
ABSTRACT
Small molecule therapy is a critical component of targeted anticancer treatment, with tyrosine kinase inhibitors (TKIs) being the first compounds to treat the clonal Chronic Myelogenous Leukaemia (CML) translocation t (9;22) (q34; q11) effectively since 2001. TKIs, such as imatinib, have improved the 10-year survival rate of CML patients to 80%. They bind the BCR::ABL1 kinase and inhibit downstream signaling pathways. However, therapy failure may be seen in 20-25% of CML patients due to intolerance or inadequacy related to BCR::ABL1 dependent or independent mechanisms. This review aimed to summarize current treatment options involving TKIs, resistance mechanisms and the prospective approaches to overcome TKI resistance. We highlight BCR::ABL1-dependent mechanisms of TKI resistance by reviewing clinically-documented BCR::ABL1 mutations and their consequences for TKI binding. In addition, we summarize BCR::ABL1 independent pathways, including the relevance of drug efflux, dysregulation of microRNA, and the involvement of alternative signaling pathways. We also discuss future approaches, such as gene-editing techniques in the context of CML, as potential therapeutic strategies.
1. Introduction
Chronic myelogenous leukaemia (CML) is associated with the presence of the Philadelphia (Ph) chromosome, resulting from a reciprocal chromosomal translocation between chromosome 22 and chromosome 9. In this translocation, regions from the first exon of ABL1 (c-Abl) are translocated into the BCR gene, generating a fusion oncogene referred to as BCR::ABL1 [Citation1]. This fusion gene encodes for a hybrid truncated protein with uncontrollable tyrosine kinase activity. It accounts for approximately 95% of all CML cases [Citation2].
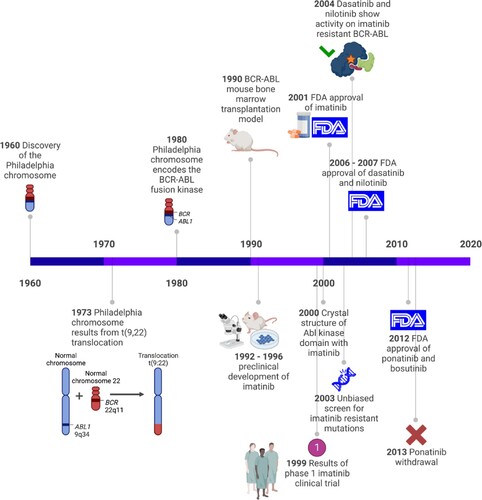
For many years, CML was considered a fatal disease that could solely be cured through hematopoietic stem cell transplantation [Citation3]. However the discovery of Imatinib which targets BCR::ABL1 kinase activity, the CML 10-year survival rates was found to be improved to 83.3% [Citation4] and tyrosine kinases became feasible targets for anti-cancer therapy. A timeline that highlights some of the important breakthroughs that led to the development of BCR::ABL1 TKIs is depicted in .
Figure 1. Key events in the development of BCR::ABL1 TKIs. A timeline highlighting significant research breakthroughs that led to the development of TKIs for CML treatment.
As homologs of adenosine triphosphate (ATP), tyrosine kinase inhibitors (TKIs) occupy the ATPs-binding site of protein tyrosine kinases (PTKs) competitively and block PTK-mediated signaling pathways, reducing the growth and proliferation of cancer cells. TKIs have substantial advantages over conventional chemotherapeutic drugs, including high efficacy, low toxicity, and good specificity. However, therapeutic resistance may occur [Citation5]. In fact, TKI resistance is the major cause of recurrence, progression, therapy failure, low compliance, and death. Resistance mechanisms to TKIs are complicated. Previous research has confirmed the association between TKI resistance and mutations in the target gene. Growing research shows that other variables, such as epigenetic alterations and activation of alternative survival pathways, also contribute to TKI resistance [Citation6].
Despite the extraordinary success of TKIs in targeted therapy, TKI resistance is on the rise. Here, we summarize the underlying mechanisms of TKI resistance and discuss potential approaches to overcome TKI resistance, and future treatment options aiming to establish a theoretical foundation for CML management.
2. Current treatment strategies: TKIs in CML
2.1. Overview of TKIs in CML
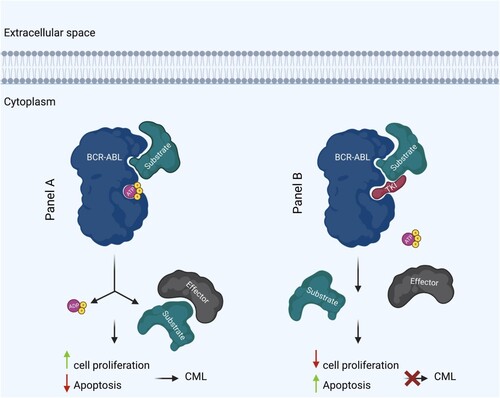
Ph + cells have a constitutively active BCR::ABL1 gene, which causes malignant CML formation. Due to the structural similarity between TKIs and ATPs, TKIs can bind the tyrosine kinase domain of BCR::ABL1 and prevent ATP from binding to the ATP binding domain, thus blocking downstream target protein phosphorylation and signaling pathway activation, inhibiting tumor growth (). Through this mechanism, imatinib, among other TKIs, targets CML and other cancers [Citation7]. A detailed list of currently available TKIs for the treatment of CML is presented in Table S1.
Figure 2. Mechanism of action of BCR::ABL1 tyrosine kinase and TKI in CML. BCR::ABL1 tyrosine kinase in CML (left). TKI in treatment of CML (right). P indicates phosphate. Figure was created with BioRender.com.
Besides BCR::ABL1, imatinib interacts with several other proteins, including ABL1 and ABL2. Suppressing both ABL paralogs has been reported to contribute to imatinib adverse effects. Side effects, such as nausea, vomiting, eczema, leukopenia (in severe cases), heart failure, and liver disease, are among the most frequent side effects of TKI treatment [Citation8]. Compared to conventional chemotherapeutic drugs, these side effects are less severe, and no definitive contraindications or life-threatening issues have been observed. Nevertheless, some patients may experience specific side effects that result in therapy interruption or termination. In fact, research has shown that up to 10% of patients may discontinue TKI treatment due to side effects. In addition, as a manifestation of TKIs resistance, some patients may exhibit weak response to TKIs, and nearly a quarter of CML would gradually lose their cytogenetic or substantial molecular response after five years of treatment [Citation9–11]. TKIs resistance results in recurrence or rapid tumor progression, which has a detrimental effect on patient survival. Therefore, finding TKI-resistance solutions is an urgent priority.
Second and third-generation TKIs were designed to address this challenge. To date, the second-generation TKIs (dasatinib, nilotinib, and bosutinib), and the third-generation ponatinib have been developed for CML treatment with different side effects, targets, potencies, and effectiveness against different mutations in BCR::ABL1 (Table S1). Nilotinib, for example, is 20 times more potent than imatinib and poses a lower risk of arterial occlusive events (AOEs) in dyslipidemic CML patients via lowering both LDL and cholesterol plasma levels but may cause cardio-vascular events (CVEs) in 20% of patients via binding the Mitogen Activated Protein (MAP)-kinases in comparison to the (5%) incidence of CVEs seen with imatinib.
