2-PRKAA1 pathway in CSF1-treated monocytes.
Figure 5. For figure legend, see page 1122. Figure 5 (See previous page). P2RY6 engagement activates a PLCB3-CAMKK2-PRKAA1 pathway that promotes human monocyte differentiation. (A) Human monocytes were exposed for one d to 100 ng/mL CSF1 or 100 μM UDP alone or in combination with either 10 μM STO-609 or MRS2578 (3 or 5 μM), which were added 45 min before CSF1 treatment. The expression of PRKAA1 and Phospho-PRKAA1 was analyzed by immunoblotting. (B and C) Human monocytes were exposed for 2 d to 100 ng/mL CSF1 alone or in combination with 5 μM MRS2578, which was added 45 min before CSF1 treatment. (B) Differentiation was examined as described previously. Percentages indicate cells that express both TFRC/CD11b and CD163. (C) Functional assay of monocytes exposed for 2 d to 100 ng/mL CSF1 alone or in combination with 5 μM MRS2578. The results are expressed as the number of phagocytosed bacteria per cell and represent the mean ± SD of 4 independent experiments performed in triplicate. ***P < 0.001 (vs CSF1 treated cells) according to a paired Student t test. (D and E) Monocytes were transfected with siRNA targeting LUCIFERASE (LUC), P2RY6 or PLCB3 and exposed for one d to 100 ng/mL CSF1. (D) The expression of PLCB3, Phospho-PRKAA1 and PRKAA1 was analyzed by immunoblotting. The ratio between phospho-PRKAA1 protein and ACTB was determined from 3 independent experiments using the ImageJ software. *P < 0.05 (vs d1 LUC) according to a paired Student t test. (E) The expression of P2RY6 in transfected monocytes was analyzed by immunofluorescence and flow cytometry after one d of treatment with 100 ng/mL CSF1. Two representative pictures are shown (nuclear staining in blue and P2RY6 in green, left panel). The results are expressed as the fold induction compared to LUC siRNA and represent the mean ± SD of 3 independent experiments performed in duplicate (Right panel). **P < 0.01 (vs d2 siLUC) according to a paired Student t test. (F) Monocytes were transfected with siRNAs targeting LUCIFERASE (LUC), P2RY6 or PLCB3 and exposed for 2 d to 100 ng/mL CSF1. Differentiation was examined as previously described. Each panel is representative of at least 3 independent experiments.
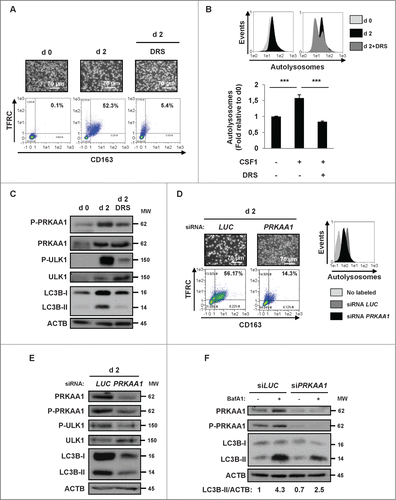
The P2RY6-PRKAA1 pathway mediates autophagy induction and monocyte differentiation
Autophagy induction by PRKAA requires ULK1-dependent phosphorylation and activation of PIK3C3/VPS34.Citation14,25 Therefore, we investigated whether ULK1 was involved in autophagy induction and monocyte differentiation mediated by CSF1. To this aim, we used pharmacological and siRNA approaches. As expected from the results presented in , DRS (2 μM) prevented an accumulation of the double-positive TFRC+ and CD163+ cell population that corresponds to macrophages (). Inhibition of differentiation by DRS also correlated with a reduction in the number of autolysosomes in cells treated with CSF1 and DRS, as assessed by the Cyto-ID assay (). Of note, DRS also reduced CSF1-induced PRKAA1 phosphorylation on Thr172 and phosphorylation of ULK1 on Ser555 (). We also confirmed that PRKAA1 knockdown with a specific siRNA significantly reduced CSF1-induced monocyte differentiation and autolysosome formation (). Importantly, knockdown of PRKAA1 reproduced the effect of DRS, as shown by the inhibition of PRKAA1 expression and phosphorylation on Thr172, the reduction of ULK1 phosphorylation on Ser555 and the decrease in LC3B-I conversion into LC3B-II (). Moreover, knockdown of PRKAA1 in mice also dampened the phagocytic capacity of macrophages and the autophagy activation (Fig. S5C). To further confirm the linear relation from CSF1R to PRKAA1-induced autophagy, we performed autophagic flux assays in monocytes treated with CSF1 with or without bafilomycin A1 in conditions in which PRKAA1 is downregulated or inhibited using siRNA or pharmacological inhibitors respectively ( and Fig. S5D). We found that both approaches reduced the conversion of LC3B-I to LC3B-II, indicating an inhibition of the autophagic flux in monocytes treated with CSF1. These findings confirm the key role of PRKAA1 regulation during differentiation of monocytes. Finally, in contrast to the P2RY1 antagonist MRS2179, the P2RY6 antagonist MRS2578 inhibited both monocyte differentiation and LC3B-II conversion, highlighting the specific role of P2RY6 in the regulation of autophagy (Fig. S5B). All together, our findings established that the P2RY6- PRKAA1-ULK1 pathway is required for CSF1-mediated induction of autophagy and differentiation of human monocytes.