On the other hand, the second-generation TKI, dasatinib, is likely to induce pleural effusion toxicity in ∼30% of patients [Citation12]. Moreover, bosutinib (SRC/ABL inhibitor) was designed to inhibit SRC in SRC-overexpressing tumors and was found to have high activity against ABL and BCR::ABL1 [Citation13] It binds the inactive state of BCR::ABL1, namely the DFG-out motif independent of its conformation. It may lead to increased transaminase levels, and is associated with transient diarrhea in ∼30% of patients [Citation14]. Ponatinib, the only approved third-generation TKI, is utilized for first and second-generation resistant diseases due to the T315I mutation [Citation15]. Its most adverse events occur during therapy, with cardiovascular damage and cardiovascular risk factors accounting for 30% of contraindications [Citation16].
2.2. TKI resistance
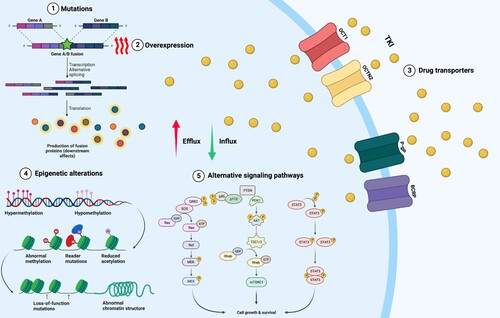
Within this complex interplay between different cells; CML, LSCs, drugs; TKIs, and homeostatic microenvironment, resistance to therapy develops. Drug resistance mechanisms have been thoroughly explored and are either BCR::ABL1 dependent or independent. A fundamental understanding of the tyrosine kinase domain of BCR::ABL1 and mechanisms of resistance are summarized in .
Figure 3. Mechanisms of TKI resistance in CML. BCR::ABL1 dependent mechanisms include (1) mutations that affect TKI binding. (2) overexpression or amplification of BCR::ABL1 leading to elevated kinase activity. BCR::ABL1 independent mechanisms include (3) increased expression of drug efflux pumps, including P-glycoprotein (Pgp), and reduced expression of drug influx pumps, including the organic cation transporter hOCT1. Other mechanisms that play a role BCR::ABL1 independent TKI resistance include (4) epigenetic alterations and (5) activation of alternative signaling pathways downstream of BCR::ABL1. Figure was created with BioRender.com.
2.2.1. BCR::ABL1-dependent TKI resistance mechanisms
2.2.1.1. Point mutations in BCR::ABL1
According to the types of mutations that confer resistance to a particular TKI medication, TKIs are classified into two primary groups, both of which overlap with the ATP-binding site. Type 1 inhibitors target the active conformation, which is catalytically competent, whereas type 2 inhibitors target the inactive conformation. Dasatinib is a type I inhibitor, whereas Imatinib, nilotinib, ponatinib, and ponatinib are type II inhibitors. Bosutinib has traits of both kinds of TKIs [Citation17]. Type II inhibitors have better selectivity but are subjected to larger mutational vulnerabilities due to their strict binding requirements [Citation18], whereas type I inhibitors are often less prone to mutational escape and more promiscuous. Despite being less prone to mutational escape, type I inhibitors are more promiscuous than type II inhibitors.
Imatinib has to engage with the activation domain and P-loop conformation switch and make six hydrogen bonds with it to prevent the transition from the inactive to active conformation of the ATP binding pocket of BCR::ABL1. The effectiveness and safety of the drug may thus be significantly impacted by mutations that change the required amino acids. It is known that approximately 50% of patients who relapse on imatinib have mutations within the ABL kinase domain, affecting imatinib binding within the kinase pocket [Citation19].
When imatinib was still in the early stages of development, it was reported that BCR::ABL1 might escape inhibition via substitution of threonine for isoleucine at position 315 (T315I), which was the first identified mutation associated with drug resistance [Citation20]. Later, however, it was shown that, considering the disease course, the sooner the TKI treatment is started, the lower the relapse rate and the degree of genetic instability responsible for mutation acquisition. In other words, although TKI-resistant mutations continue to be a problem in patients with accelerated-phase CML (AP-CML) and blast-phase CML (BP-CML), they are considerably less common in patients with CP who are appropriately treated with TKIs as early as possible. Being the target of a key mutation (T315I), Threonine 315 residue was subsequently named ‘the gatekeeper’ residue because of its strategic placement in the ATP-binding pocket, which allows it to regulate the accessibility of the pocket. While in contact with imatinib, the hydroxyl group of the amino acid threonine 315 binds to the inhibitor via a hydrogen bond, and the side chain at position 315 allosterically regulates the inhibition of the inhibitor’s binding to hydrophobic regions adjacent to the ATP-binding site. Thus, imatinib binding is eliminated [Citation21,Citation22].
Imatinib failure can also result from mutations in the P-loop, such as G250E or Y253H, which impair imatinib binding, or in the activation loop, such as H396R, which prevents the activation loop from being closed [Citation23]. Additional BCR::ABL1 mutations in CML patients have been found by our team, including the E459K mutation, which manifested in one patient with a poor response to TKI [Citation24].
Dasatinib, nilotinib, and/or bosutinib have activity against the majority of imatinib-resistant mutants, except T315I. Although the development of a TKI active against the T315I mutant has proven challenging, ponatinib (AP24534), a third-generation TKI and a pan BCR::ABL1 inhibitor, seems to induce potent and durable responses in CML patients, regardless of their mutational status. It binds to the ATP binding domain without requiring the formation of a hydrogen bond with the mutation T315I. Ponatinib was tested in the PACE clinical trial in patients with the T315I mutation or who are resistant/intolerant to either dasatinib or nilotinib. Findings from PACE show that major molecular response (MMR) is achieved in 56% of CP patients with the T315I mutation. Nevertheless, the presence of this mutation necessitates an increase in the ponatinib dose, and a proportion of patients might ultimately develop ponatinib-resistance. Therefore, it is crucial to categorize patients in accordance with their BCR::ABL1 mutation pattern to select the most effective TKI for treatment.Conflicting results have been reported regarding the involvement of many of the discovered BCR::ABL Kinase Point Mutations in TKI resistance [Citation25]. This makes it challenging to identify the most appropriate treatment course for each patient, which further emphasize the need for continued research in this area to improve the effectiveness of treatment options for CML patients. This will not only help to identify the mutations responsible for resistance but also enable the development of new therapies and treatment strategies that target these mutations effectively. The most common BCR::ABL1 mutations and how they affect individuals with CML’s responsiveness to TKIs are shown in Table S2. Our group previously documented BCR::ABL1 mutation and additional chromosomal abnormalities in CML patients [Citation26,Citation27].