Figure 6. For figure legend, see page 1124. Figure 6 (See previous page). The P2RY6-PRKAA1 pathway mediates autophagy induction and monocyte differentiation. (A–C) Human monocytes were exposed for 2 d to 100 ng/mL CSF1 alone or in combination with 2 μM DRS, which was added 45 min before CSF1 treatment. (A) Differentiation was visualized as described previously. (B) The autolysosome number was quantified by flow cytometric analysis using a Cyto-ID® Autophagy Detection Kit. A representative cytometry profile is shown in the upper panel. The results are expressed as the fold induction compared with untreated cells and represent the mean ± SD of 3 independent experiments performed in duplicate (lower panel). ***P < 0.001 (vs untreated cells) according to a paired Student t test. (C) Immunoblot analysis of the indicated protein is shown. (D and E) Monocytes were transfected with siRNA targeting LUCIFERASE (LUC) or PRKAA1 and exposed for 2 d to CSF1. (D) Differentiation was examined as described in . Autolysosomes were quantified by flow cytometric analysis using the Cyto-ID® Autophagy Detection Kit. A representative cytometry profile is shown (right panel). (E) The expression of PRKAA1, Phospho-PRKAA1, Phospho-ULK1, ULK1 and LC3B was analyzed by immunoblotting. (F) Monocytes were transfected with siRNA targeting LUCIFERASE (LUC) or PRKAA1 and exposed for 2 d to 100 ng/mL CSF1 alone or in association with 15 nM bafilomycin A1 (BafA1) added 3 h before the end of CSF1 treatment and protein expression was analyzed by immunoblot. The ratio between LC3B-II protein and ACTB was determined using the ImageJ software. For all western blot experiments, ACTB was detected as the loading control. Each panel is representative of at least 3 independent experiments.
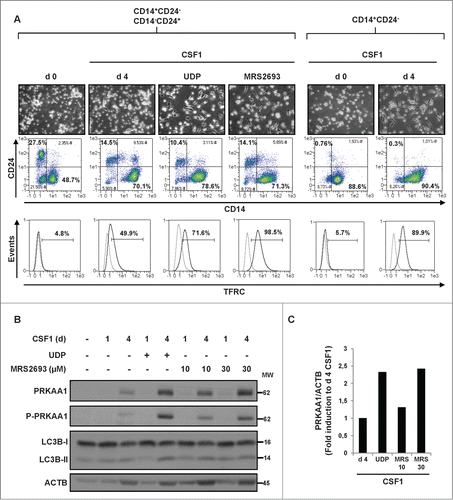
Impaired monocyte differentiation in CMML patients can be overcome by UDP and P2RY6 agonists through PRKAA1 reactivation
We previously established the occurrence of an immature CD14− and CD24+ granulocytic subpopulation that represses the differentiation of the CD14+ and CD24− patient's blasts in CMML patients.Citation17 This CD14− and CD24+ population, which arose from the same leukemic clone than as the CD14+ and CD24− population produces high levels of α defensins that in turn block the differentiation of CD14+ and CD24− monocytes through inhibition of P2RY6 signaling. As shown in , incubation of both CD14+ and CD24−, as well as CD14− and CD24+ (representing 27.5% of total cells) cells isolated from a CMML patient with CSF1 resulted in altered monocyte differentiation of CD14+ and CD24− cells (49.9% TFRC+ cells versus 89.9% in cells depleted from the suppressive CD14− and CD24+ subpopulation). Normal CSF1-induced monocyte differentiation was restored by the physiological P2RY6 ligand UDP (100 μM) (71.6% TFRC+ cells) or the P2RY6 agonist MRS2693 (30 μM) (98.5% TFRC+ cells). To investigate the potential role of PRKAA1 in the effect of P2RY6 agonists, we searched for PRKAA1 expression and autophagy induction in monocytes from a CMML patient treated with CSF1. After 4 d of treatment, there was a significant increase in both PRKAA1 expression and phosphorylation on Thr172 in monocytes from CMML patients treated with the combination of CSF1 and UDP compared with CMML patients treated with CSF1 alone (). This increase in PRKAA1 activation correlated both with a higher level of differentiation () and an increase in the conversion of LC3B-I into LC3B-II (). A similar situation was observed when monocytes from CMML patients were incubated with a combination of CSF1 and MRS2693 (). Increased PRKAA1 expression was confirmed by quantification of the PRKAA1/ACTB ratio in cells from CMML patients treated with a combination of CSF1 plus UDP or MRS2693 (). Taken together, our data show that the defect in CSF1-mediated autophagy and monocyte differentiation found in CMML patients can be overcome by P2Y6 engagement.
Figure 7. Differentiation defect in CMML patients can be restored by UDP and P2RY6 agonists through PRKAA1 reactivation. (A–C) Following negative sorting, CD14+ and CD24− (monocytes), as well as CD14− and CD24+ (immature granulocytes) populations from one CMML patient were cultured separately or together with CSF1 alone or in the presence of 100 μM UDP or 30 μM MRS2693 for the indicated times. (A) Flow cytometric analysis of CD14 and CD24, identifying CD14+ and CD24−, as well as CD14− and CD24+ cells in the CMML patient. Differentiation was only examined for CD14+ cells. (B) CD14+ and CD24− (monocytes), as well as CD14− and CD24+ (immature granulocytes) populations were cultured together with CSF1 alone or in the presence of 100 μM UDP or MRS2693 (10 or 30 μM) for the indicated times. The expression of PRKAA1, Phospho-PRKAA1, and LC3B was analyzed by immunoblotting. ACTB was detected as a loading control. (C) The ratio of the phospho-PRKAA1 protein level to that of ACTB protein level was measured from the protein band signals of , using the ImageJ software.
The same experiments were reproduced with 2 other CMML patients. Figure S6 illustrated the result obtained with 2 representative patients that exhibited a high level of CD14− and CD24+ granulocytic subpopulation (44% and 67%). The coculture of CD14+ and CD24− monocytes with CD14− and CD24+ granulocytic subpopulation was found to abolish PRKAA1 expression and phosphorylation, as is also the case for the first patient (). However, UDP or MRS was unable to restore significant PRKAA1 expression and phosphorylation in this patient. Globally, it appears that a contingent, higher than 30%, of the CD14− and CD24+ granulocytic subpopulation exerted an inhibition of PRKAA1 activity and differentiation that cannot be overcome with P2RY6 agonists.
Discussion
Autophagy is an evolutionarily conserved catabolic process for the degradation of long-lived molecules and organelles that also plays a crucial role during the differentiation of a wide range of cell types.Citation26–28 Notably, the differentiation of hematopoietic cells requires intense energy consumption, membrane remodeling and/or organelles elimination, as exemplified for myeloid, megakaryocytic, or erythroid differentiation.Citation7,11,29,30 Physiological monocyte differentiation triggered by CSF1R engagement requires the formation of a multimolecular complex consisting of FADD, CFLAR/FLIP, CASP8, RIPK1 and other protein partners; and CASP8 acts as the initiatory caspase to induce the cleavage of key protein substrates (such as RIPK1 and NUCLEOPHOSMIN, among others) that orchestrate the differentiation process.Citation6,31 Accordingly, inhibition of caspase activation with pancaspase inhibitors or by CASP8 silencing is sufficient to dampen the differentiation of human monocytes into macrophages. More recently we established that the induction of autophagy is also a prerequisite for CSF1-induced monocyte differentiation and the acquisition of phagocytic functions.Citation7,8 In the same line, it has been previously reported that induction of autophagy is essential for monocyte-macrophage differentiation upon CSF2/GM-CSF stimulation. Indeed, CSF2 induced MAPK8/JNK1 activation to mediate the induction of autophagy and monocyte survival. The authors also showed that CSF1 triggered autophagy induction in both human and murine monocytes and that inhibition of MAPK8 by SP600125 hampered CSF1-mediated autophagy and SQSTM1/p62 accumulation.Citation32 Therefore, it would be interesting in further studies to decipher the interrelation between PRKAA1 and MAPK8 activation. Nevertheless, the molecular mechanisms that link the CSF1R to the induction of autophagy remained ill defined. In the present study we deciphered the complete signaling pathway from CSF1 receptor engagement to the induction of autophagy and monocyte differentiation. Using both pharmacological and siRNA approaches, we demonstrate that the physiological differentiation of human monocytes into macrophages upon CSF1R activation requires an induction of autophagy through the purinergic receptor P2RY6. Accordingly, stimulation of human monocytes with CSF1 induced P2RY6 mRNA accumulation and increased P2RY6 protein expression at the monocyte cell surface. Among all of the P2Y receptors analyzed in the present study, the expression of P2RY6 mRNA was solely induced by CSF1, whereas expression of all other P2Y mRNA subtypes was reduced, which reinforces the notion of a specific role of P2RY6 during human monocyte differentiation. There are few studies in the literature that show that P2Y6 receptor activation is involved in differentiation processes such as osteogenic, monocytic, or dopaminergic differentiation.Citation17,33–35 Moreover, it has been previously reported that P2RY13 activation by ADP induces autophagy in hepatoma cell lines.Citation36 However to the best of our knowledge, this is the first description of purinergic receptor-mediated differentiation being dependent on autophagy induction.