2.2.1.2. Amplification of BCR::ABL1 oncogene
BCR::ABL1 dependent mechanisms of resistance include amplification or overexpression of the BCR::ABL1 oncogene. High BCR::ABL1 transcript levels in the advanced stage have been reported to be linked to TKI resistance [Citation28,Citation29], mainly because BCR::ABL overexpression can impair TKI binding [Citation30], and thus lead to elevated kinase activity [Citation20,Citation31]. Despite the increased rate of amplification per cell division, it is more common to identify point mutations in a clinical context, mainly because overexpression of BCR::ABL1 promotes cell injury [Citation32].
2.2.2. BCR::ABL1 -Independent TKI resistance mechanisms
Besides alterations in BCR::ABL1, it is plausible that resistance occurs independently of the kinase, which may involve mechanisms related to genomic instability, epigenetic alterations, overexpression of multidrug resistance (MDR) proteins involved in the efflux of anti-cancer drugs, activation of survival pathways, and interactions with the LSC microenvironment.
2.2.2.1. Impact of drug metabolism
Pharmacogenetic variants in cytochrome P450 enzymes have been linked to TKI resistance CML. TKIs are cytochrome P450 substrates, namely CYP3A4 and CYP3A5 [Citation33] (Table S3). Consequently, it is reasonable to predict drug–drug interactions with a broad variety of drugs to occur, resulting in the activation or inhibition of TKI metabolism. These treatments include rifampicin, anticonvulsants, and herbal remedies, all of which are known to activate certain cytochrome P450 enzymes. As a consequence of increased TKI metabolism, the desired TKI plasma concentration is reduced, resulting in chemoresistance [Citation34]. Imatinib, for example, is both a moderate CYP3A4 inhibitor and a substrate [Citation35. Imatinib is converted by CYP3A4 to the active but less cytotoxic metabolite CPG74588 [Citation36]. Autoinhibition of CYP3A4 leads to the identification of a second elimination route, CYP2C8, which is involved in the hepatic clearance. Furthermore, CYP3A4 and CYP3A5 enzyme activity have been demonstrated to be greater in CML patients who achieve full molecular remission compared to poor responders . Because the potency of the significant metabolite CGP74588 is equivalent to that of imatinib, CYP3A and its products have demonstrated therapeutic significance in imatinib-treated-CML patients [Citation37].
In the presence of pharmacogenetic differences, enzyme activity may be restricted or enhanced, which may have an impact on TKI metabolism. The most clinically significant polymorphisms include CYP3A4*20 and CYP3A4*22, both of which may result in a reduction of up to 20% of enzyme activity [Citation38,Citation39]. CYP3A4 and CYP3A5 have a high degree of sequence similarity, and their substrate spectra overlap [Citation40]. The major variations of CYP3A5 include nonfunctional CYP3A5*3, CYP3A5*6, and CYP3A5*7, which vary in their expression patterns across ethnicities [Citation38]. Patients with CML who have known CYP3A4 polymorphisms may have decreased TKI metabolism, resulting in greater side effects, but their response to TKI may be enhanced. However, there is contradictory evidence regarding the significance of CYP450 variations in TKI responsiveness.
Contrary to expectations of an enhanced imatinib response in the presence of a non-functional protein, multiple investigations have shown that CYP3A5*3-harbouring patients had a worse response to the drug [Citation41]. On the other hand, a link between imatinib therapy and greater complete cytogenetic response rates has been observed in some populations, including Asians [Citation42]. Although the relationship between the TKI response and many metabolism-related genetic variants has not been fully established, some evidence suggests that CYP2C8*2 and CYP3A4*7 play a role in alterations in imatinib levels in homozygous carriers [Citation43,Citation44]. Table S4 summarizes the major genetic variations in cytochrome P450 enzymes and their relevance to TKI response in CML.
CYP3A4 interactions have a more significant impact on the development of adverse events or the inability to react to TKIs [Citation45]. A consequence of this is that genotyping and measuring cytochrome P450 activity or function are no longer routinely performed in clinical practice [Citation45]. Therefore, strong CYP3A4 inducers should be avoided when beginning the imatinib treatment [Citation45]. Patients who must be co-administered with a strong CYP3A4 inducer should have their imatinib dosage raised by ∼50%, and their clinical response should be closely monitored. Because the concomitant treatment of imatinib and strong CYP3A4 inducers may result in a reduction in overall exposure to IM, alternative agents should be investigated [Citation46]. A substantial increase in imatinib exposure may occur when imatinib is administered concurrently with strong CYP3A4 inhibitors, on the other hand. Imatinib will raise the plasma levels of medicines that are metabolized by CYP3A4. As a result, care should be used when giving imatinib in conjunction with CYP3A4 substrates that have a limited therapeutic window. Patients should be given LMWH instead of warfarin when imatinib is administered, because warfarin is metabolized by CYP2C9 and CYP3A4, and because imatinib interacts with medications metabolized by CYP2D6 and has a limited therapeutic window. Other factors such as patients' adherence, it was found that high rate of TKIs failure is explained by poor adherence [Citation47,Citation48].
2.2.2.2. Impact of drug transporters
There is evidence that drug transporters contribute to drug resistance by either decreasing drug concentrations inside cells or limiting a drug’s bioavailability in certain tissues. Many drug efflux transporters and drug importers have been linked to TKI resistance in CML (Table S3, S4).
The efflux proteins belonging to the ATP-binding cassette (ABC) superfamily are represented by 49 genes, of which some ABC members are associated with CML, among other diseases [Citation49]. Particularly, members of the ABCB, ABC subfamilies (Table S4).
In recent years, ABCB1 has emerged as one of the most extensively researched drug transporters, with pharmacogenetic variants being investigated in considerable depth (Table S3, S4). The ABCB1/pump P-glycoprotein (Pgp) was first described by Juliano and Ling [Citation50], as a 170-kDa cell surface glycoprotein which showed a membrane permeability barrier ‘function’ in drug-resistant cells. Studies have shown that Pgp, play an important role in drug efflux, by pumping out a wide range of substrates, including TKIs [Citation51]. It is conceivable that TKI responsiveness will be enhanced by loss-of-function mutations or variants with lower protein function, resulting in decreased efflux ability. Notwithstanding, 40 years after the discovery of Pgp [Citation52], there is still no solid confirmation regarding the role of ABCB1/Pgp in drug resistance. Most importantly, ongoing controversy surrounds the relevance of ABCB1 polymorphisms in the development of TKI resistance among CML patients, with no compelling evidence that ABCB1 variants may be utilized as predictive biomarkers in pharmaceutical therapy. As a result, we believe it is important to review the pre-clinical approaches involving ABCB1 expression, activity, and genetic variants in CML to better understand the role of ABCB1 transporters in TKI resistance among CML patients in a clinical context. While P-gp inhibitors can help overcome drug resistance, they can also significantly increase the toxicity of P-gp substrate cytotoxics [Citation53]. Thus, studying TKI-transporter interactions may assist in discovering elusive TKI resistance mechanisms, guide the creation of combination scheduling trials, or reduce undesirable side effects ].