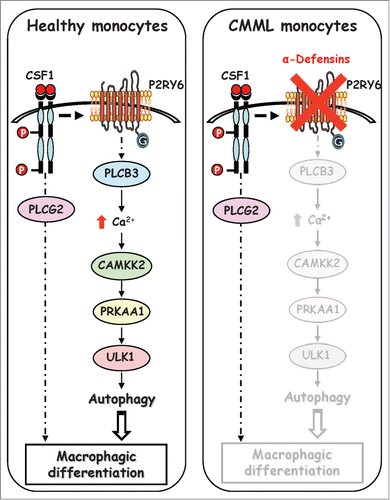
Understanding how physiological monocyte differentiation occurs at the molecular and cellular levels is obviously a key question in cell biology. Here we demonstrate that PLCB3 interacts with the P2RY6 receptor to induce intracellular Ca2+ production and CAMKK2 activation. CAMKK2 in turn phosphorylated PRKAA on Thr172, leading to its activation. Once activated, PRKAA1 is phosphorylated and activates the serine/threonine kinase ULK1 on Ser555, resulting in the induction of autophagy. Importantly, inhibition of P2RY6-mediated autophagy induction using different pharmacological approaches or specific siRNA directed against PRKAA1 reduced autophagy induction and inhibited human monocyte differentiation into macrophages (, left panel). Moreover, inhibition of the P2RY6 pathway by specific inhibitors (DRS, STO-609, U73122 and MRS2578) prevented CSF1-induced differentiation of human monocytes without increasing the rate of cell death, as assessed by ANXA5/annexin-V staining and PARP1 cleavage (Fig. S7A and S7B). Furthermore, we also showed that PRKAA1 silencing neither affected the proliferation status nor the cell cycle of monocytes, strongly suggesting that the effect of PRKAA1 resides essentially on its ability to modulate autophagy during monocyte differentiation (Fig. S7C and S7D). The characterization of the molecular pathways that control monocyte differentiation and more precisely the discovery of the implication of the P2RY6 signaling pathway in the regulation of PRKAA1 activation and autophagy could be of highest interest for the treatment of myeloid malignancies, including CMML. Of note, we have recently identified a granulocytic subpopulation in the blood of CMML patients that produces high levels of α-defensins that inhibit the differentiation of monocytes by binding to P2RY6 (, right panel). Accordingly, depletion of this granulocytic subpopulation allowed monocytes from CMML patients to fully differentiate into macrophages. In light of this finding, we show here that UDP or P2RY6 agonists could restore physiological monocyte differentiation ex vivo in a blood sample of one CMML patient. In the other 2 patients tested, that both exhibit a high contingent of granulocytic subpopulation in the blood (44% and 67%), PRKAA1 expression, and phosphorylation were drastically reduced in coculture conversely to isolated CD14+ and CD24− cells. Importantly in those patients P2RY6 agonists failed to restore normal monocyte differentiation. Nevertheless, our findings show that in CMML patients, the granulocytic subpopulation exerted a negative constraint on the differentiation of monocytes that is strikingly correlated to the lack of PRKAA1 expression and activation.
Figure 8. Schematic view of the signaling pathways involved in healthy and CMML monocytes during CSF1-induced differentiation. Upon CSF1 binding, the CSF1R triggers the activation of the P2RY6-PLCB3-CAMKK2-PRKAA1-ULK1 pathway leading to induction of autophagy and human monocyte differentiation. In a CMML-dependent context, immature dysplastic granulocytes synthesize and secrete large amounts of α-defensins DEFA1 and DEFA3, which antagonize P2RY6 and inhibit CSF1-induced differentiation of monocytes.
The reinduction of differentiation has been shown to be a valuable therapeutic strategy in some hematopoietic malignancies and has particularly been well exemplified in acute promyelocytic leukemia (APL). APL is caused by an arrest of leukocyte differentiation at the promyelocyte stage and all-trans retinoic acid (ATRA) treatment, which is able to reinduce differentiation, has transformed APL from a highly fatal to a highly curable disease.Citation37 Importantly, the antileukemic and differentiation effect of ATRA in APL has been recently linked to its ability to induce autophagy-dependent degradation of the PML-RARA/RARα oncoprotein.Citation38–40
The discovery that P2RY6 ligands such as UDP or P2RY6 agonists are able to restore normal differentiation in some CMML patients through the PRKAA1 reactivation is exciting. In agreement with these findings, it has been recently reported that 5-aminoimidazole-4-carboxamide ribonucleoside, an PRKAA activator, enhances ATRA-mediated differentiation of promyelocytic NB4 cells and, as a single agent induces the expression of cell surface markers associated with mature monocytes and macrophages in the acute myeloid leukemia cell line U937.Citation41 Decitabine and azacitidine are 2 nucleoside analogs that are currently used as first-line treatments for high-risk MDS and CMML patients. Indeed, both agents exert potent antileukemic effects in MDS and CMML.Citation42,43 Therefore, it is tempting to speculate that part of their antileukemic effect might also involve an induction of autophagy and myeloid differentiation. Nevertheless, the data presented herein deciphers the signaling pathways involved in CSF1-mediated induction of autophagy and monocyte differentiation, as well as paves a new research avenue for future therapeutic interventions through PRKAA reactivation in the context of CMML.
Materials and Methods
Reagents and antibodies
Human CSF1 was purchased from Miltenyi (130–096–493). Cycloheximide (C1988) and propidium iodide (P4170) and RNase A (R6513) were purchased from Sigma-Aldrich. Dorsomorphin, STO-609, MRS2578, UDP, MRS2179, MRS2693 and bafilomycin A1 were from Tocris (3093, 1551, 2146, 3111, 0900, 2502, and 1334, respectively) and U73122 (BML-ST391–0005) was from ENZO Life Sciences. Antibodies to PRKAA1/2 (detects the α1 and α2 isoforms of the catalytic subunit), PRKAA2, phospho-PRKAA1/2 (Thr172), phospho-(Ser/Thr)-PRKAA1/2 substrates (P-S/T2–102), PLCG2/PLCγ2, phospho-PLCG2 (Y759), PLCB3, ULK1, phospho-ULK1 (Ser555), and LC3B were purchased from Cell Signaling Technology (2532, 2757, 2535, 5759, 3872, 3874, 2482, 8054, 5869 and 2775, respectively). ACTB, CSF1R, P2RY6 (SC-20127), STK11/LKB1 and HSP90 antibodies were from Santa Cruz Biotechnology (sc-1616, sc-692, sc-20127, sc-32245 and sc-13119, respectively). PRKAG1/AMPKγ1 antibody was from Abcam (ab32508). HRP-conjugated rabbit anti-goat or anti-mouse was purchased from Dako (P0449 and P0260) and HRP-conjugated goat anti-rabbit was from Cell Signaling Technology (5127).
Patient samples
Patients and volunteers signed an informed consent according to the Declaration of Helsinki and recommendations of an independent scientific review board. Chronic-phase CMML diagnosis was based on WHO criteria. Patients were newly diagnosed or had previously diagnosed hematopoietic disease and were followed every 3 mo. They were either untreated or received supportive care or cytotoxic treatment, in most cases hydroxyurea.
Cell culture, cell sorting, and differentiation
Blood samples were collected using ethylene diamine tetraacetic acid–containing tubes. Mononucleated cells were first isolated using Ficoll Hypaque (Eurobio, CMSMSL0101). Then, we used the autoMACS® Pro Separator (Miltenyi, Paris, France) to perform cell enrichment. An initial negative selection, which included antibodies targeting CD3, CD7, CD16, CD19, CD56, CD123, and GYPA/glycophorin A, was used for monocyte enrichment (Miltenyi, 130–091–153). In CMML samples, CD14+ and CD14− populations were further enriched using an anti-CD14 antibody (Miltenyi, 130–050–201). Mouse monocytes were obtained from C57BL6 WT and prkag1−/-. Briefly, cells were extracted from tibia and incubated with PE-labeled anti-ITGA2/CD49b, -PTPRC/CD45R, -CD3E, and -LY76/Ter119 antibodies (Miltenyi, 130–102–337, 130–102–292, 130102600, 130–102–336, respectively). Cells were then separated with an autoMACS™ and the negative fraction containing purified monocytes was treated with murine CSF1 (100 ng/mL). Purified monocytes from humans and mice were grown in RPMI 1640 medium with glutamax-I (Life Technologies, 61870044) supplemented with 10% (vol/vol) fetal bovine serum (Life Technologies, 10270106). Macrophage differentiation (adhesion to culture flasks and fibroblast-like shape) was visualized using standard optics (Leica, Paris, France) equipped with a Moticam 2500 camera (Motic, Wetzlar, Germany). Phase images of the cultures were recorded with a 20×/0.30 PH1 objective with the Motic Image Plus software (Motic). The human leukemic cell line K562 (ACC 10, DSMZ) and human prostate carcinoma PC3 cells (ACC 465, DSMZ) cells were respectively grown in RPMI 1640 medium with glutamax-I and in 45% Ham's F12 + 45% RPMI 1640 (Life Technologies) supplemented with 10% (vol/vol) fetal bovine serum.
Flow cytometry
The monocyte differentiation, the cell surface expression of P2RY6 and autophagy were studied by flow cytometry.Citation44 To analyze the macrophagic differentiation of monocytes, cells were washed with ice-cold phosphate-buffered saline (PBS, Life Technologies, 14190169), and incubated at 4°C for 10 min in PBS/bovine serum albumin (BSA 0.5%; Dutscher, 871002) with anti-TFRC and anti-CD163 or with anti-ITGAM/CD11b and anti-EMR1/F4/80 or an isotype control (Miltenyi, 130-091-727, 130-097-628, 130-091-240 and 130-102-379, respectively). Finally, the cells were washed and fixed in 2% paraformaldehyde (EMS, 15710). To analyze the cell surface expression of P2RY6, cells were washed with ice-cold PBS, incubated at 4°C for 1 h in PBS/ BSA (0.5%) with P2RY6 (1:100) or an isotype control (Santa Cruz Biotechnology, sc-2027), washed and incubated with secondary antibody (1:1500) for 30 min. Finally, the cells were washed and fixed in 2% paraformaldehyde. A Cyto-ID® Autophagy Detection Kit was used to monitor autophagic activity at the cellular level according to the manufacturer's instructions (Enzo Life Sciences, ENZ-51031-K200). Briefly, the 488 nm-excitable green fluorescent detection reagent supplied in the Cyto-ID® Autophagy Detection Kit becomes brightly fluorescent in vesicles produced during autophagy. Fluorescence was measured by the use of a MACSQuant® Analyzer (Miltenyi, Paris, France).