Additionally, ABCG2 is a drug efflux transporter and is extensively expressed in hematopoietic precursors and stem cells. ABCG2 is a stem cell factor that is highly expressed in hematopoietic precursors and stem cells. Several studies have shown that ABCG2 is linked to the development of leukaemia [Citation54]. In regard to changing the transport capacity of the protein, ABCG2 polymorphisms are addressed in the same manner as ABCB1 polymorphisms are discussed to alter the protein’s transport capacity. One of the most essential variants is 34G > A (V12M, exon 2, rs2231137), and the other is 421C > A (V12M, exon 2, rs2231137) (Q141K, rs2231142) (Table S3). The homozygous 34G > A mutation, which results in an amino acid switch from valine to methionine, has been associated to a greater response to imatinib, perhaps due to decreased ABCG2 expression . Evidence on the effects of 421C > A, which is believed to change the conformation of the ATP binding domain remains inconsistent. The expression of the variant was shown to decrease imatinib bioavailability in some studies, while others indicated that it had no impact on imatinib pharmacokinetics in vivo [Citation55–57]. Nonetheless, Jiang and colleagues suggested that this variation might be utilized to predict imatinib response in individuals with chronic lymphocytic leukaemia [Citation58]. The −15,622C > T promoter SNP (rs7699188) has also been linked to lower BCRP expression in a number of tissues, including the liver, suggesting that imatinib clearance from the cell is reduced [Citation59]. Additional ABCG2 variants were examined, but they did not affect TKI clearance or responsiveness, as previously reported []. As a result, there is no apparent connection between pharmacogenetic polymorphisms and imatinib response for either ABCB1 or ABCG2, and further study is required to determine their relevance in drug resistance [Citation44]. Many studies have revealed that the expression of ABCB1 and ABCG2 in drug resistance seems to be dose-dependent. Following imatinib cessation, ABCG2 expression in peripheral blood leukocytes was shown to be a good predictor of treatment-free remission. More study is needed to establish the effect of ABC transporter polymorphisms on neoplastic disorders like CML. In our investigation, there was no significant pattern of AGP and PGP expression, regardless of whether the patient responded to or was resistant to medication. AGP and PGP levels were not linked with resistance in our CML patient group, which was unanticipated [Citation60–62].
Some TKIs are believed to be transported into tumor cells via the organic cation transporter 1 (OCT1/SLC22A1), found on the surface of tumor cells. Although there are contradicting data regarding the significance of OCT1 in CML, some studies have demonstrated overexpression of OCT1 in patients with imatinib resistance [Citation63], while others have shown that OCT1 is not regulated [Citation64,Citation65]. It was found that OCT1 expression and activity may be used as a predictive predictor for long-term imatinib response in patients with CML [Citation66]. However, no evidence that OCT1 variation impacts imatinib response in various clinical studies is available to date [Citation66].
2.2.2.3. Impact of protein-tyrosine phosphatases
Among the TKI resistance mechanisms, there has been a growing body of evidence regarding the mutual interplay between protein-tyrosine phosphatases (PTP) receptor type γ (PTPRG) and BCR::ABL1 [Citation67]. Our group previously documented that the expression level of PTPRG was considerably higher among optimal TKI responders compared to non-responders. Also, we reported that the protein level of PTPRG is drug dependent [Citation68], suggesting that approaches aiming at enhancing PTPRG expression/activity might benefit CML patients [Citation69,Citation70].
Our team also identified an antagonistic relationship between β-catenin and PTPRG in CML cells [Citation71,Citation72]. Our findings showed that PTPRG can dephosphorylate β-catenin, causing cytosolic instability and protein degradation. Furthermore, we found that increased β-catenin expression is associated with DNA (cytosine-5)-methyl transferase 1 (DNMT1) overexpression, whereas its inhibition or down-regulation is associated with PTPRG re-expression [Citation71,Citation72]. Furthermore, we found that PTPRG plays a role in controlling BCR::ABL1 and β-catenin phosphorylation in primary human CML samples. These findings suggest a regulatory loop involving PTPRG and β-catenin, the imbalance of which alters the kinetics of the CML [Citation73]. In addition, we recently studied the impact of PTPRG SNPs on TKI response prediction [Citation68,Citation74]. We discovered three novel SNPs (c.1602 1603insC, c.85 + 14412delC, and c.2289-129delA). Compared to responders and healthy persons, these SNPs had significantly different genotypes and allele frequencies in CML patients who failed imatinib therapy [Citation74,Citation75]. Other phosphatases have been reported to be involved in CML pathogenesis [Citation76].
2.2.2.4. Epigenetic alterations
Epigenetic factors play a role in regulating gene expression. There has been growing body of evidence that aberrant DNA methylation plays a key role in CML progression. Our group previously reported that methylated CpG sites were dramatically increased in CML patients compared to healthy subjects, suggesting the potential involvement of CpG methylation sites in the molecular processes leading to CML. These results support the hypothesis that CML etiology may involve a combination of factors, not only the BCR::ABL1 translocation [Citation73,Citation77].
Furthermore, microRNA expression and epigenetic control may have a role in the pathogenesis of CML and treatment resistance. The presence of TKIs in CML patients’ blood samples has been found to affect the microRNA expression pattern. Furthermore, the global microRNA expression pattern appears to vary between responding and non-responding CML patients [Citation78–80]. In addition, it has been reported that the dysregulation of some microRNAs, such as miR-203 or −30a/e, may lead to an altered TKI response [Citation81–83]. Even if BCR::ABL1 is not active, microRNAs such as miR-144/451 or miR-212/ABCG2 may be engaged in regulating several downstream target genes in the signaling cascade, thus contributing to drug resistance []. As a result, it has been debated whether microRNA expression may be utilized for monitoring TKI therapy response [Citation84].
2.2.2.5. Alternative pathways
The activation of the BCR::ABL1 fusion protein has been reported to trigger the PI3KAkt, JAK-STAT, and MAPkinase signaling pathways, among others, leading to enhanced cell proliferation, antiapoptotic signaling, alterations in cell motility, and changes in stromal cell adhesion [Citation85]. Many of the signaling proteins in these pathways function as BCR::ABL1 activation-dependent oncogenes. When different stimuli activate these pathways in cancer cells, such as WNT/ β-catenin signaling in leukemic stem cells or JAK2 activation by external stimuli, the tumor cell becomes at least partially independent of BCR::ABL1, potentially facilitating therapy failure or low response rates. Many signaling system adaptations come not just from mutations downstream of BCR::ABL1 or in alternate signaling pathways but also from changes in gene transcription. Autophagy activates or reactivates proliferative pathways such as hedgehog and PI3K/Akt signaling. Discovering and combining critical signaling pathways to design an optimal target to overcome resistance and cause synthetic lethality, particularly in leukemic stem cells, is a significant issue. TKIs promote mutations, clonal evolution, and selection, all of which contribute to the progression of CML. Still, they also promote TKI resistance and, as a result, the alteration of treatment regimens, given that TKIs must be taken for the rest of one’s life. In most subjects who failed to respond to imatinib, switching to newer generation TKIs in compliance with the recommendations resulted in positive outcomes.