Immunoblot assays
Cells were lysed for 30 min at 4°C in lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 20 mM EDTA, 100 mM NaF, 10 mM Na3VO4, complete protease inhibitor mixture [Roche, 11836153001], 1% Triton X-100 [Sigma, T9284]). Lysates were centrifuged at 20,000 g (15 min, 4°C) and supernatant fractions were supplemented with concentrated sodium dodecyl sulfate (Euromedex, EU0660). Fifty micrograms of proteins were separated and transferred following standard protocols before analysis with a chemiluminescence detection kit (GE Healthcare, RPN2105).
Reverse-transcription and real-time polymerase chain reaction
RNA was prepared from 5 × 106 cells using the RNeasy Mini Kit according to the manufacturer's protocol (Qiagen, 74104). Each cDNA sample was prepared using superscript II RT and random primers (Life Technologies, 18064–014 and 48190–011). Real-time polymerase chain reaction (PCR) was performed using the SyBR Green detection protocol (Life Technologies, 4367659). Briefly, 5 ng of total cDNA, 125 nM (each) primers, and 10 μL SyBR Green mixture were used in a total volume of 20 μL. Detection of multiple endogenous controls (ACTB, RPL32/L32 and UBB) were used to normalize the results. Specific forward and reverse primers are accessible upon request.
siRNA knockdown
Small interfering (si) RNAs were introduced into monocytes or K562 by nucleoporation (Amaxa, VPA-1007, VACA-1003) of 5 × 106 monocytes or K562 in 100 μL of nucleofector solution with 15 nmol of siRNA. Cells were incubated for 24 h with 5 mL of prewarmed complete medium, and CSF1 was subsequently added. We used siRNAs (Life Technologies) targeting PRKAA1/AMPKα1 (HSS108454), PLCG2 (HSS108098), CAMKK2/CaMKKβ (HSS173805), P2RY6 (HSS143211), PLCB3 (HSS108082), CSF1R (HSS102358), STK11/LKB1 (VHS50411) and, LUCIFERASE as a negative control (Sense: 5'-CUUACGCUGAGUACUUCGAtt-3').
Functional assay - (Gentamicin protection assay)
Monocytes (106) were infected for 20 min in RPMI 1640 medium supplemented with 10% fetal calf serum with ampicillin resistant E. coli K12 (MOI = 50)(Life Technologies, C4040). The cells were washed 3 times before incubation for 20 min with RPMI 1640 medium supplemented with 10% fetal calf serum and gentamicin (50 μg/mL; Life Technologies, 15750–045) to measure internalized bacteria. The cells were lysed in PBS with 0.1% Triton X-100, and the number of bacteria on LB plates containing ampicillin (100 μg/mL) were counted. The mean values of triplicates that were representative of 4 independent experiments were calculated.
Calcium flux assay
Monocytes were loaded using a Fluo-4 Direct Calcium Assay Kit according to the manufacturer's instructions (Molecular Probes, F10471). Calcium release was measured by the use of an autoMACS® Pro Separator (Miltenyi, Paris, France).
Immunofluorescence
Cells were washed with ice-cold PBS, incubated at 4°C for 1 h in PBS/ BSA (0.5%) with P2RY6 (Santa Cruz Biotechnology, 1:100) and/or CSF1R (BioLegend, 1:50) antibodies and, washed and incubated with secondary antibody (1:1500) for 30 min. Finally, the cells were washed and fixed in 2% paraformaldehyde and spun on to a microscope slide for 4 min at 800 g in a Cytospin 3 apparatus (Shandon Thermo Electron Corp, Paris, France). The cells were then mounted on coverslips and analyzed with a confocal microscope (Carl Zeiss, Paris, France).
Statistical analysis
Statistical analysis was performed using a paired Student t test and significance was considered when P values were lower than 0.05. The results are expressed as the mean ± SEM.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1034406_supplemental_data.pptx.zip
Download Zip (1.1 MB)Acknowledgments
The authors are indebted to the Etablissement francais du Sang for providing us with human blood from healthy donors. We also thank the C3M facilities (ISO-9001 certified imagery platform and animal facilities). S.O. was supported by a fellowship from the Inserm-Region PACAC. Z.H., M.A.H. and M.C. were supported by a fellowship from the French Government. A.P and DG were awarded fellowships from the Ligue Nationale Contre le Cancer and the Fondation pour la Recherche Médicale, respectively.
Funding
This work was supported by Inserm, the Ligue Nationale Contre le Cancer (Equipe labellisée 2011–2013), the Association pour la Recherche contre le Cancer, the Fondation pour la Recherche Médicale, the Association Laurette Fugain and the French Government (National Research Agency, ANR) through the “Investments for the Future” Labex Signalife (ANR-11-LABX-0028–01).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol 2009; 27:669–92; PMID:19132917; http://dx.doi.org/10.1146/annurev.immunol.021908.132557
- Kikuta J, Ishii M. Osteoclast migration, differentiation and function: novel therapeutic targets for rheumatic diseases. Rheumatology 2013; 52:226–34; PMID:23024017; http://dx.doi.org/10.1093/rheumatology/kes259
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science 2010; 327:656–61; PMID:20133564; http://dx.doi.org/10.1126/science.1178331
- Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol 2013; 34:81–9; PMID:23000011; http://dx.doi.org/10.1016/j.it.2012.08.006
- Jacquel A, Benikhlef N, Paggetti J, Lalaoui N, Guery L, Dufour EK, Ciudad M, Racoeur C, Micheau O, Delva L, et al. Colony-stimulating factor-1-induced oscillations in phosphatidylinositol-3 kinase/AKT are required for caspase activation in monocytes undergoing differentiation into macrophages. Blood 2009; 114:3633–41; PMID:19721010; http://dx.doi.org/10.1182/blood-2009-03-208843
- Rebe C, Cathelin S, Launay S, Filomenko R, Prevotat L, L'Ollivier C, Gyan E, Micheau O, Grant S, Dubart-Kupperschmitt A, et al. Caspase-8 prevents sustained activation of NF-kappaB in monocytes undergoing macrophagic differentiation. Blood 2007; 109:1442–50; PMID:17047155; http://dx.doi.org/10.1182/blood-2006-03-011585
- Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P, Lemichez E, Luciano F, Solary E, Auberger P. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012; 119:4527–31; PMID:22452982; http://dx.doi.org/10.1182/blood-2011-11-392167
- Jacquel A, Obba S, Solary E, Auberger P. Proper macrophagic differentiation requires both autophagy and caspase activation. Autophagy 2012; 8:1141–3; PMID:22751215; http://dx.doi.org/10.4161/auto.20367
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15:713–20; PMID:23817233; http://dx.doi.org/10.1038/ncb2788
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814–22; PMID:20811353; http://dx.doi.org/10.1038/ncb0910-814
- Colosetti P, Puissant A, Robert G, Luciano F, Jacquel A, Gounon P, Cassuto JP, Auberger P. Autophagy is an important event for megakaryocytic differentiation of the chronic myelogenous leukemia K562 cell line. Autophagy 2009; 5:1092–8; PMID:19786835; http://dx.doi.org/10.4161/auto.5.8.9889
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823–30; PMID:20811354; http://dx.doi.org/10.1038/ncb0910-823
- Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A 2010; 107:832–7; PMID:20080761; http://dx.doi.org/10.1073/pnas.0913170107
- Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 2013; 9:124–37; PMID:23295650; http://dx.doi.org/10.4161/auto.23323
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741–50; PMID:23685627; http://dx.doi.org/10.1038/ncb2757
- Itzkson R, Fenaux P, Solary E. Chronic myelomonocytic leukemia: Myelodysplastic or myeloproliferative? Best Pract Res Clin Haematol 2013; 26:387–400; PMID:24507815; http://dx.doi.org/10.1016/j.beha.2013.09.006
- Droin N, Jacquel A, Hendra JB, Racoeur C, Truntzer C, Pecqueur D, Benikhlef N, Ciudad M, Guery L, Jooste V, et al. Alpha-defensins secreted by dysplastic granulocytes inhibit the differentiation of monocytes in chronic myelomonocytic leukemia. Blood 2009; 115:78–88; PMID:19864642; http://dx.doi.org/10.1182/blood-2009-05-224352
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2005; 2:9–19; PMID:16054095; http://dx.doi.org/10.1016/j.cmet.2005.05.009
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003; 13:2004–8; PMID:14614828; http://dx.doi.org/10.1016/j.cub.2003.10.031
- Foretz M, Hebrard S, Guihard S, Leclerc J, Do Cruzeiro M, Hamard G, Niedergang F, Gaudry M, Viollet B. The AMPKgamma1 subunit plays an essential role in erythrocyte membrane elasticity, and its genetic inactivation induces splenomegaly and anemia. FASEB J 2011; 25:337–47; PMID:20881209; http://dx.doi.org/10.1096/fj.10-169383
- Tokumitsu H, Soderling TR. Requirements for calcium and calmodulin in the calmodulin kinase activation cascade. J Biol Chem 1996; 271:5617–22; PMID:8621423; http://dx.doi.org/10.1074/jbc.271.10.5617
- Bourgin-Hierle C, Gobert-Gosse S, Therier J, Grasset MF, Mouchiroud G. Src-family kinases play an essential role in differentiation signaling downstream of macrophage colony-stimulating factor receptors mediating persistent phosphorylation of phospholipase C-gamma2 and MAP kinases ERK1 and ERK2. Leukemia 2008; 22:161–9; PMID:17972959; http://dx.doi.org/10.1038/sj.leu.2404986