2.2.2.6. TKIs and leukemic Stem Cells (LSCs)
CML leukemic stem cells (LSCs) represent a low-frequency subpopulation of leukaemia cells that are able to self-renew and are drug-resistant. Although they seem to express BCR::ABL1, they can escape the TKIs effect and increase the chances of relapse [Citation86], even after BCR::ABL1 activity has been shut down, and thus posing as a BCR::ABL1 -independent TKI resistance mechanism [Citation87–90]. To overcome quiescence and induce an LSC-free state, combined strategies to target LSCs are currently being evaluated, such as the combination of the anti-TNFα antibody infliximab (IFX) with TKIs, to induce the susceptibility of LSC to TKIs [Citation91] and the combination of the CXCR4 antagonist BKT140 with TKIs to overcome the protective effect of bone marrow milieu [Citation90]. CML LSCs can also be targeted through signaling pathways such as Wnt/b-catenin by C82, Hedgehog by LDE225, JAK/STAT by Pioglitazone, among others. Moreover, LSCs could be targeted via BCL-2 and P53 modulation, targeting lysosomal mediated autophagy by hydroxychloroquine or Lys05. Epigenetic targeting by EZH2 histone methyltransferase inhibitor and targeting LSCs via surface markers are potential strategies to achieve LSC treatment-free remissions [Citation90].
3. Future strategies
3.1. Discontinuation of TKI Therapy in CML
TKI-treated CML patients have a relative survival rate that is on par with the general population [Citation92]. Although TKI therapy is generally safe and effective, there may be certain risks associated with long-term use of these medications. Patients regularly encounter low-grade, persistent adverse events that are common but may have an effect on their overall health. Additionally, late adverse effects such arterio-thrombotic events [Citation93] and the potential elevated risk of second malignancies identified in some [Citation93] reports are becoming more recognized. The financial burden that prolonged TKI treatment places on patients and society is another factor that is becoming more significant [Citation94]. Several studies have been conducted to determine if TKIs may be safely discontinued in individuals who have established long-term deep molecular responses or patients with less deep but stable remission [Citation95–99]. Currently, only patients with at least 5 years of therapy, 2 years of deep molecular response, no past history of resistance or suboptimal/warning response, no hematopoietic stem cell transplantation with a typical BCR::ABL1 transcript, and aged ≥ 18 years may be considered for treatment discontinuation [Citation100]. Many aspects, however, still remain unaddressed, such as how to more accurately identify individuals who will have deep molecular responses and treatment-free remission, and if these people should be deemed ‘cured’? How can we help more patients experience deep molecular response so they will be ready to try a treatment-free remission trial? Moreover, the effects of gradual treatment withdrawal and whether it’s feasible in patients with less deep but stable remission is a promising strategy but requires further investigation [Citation99,Citation101,Citation102].
3.2. TKI dose individualization
Drug personalization is an ultimate goal for doctors to reach; the aim is to tailor patient therapy to optimize efficacy and decrease toxicity. There are currently several ways of individualization such as therapeutic drug monitoring, population pharmacokinetics, selecting antibiotics according to bacterial culture sensitivity, and pharmacogenomics. Nowadays, building simulation models using pharmacometrics science provides a great tool for health care workers to integrate information from different domains into one model to make medical decisions. Different techniques are currently used to personalize TKI dose in clinical practice [Citation103].
Currently, the most common tool for dose individualization is therapeutic drug monitoring (TDM) [Citation103]. TDM is involved in measuring drug levels in biological fluids at steady to improve patient care. TDM has been shown to be useful in predicting the efficacy of imatinib in CML [Citation104]. Multiple studies have shown that imatinib (Trough level) 1,000 ng/mL is currently considered a target to reach. This level (1000 ng/mL) in CML patients is considered a guarantee for a higher chance of attaining a complete cytogenetic response and reaching MMR. Nevertheless, further research is needed to determine its validity with other TKIs [Citation105,Citation106].
Moreover, pharmacogenetics (PGx) is important for dose personalization in cancer patients. Worldwide, there are few organizations responsible for guideline development and release, such as the French National Network of Pharmacogenetics (RNPGx) and the Clinical Pharmacogenetics Implementation Consortium (CPIC). Currently, more than 10 chemotherapy drugs have clinical PGx guidelines, but TKI is not one of them. Although there are different pharmacogenetic variants that influence TKI efficacy and toxicity, clinical PGx guidelines are still not available for TKI due to contradicting data [Citation107,Citation108].
In addition to PGx and TDM, other dose personalization techniques are evolving, such as pharmacometrics or population pharmacokinetics models (PPK). It has been previously reported that there is variation in imatinib disposition after administration of the standard dose [Citation109–113]. The patients’ characteristics that may impact the interindividual imatinib variability were identified by those studies [Citation109–113]. Those reported variables included chronic imatinib exposure, genetic polymorphisms, age, and weight. Population pharmacokinetics models can identify the different variables that affect patient drug response variability [Citation109–113]. A recent study on the Nigerian population reported wide variability in the imatinib area under the curve and clearance compared to other studies done on different populations [Citation113]. The genetic and Pk results can help optimize patients’ imatinib doses. Most importantly, they will be a powerful tool in describing those variabilities if combined in one mathematical model [Citation114]. After building the model, it can predict the patient optimized dose.
3.3. Gene editing
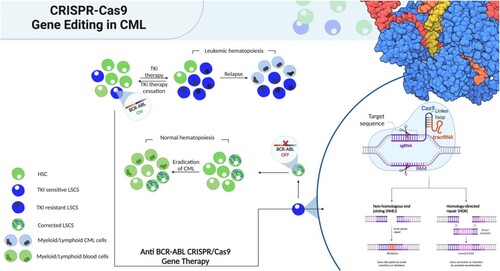
Over the past few years, gene editing techniques, particularly CRISPR/Cas9 have gained considerable attention in the context of leukaemia [Citation115–120]. CRISPR/Cas9 system was reported to successfully silence the BCR::ABL1 oncogene, reversing its tumor-promoting function [Citation119]. In addition, in a CML xenograft model, subcutaneous injection of edited single cell derived clones failed to develop tumors [Citation119]. These findings suggest that CRISPR/Cas9 technology could serve as a successful promising new therapeutic option for CML patients who experience TKI resistance. However, it should be noted that studies on CML specifically are limited, and further research is needed. A precision medicine approach utilizing clinically validated biomarkers and population health analyses would aid in the successful implementation of gene therapy for CML [Citation121–123]. illustrates the comparison between TKI therapy and gene therapy for CML, where gene therapy can eradicate the oncogene at the genome level and replenish the bone marrow niche with corrected LSCs, enabling regular hematopoiesis.
Figure 4. Gene therapy vs conventional therapy for CML. Conventional therapy based on tyrosine kinase inhibitors (TKI) effectively silences BCR::ABL1 in leukemic stem cells (LSCs). Because there are still cells that are BCR::ABL1 positive after treatment has ended, recurrence is possible. Recurrence of the disease may result from the emergence of TKI-resistant LSCs during therapy. However, the oncogene would be eradicated at the genome level via anti- BCR::ABL1 gene therapy. Corrected LSCs could replenish the bone marrow niche, enabling regular hematopoiesis. Figure was created with BioRender.com.