- Roach TI, Rebres RA, Fraser ID, Decamp DL, Lin KM, Sternweis PC, Simon MI, Seaman WE. Signaling and cross-talk by C5a and UDP in macrophages selectively use PLCbeta3 to regulate intracellular free calcium. J Biol Chem 2008; 283:17351–61; PMID:18411281; http://dx.doi.org/10.1074/jbc.M800907200
- Yu W, Hill WG. Lack of specificity shown by P2Y6 receptor antibodies. Naunyn Schmiedebergs Arch Pharmacol 2013; 386:885–91; PMID:23793102; http://dx.doi.org/10.1007/s00210-013-0894-8
- Zhao M, Klionsky DJ. AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab 2011; 13:119–20; PMID:21284977; http://dx.doi.org/10.1016/j.cmet.2011.01.009
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24–41; PMID:24366339; http://dx.doi.org/10.1038/cr.2013.168
- Chen P, Cescon M, Bonaldo P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 2014; 10:192–200; PMID:24300480; http://dx.doi.org/10.4161/auto.26927
- Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity 2013; 39:211–27; PMID:23973220; http://dx.doi.org/10.1016/j.immuni.2013.07.017
- Mortensen M, Ferguson DJ, Simon AK. Mitochondrial clearance by autophagy in developing erythrocytes: clearly important, but just how much so? Cell Cycle 2010; 9:1901–6; PMID:20495377; http://dx.doi.org/10.4161/cc.9.10.11603
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 2009; 119:3329–39; PMID:19855132; http://dx.doi.org/10.1172/JCI35541
- Guery L, Benikhlef N, Gautier T, Paul C, Jego G, Dufour E, Jacquel A, Cally R, Manoury B, Vanden Berghe T, et al. Fine-tuning nucleophosmin in macrophage differentiation and activation. Blood 2011; 118:4694–704; PMID:21876121; http://dx.doi.org/10.1182/blood-2011-03-341255
- Zhang Y, Morgan MJ, Chen K, Choksi S, Liu ZG. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012; 119:2895–905; PMID:22223827; http://dx.doi.org/10.1182/blood-2011-08-372383
- Noronha-Matos JB, Costa MA, Magalhaes-Cardoso MT, Ferreirinha F, Pelletier J, Freitas R, Neves JM, Sévigny J, Correia-de-Sá P. Role of ecto-NTPDases on UDP-sensitive P2Y(6) receptor activation during osteogenic differentiation of primary bone marrow stromal cells from postmenopausal women. J Cell Physiol 2012; 227:2694–709; PMID:21898410; http://dx.doi.org/10.1002/jcp.23014
- Milosevic J, Brandt A, Roemuss U, Arnold A, Wegner F, Schwarz SC, Storch A, Zimmermann H, Schwarz J. Uracil nucleotides stimulate human neural precursor cell proliferation and dopaminergic differentiation: involvement of MEK/ERK signalling. J Neurochem 2006; 99:913–23; PMID:17076658; http://dx.doi.org/10.1111/j.1471-4159.2006.04132.x
- Orriss IR, Wang N, Burnstock G, Arnett TR, Gartland A, Robaye B, Boeynaems JM. The P2Y(6) receptor stimulates bone resorption by osteoclasts. Endocrinology 2011; 152:3706–16; PMID:21828185; http://dx.doi.org/10.1210/en.2011-1073
- Chatterjee C, Sparks DL. Extracellular nucleotides inhibit insulin receptor signaling, stimulate autophagy and control lipoprotein secretion. PloS One 2012; 7:e36916; PMID:22590634; http://dx.doi.org/10.1371/journal.pone.0036916
- de The H, Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer 2010; 10:775–83; PMID:20966922; http://dx.doi.org/10.1038/nrc2943
- Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARalpha oncoprotein. Autophagy 2011; 7:401–11; PMID:21187718; http://dx.doi.org/10.4161/auto.7.4.14397
- Auberger P. BCR-ABL/p62/SQSTM1: a cannibal embrace. Blood 2012; 120:3389–90; PMID:23100300; http://dx.doi.org/10.1182/blood-2012-08-451492
- Ablain J, Nasr R, Bazarbachi A, de The H. The drug-induced degradation of oncoproteins: an unexpected Achilles' heel of cancer cells? Cancer Dis 2011; 1:117–27; PMID:22586354; http://dx.doi.org/10.1158/2159-8290.CD-11-0087
- Lalic H, Dembitz V, Lukinovic-Skudar V, Banfic H, Visnjic D. 5-Aminoimidazole-4-carboxamide ribonucleoside induces differentiation of acute myeloid leukemia cells. Leuk Lymphoma 2014; 55:2375–83; PMID:24359245; http://dx.doi.org/10.3109/10428194.2013.876633
- Braun T, Itzykson R, Renneville A, de Renzis B, Dreyfus F, Laribi K, Bouabdallah K, Vey N, Toma A, Recher C, et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood 2011; 118:3824–31; PMID:21828134; http://dx.doi.org/10.1182/blood-2011-05-352039
- Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Gattermann N, Sanz G, List A, Gore SD, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009; 10:223–32; PMID:19230772; http://dx.doi.org/10.1016/S1470-2045(09)70003-8
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445–544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496