Conclusion
Despite considerable advances in CML treatment with the development of TKIs, many patients acquire TKI resistance, some progress to blast crisis, and the majority relies on TKI therapies for long-term disease management. Current strategies to overcome TKIs resistance have mostly focused on increasing the potency and specificity of drugs that target BCR::ABL1 or overcoming resistance caused by mutations in the BCR::ABL1 oncogene. This strategy, however, may be less successful in many CML patients who acquire resistance despite effective BCR::ABL1 suppression with TKIs, i.e. BCR::ABL1 -independent resistance mechanisms. To enhance treatment results in such individuals, novel therapeutic options, such as combining TKIs with other therapeutics that target BCR::ABL1 independent pathways, may be required. A combined therapy strategy might eliminate LSCs, maximizing treatment-free remission in CML and improving sensitivity to TKIs in patients who are currently unresponsive to TKIs. Furthermore, CML patients who fail TKIs without evidence of kinase domain mutations should be screened for alternative mechanisms of resistance that can activate alternative survival pathways in order to determine an appropriate therapeutic approach. Most importantly, novel therapeutic strategies such as gene editing techniques need to be further explored in the context of CML as potential therapeutic strategies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Rowley JD. A New consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and giemsa staining. Nature. 1973;243:290–293. doi:10.1038/243290a0.
- Sandberg AA, Gemmill RM, Hecht BK, etal. The Philadelphia chromosome: a model of cancer and molecular cytogenetics. Cancer Genet Cytogenet. 1986;21:129–146.
- Nair AP, Barnett MJ., Broady RC, et al. Allogeneic hematopoietic stem cell transplantation is an effective salvage therapy for patients with chronic myeloid leukemia presenting with advanced disease or failing treatment with tyrosine kinase inhibitors. Biology of Blood and Marrow Transplantation. 2015;21(8):1437–1444. doi:10.1016/j.bbmt.2015.04.005.
- Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. New England Journal of Medicine. 2017;376(10):917–927. doi:10.1056/NEJMoa1609324.
- Rumjanek VM, Vidal RS, Maia RC. Multidrug resistance in chronic myeloid leukaemia: how much can we learn from MDR–CML cell lines? Biosci Rep. 2013;33; doi:10.1042/bsr20130067.
- Soverini S, Branford S, Nicolini FE, et al. Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leukemia. 2011;25(2):161–170.
- Nader A-D, Andrew J, Yassin MA, et al. Molecular monitoring of patients with Chronic Myeloid Leukemia (CML) in the state of Qatar: optimization of techniques and response to Imatinib. QScience Connect. 2014;2014(1).
- Kalmanti L, Saussele S, Saussele S, et al. Safety and efficacy of imatinib in CML over a period of 10 years: data from the randomized CML-study iv. Leukemia. 2015;29:1123–1132. doi:10.1038/leu.2015.36.
- Milojkovic D, Apperley J. Mechanisms of resistance to imatinib and second-generation tyrosine inhibitors in chronic myeloid leukemia. Clin Cancer Res. 2009;15:7519–7527. doi:10.1158/1078-0432.CCR-09-1068.
- Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971–979.
- Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575:299–309.
- Jain AG, Gesiotto QJ, Ball S, et al. Incidence of pleural effusion with Dasatinib and the effect of switching therapy to Bosutinib in patients with chronic phase CML. Blood. 2021;138(Supplement 1):1484–1484. doi:10.1182/blood-2021-152017.
- Keller G, Schafhausen P, Brummendorf TH. Bosutinib: a dual SRC/ABL kinase inhibitor for the treatment of chronic myeloid leukemia. Expert Rev Hematol. 2009;2:489–497. doi:10.1586/ehm.09.42.
- Cortes JE, Kim D-W, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783–1796. doi:10.1056/NEJMoa1306494.
- Zhou T, Commodore L, Huang WS, et al. Structural mechanism of the Pan-BCR-ABL inhibitor ponatinib (AP24534): lessons for overcoming kinase inhibitor resistance. Chem Biol Drug Des. 2011;77(1):1–11.
- E Dianne Pulte H, Lauren SL, Price R, et al. FDA approval summary: revised indication and dosing regimen for Ponatinib based on the results of the OPTIC trial. The Oncologist. 2022;27(2):149–157.
- Levinson NM, Boxer SG. Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain. PLoS One. 2012;7.
- Davis MI, Hunt JP, Herrgard S. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051.
- Quintas-Cardama A, Kantarjian HM, Cortes JE. Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control. 2009;16(2):122–131.
- Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–880.
- Chindler T, Bornmann W, Pellicena P, et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289:1938–1942.
- Wang D, Pan H, Wang Y. T315L: a novel mutation within BCR-ABL kinase domain confers resistance against ponatinib. Leuk Lymphoma. 2017;58:1733–1735.
- Valent P. Imatinib-resistant chronic myeloid leukemia (CML): Current concepts on pathogenesis and new emerging pharmacologic approaches. Biologics : targets and therapy. 2007;1(4):433–448.
- Al-Dewik NI, Jewell AP, Yassin MA, et al. Studying the impact of presence of point mutation, insertion mutation and additional chromosomal abnormalities in chronic myeloid leukemia patients treated with imatinib mesylate in the State of Qatar. QScience Connect. 2014;2014(1).
- Al-Dewik N I E. BCR-ABL Kinase Point Mutations don't Correlate with the Resistance of Chronic Myelocytic Leukemia (CML) to Imatinib Mesylate (IM); A Study on CML Patient Population in Qatar. 2011.
- Al-Dewik N, Jewell A, Yassin MA, et al. Molecular monitoring of patients with Chronic Myeloid Leukemia (CML) in the state of Qatar: optimization of techniques and response to Imatinib. QScience Connect. 2014;2014(1).
- Izz EA-DN. BCR-ABL kinase point mutations don't correlate with the resistance of Chronic Myelocytic Leukemia (CML) to Imatinib Mesylate (IM); a study on CML patient population in Qatar. Qatar Foundation Annual Research Forum Proceedings. 2011.
- Barnes D J, Palaiologou D, Panousopoulou E. BCR-ABL expression levels determine the rate of development of resistance to imatinib mesylate in chronic myeloid leukemia. Cancer Res. 2005;65:8912–8919.
- Hochhaus A, Kreil S, Corbin AS. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16(11):2190–2196.
- Jabbour E, Kantarjian H, Jones D. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia. 2006;20:1767–1773.
- Hochhaus A, Kreil S, Corbin AS. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16(11):2190–2196.
- Amarante-Mendes G P, Datoguia Rana A. BCR-ABL1 Tyrosine Kinase Complex Signaling Transduction: Challenges to Overcome Resistance in Chronic Myeloid Leukemia. Pharmaceutics. 2022;14(1).
- Haouala A, Widmer N, Duchosal MA, et al. Drug interactions with the tyrosine kinase inhibitors imatinib, dasatinib, and nilotinib. Blood. 2011;117:e75–e87. doi:10.1182/blood-2010-07-294330.
- Tian X, Zhang H, Heimbach T, et al. Effect of vemurafenib on the pharmacokinetics of a single dose of digoxin in patients with BRAFV600mutation-positive metastatic malignancy. J Clin Pharmacol. 2018;58:1067–1073. doi:10.1002/jcph.1111.
- Filppula A, Laitila J, Neuvonen P, et al. Potent mechanism-based inhibition of CYP3A4 by imatinib explains its liability to interact with CYP3A4 substrates. Br J Pharmacol. 2012;165:2787–2798. doi:10.1111/j.1476-5381.2011.01732.x.
- Mlejnek P, Dolezel P, Faber E, et al. Interactions of N-desmethyl imatinib, an active metabolite of imatinib, with P-glycoprotein in human leukemia cells. Ann Hematol. 2011;90:837–842. doi:10.1007/s00277-010-1142-7.
- Gréen H, Skoglund K, Rommel F, etal. CYP3A activity influences imatinib response in patients with chronic myeloid leukemia: a pilot study on in vivo CYP3A activity. European journal of clinical pharmacology. 2010;66(4):383–386.
- Werk AN, Cascorbi I. Functional gene variants of CYP3A4. Clin Pharmacol Ther. 2014;96:340–348. doi:10.1038/clpt.2014.129.
- Saiz-Rodriguez M, Almenara S, Navares-Gomez M, et al. Effect of the most relevant CYP3A4 and CYP3A5 polymorphisms on the pharmacokinetic parameters of 10 CYP3A substrates. Biomedicines. 2020;8; doi:10.3390/biomedicines8040094.
- Williams JA, Ring BJ, Cantrell VE, et al. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos. 2002;30:883–891. doi:10.1124/dmd.30.8.883.
- Harivenkatesh N, Kumar L, Bakhshi S, etal. Influence of MDR1 and CYP3A5 Genetic Polymorphisms on Trough Levels and Therapeutic Response of Imatinib in Newly Diagnosed Patients with Chronic Myeloid Leukemia. Pharmacol. Res. 2017;120:138–145.
- Cargnin S, Ravegnini G, Soverini S, et al. Impact of SLC22A1 and CYP3A5 genotypes on imatinib response in chronic myeloid leukemia: a systematic review and meta-analysis. Pharmacol Res. 2018;131:244–254. doi:10.1016/j.phrs.2018.02.005.
- Adehin A, Adeagbo BA, Kennedy MA, et al. Inter-individual variation in imatinib disposition: any role for prevalent variants ofCYP1A2,CYP2C8,CYP2C9, andCYP3A5in Nigerian CML patients? Lymphoma. 2019;60:216–221. doi:10.1080/10428194.2018.1466291.
- Kaehler M, Cascorbi I. Pharmacogenomics of impaired tyrosine kinase inhibitor response: lessons learned from chronic myelogenous leukemia. Front Pharmacol. 2021;12:696960.
- Pushpam D, Bakhshi S. Pharmacology of tyrosine kinase inhibitors in chronic myeloid leukemia; a clinician’s perspective. DARU J Pharm Sci. 2020;28(1):371–385.
- Hussaarts KG, Veerman GM, Jansman FG, et al. Clinically relevant drug interactions with multikinase inhibitors: a review. Ther Adv Med Oncol. 2019;11:1758835918818347.
- Nader A-D, Hisham M, Samara Muthanna M. Is adherence to Imatinib Mesylate treatment among patients with Chronic Myeloid Leukemia associated with better clinical outcomes in Qatar?. Clinical Medicine Insights: Oncology. 2016.
- Nader A-D, Hisham M. Ghasoub Rola, etal. Studying the Impact of Imatinib Mesylate (IM) Adherence in Chronic Myeloid Leukemia (CML) patients’ Responses in the State of Qatar. Blood. 2014;124(21):5537–5537.
- Lage H. An overview of cancer multidrug resistance: a still unsolved problem. Cell Mol Life Sci. 2008;65:3145–3167. doi:10.1007/s00018-008-8111-5.
- Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. 1976;455:152–162. doi:10.1016/0005-2736(76)90160-7.
- Klukovits A, Krajcsi P. Mechanisms and therapeutic potential of inhibiting drug efflux transporters. Expert Opin Drug Metab Toxicol. 2015;11:907–920. doi:10.1517/17425255.2015.1028917.
- Juliano R L, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. 1976;455:152–162.
- Anreddy N, Gupta P, Kathawala R, etal. Tyrosine Kinase Inhibitors as Reversal Agents for ABC Transporter Mediated Drug Resistance. Molecules. 2014;19:13848–13877.
- Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient hoechst 33342 efflux pump and Is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99:507–512. doi:10.1182/blood.V99.2.507.
- Skoglund K, Boiso Moreno S, Jönsson JI, etal. Single-nucleotide polymorphisms of ABCG2 increase the efficacy of tyrosine kinase inhibitors in the K562 chronic myeloid leukemia cell line. Pharmacogenetics and genomics. 2014;24(1):52–61.
- Gardner E, Burger H, Vanschaik R, et al. Association of enzyme and transporter genotypes with the pharmacokinetics of Imatinib. Clin. Pharmacol. Ther. 2006;80:192–201.
- Takahashi N, Miura M, Scott SA, etal. Influence of CYP3A5 and drug transporter polymorphisms on Imatinib trough concentration and clinical response among patients with chronic phase Chronic Myeloid Leukemia. J. Hum. Genet. 2010;55:731–737.
- Jiang ZP, Zhao XL, Takahashi N, et al. Trough concentration andABCG2polymorphism are better to predict imatinib response in chronic myeloid leukemia: a meta-analysis. Pharmacogenomics. 2017;18(1):35–56.
- Poonkuzhali B, Lamba J, Strom S, et al. Association of breast cancer resistance protein/ABCG2 phenotypes and novel promoter and intron 1 single nucleotide polymorphisms. Drug Metab. Dispos. 2008;36:780–795.
- Ai-Dewik NI, Jewell AP, Yassin MA, et al. Studying the impact of presence of alpha acid glycoprotein and protein glycoprotein in chronic myeloid leukemia patients treated with imatinib mesylate in the state of Qatar. Biomark Cancer. 2015;7:63–67.
- Nader A-D, Andrew J. Studying the impact of presence of Alpha Acid Glycoprotein and Protein Glycoprotein in Chronic Myeloid Leukemia patients treated with Imatinib Mesylate in state of Qatar. Blood. 2015;126(23):4846–4846.
- Eddin A-DNI. Elevation of Alpha Acid Glycoprotein (AGP) does not correlate with the resistance of Chronic Myeloid Leukaemia (CML) to Imatinib Mesylate (IM). 2011.
- White DL, Saunders VA, Dang P, et al. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but Not nilotinib (AMN107): reduced OCT-1 activity Is the cause of Low In vitro sensitivity to imatinib. Blood. 2006;108:697–704. doi:10.1182/blood-2005-11-4687.
- Davies A, Jordanides NE, Giannoudis A, et al. Nilotinib concentration in cell lines and primary CD34+ chronic myeloid leukemia cells is not mediated by active uptake or efflux by major drug transporters. Leukemia. 2009;23:1999–2006. doi:10.1038/leu.2009.166.
- Nies AT, Schaeffeler E, Van Der Kuip H, et al. Cellular uptake of imatinib into leukemic cells is independent of human organic cation transporter 1 (OCT1). Cancer Res. 2014;20:985–994. doi:10.1158/1078-0432.ccr-13-1999.
- Watkins DB, Hughes TP, White DL. BCR-ABL1 mutation development during first-line treatment with dasatinib or imatinib for chronic myeloid leukemia in chronic phase. Leukemia. 2015;29:1832–1838. doi:10.1038/leu.2015.168.
- Marzia V, Andrea M, Luisa T. A new monoclonal antibody detects downregulation of protein tyrosine phosphatase receptor type γ in chronic myeloid leukemia patients. Journal of Hematology and Oncology. 2017;10(1).
- Ismail MA, Vezzalini M, Morsi H, et al. Predictive value of tyrosine phosphatase receptor gamma for the response to treatment tyrosine kinase inhibitors in chronic myeloid leukemia patients. Sci Rep. 2021;11(1):8833.
- Ismail MA, Sorio C. Protein tyrosine phosphatase receptor gamma as potential therapeutic target for chronic myeloid leukemia patients. Cancer Control. 2022;29.
- Ismail Mohamed . A. Evaluation of protein tyrosine phosphatase receptor gamma (PTPRG) as a biomarker in chronic myeloid leukaemia patients in the State of Qatar. 2021.
- Ismail MA, Samara M, Sayab Al. Aberrant DNA methylation of PTPRG as one possible mechanism of its under-expression in CML patients in the state of Qatar. Mol Genet Genomic Med. 2020;8(10.
- Tomasello L, Vezzalini M, Boni C, et al. Regulative loop between β-catenin and protein tyrosine receptor type γ in Chronic Myeloid Leukemia. International Journal of Molecular Sciences. 2020;21(7).
- Ismail MA, Samara M, Al Sayab A, et al. Aberrant DNA methylation of PTPRG as one possible mechanism of its under-expression in CML patients in the state of Qatar. Mol Genet Genomic Med. 2020;8(10):e1319.
- Ismail MA, Nasrallah GK, Monne M, et al. Description of PTPRG genetic variants identified in a cohort of chronic myeloid leukemia patients and their ability to influence response to tyrosine kinase inhibitors. Gene. 2022;813:146101.
- Nader A-D, Maria Monne, Araby M, et al. Novel molecular findings in Protein Tyrosine Phosphatase Receptor Gamma (PTPRG) among Chronic Myelocytic Leukemia (CML) patients studied by Next Generation Sequencing (NGS): a pilot study in patients from the State of Qatar and Italy. Blood. 2016;128(22):5427–5427.
- Boni C, Sorio C. Current views on the interplay between tyrosine kinases and phosphatases in Chronic Myeloid Leukemia. Cancers. 2021;13(10):2311. doi:10.3390/cancers13102311.
- Lecca P, Sorio C. Accurate prediction of the age incidence of chronic myeloid leukemia with an improved two-mutation mathematical model. Integr Biol (Camb). 2016;8(12):1261–1275.
- San José-Enériz E, Román-Gómez J, Jiménez-Velasco A, et al. Microrna expression profiling in imatinib-resistant chronic myeloid leukemia patients without clinically significant ABL1-mutations. Mol Cancer. 2009;8:69, doi:10.1186/1476-4598-8-69.
- Turrini E, Haenisch S, Laechelt S, et al. Microrna profiling in K-562 cells under imatinib treatment. Pharmacogenet Genomics. 2012;22:198–205. doi:10.1097/FPC.0b013e328350012b.
- Stankovic M, Nikolic A, Nagorni-Obradovic L, et al. Gene–gene interactions between glutathione S-transferase M1 and matrix metalloproteinases 1, 9, and 12 in chronic obstructive pulmonary disease in serbians. J Chronic Obstructive Pulmonary Dis. 2017;14(6):581–589.
- Shibuta T, Honda E, Shiotsu H, etal. Imatinib Induces Demethylation of miR-203 Gene: an Epigenetic Mechanism of Anti-tumor Effect of Imatinib. Leuk. Res. 2013;37:1278–1286.
- Hershkovitz-Rokah O, Modai S, Pasmanik-Chor M, etal. MiR-30e induces apoptosis and sensitizes K562 cells to imatinib treatment via regulation of the BCR-ABL protein. Cancer Lett. 2014;56:597–605.
- Nader I, Salsabel A-D, Nada Elshami. Expression Profiling of Micro RNAs (MiRNAs) and Long Non-Coding RNAs (lncRNAs) in Chronic Myeloid Leukemia (CML) Patients in the State of Qatar. Blood. 2017;130.
- Litwinska Z, Machalinski B. Mirnas in chronic myeloid leukemia: small molecules, essential function. Leuk Lymphoma. 2017;58:1297–1305.
- Giulia L, Latorre R. Gene expression landscape of chronic myeloid leukemia K562 cells overexpressing the tumor suppressor gene PTPRG. International Journal of Molecular Sciences. 2022;23(17.
- Hanekamp D, Cloos J, Schuurhuis GJ. Leukemic stem cells: identification and clinical application. Int J Hematol. 2017;105(5):549–557.
- Houshmand M, Simonetti G, Circosta P, et al. Chronic myeloid leukemia stem cells. Leukemia. 2019;33(7):1543–1556.
- Morotti A, Panuzzo C, Fava C, et al. Kinase-inhibitor-insensitive cancer stem cells in chronic myeloid leukemia. Expert Opin Biol Ther. 2014;14(3):287–299.
- Soverini S, De Santis S, Monaldi C, et al. Targeting leukemic stem cells in chronic myeloid leukemia: is it worth the effort? Int J Mol Sci. 2021;22(13).
- Chen Y, Zou J, Cheng F, et al. Treatment-Free remission in chronic myeloid leukemia and new approaches by targeting leukemia stem cells. Front Oncol. 2021;11(4465).
- Herrmann O, Kuepper MK, Bütow M, et al. Infliximab therapy together with tyrosine kinase inhibition targets leukemic stem cells in chronic myeloid leukemia. BMC Cancer. 2019;19(1):658.
- Hughes TP, Hochhaus A, Branford S, et al. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the international randomized study of interferon and STI571 (IRIS). Blood. 2010;116(19):3758–3765.
- Douxfils J, Haguet H, Mullier F, et al. Association between BCR-ABL tyrosine kinase inhibitors for chronic myeloid leukemia and cardiovascular events, major molecular response, and overall survival. JAMA Oncol. 2016;2(5):625–632.
- Dusetzina SB, Winn AN, Abel GA, et al. Cost sharing and adherence to tyrosine kinase inhibitors for patients with chronic myeloid leukemia. J Clin Oncol. 2014;32(4):306–311.
- Haddad FG, Sasaki K, Issa GC, et al. Treatment-freeremission in patients with chronic myeloid leukemia following the discontinuation of tyrosine kinase inhibitors. Am J Hematol. 2022;97:856–864.
- Hochhaus A, Masszi T, Giles FJ, et al. Treatment-free remission following frontline nilotinib in patients with chronic myeloid leukemia in chronic phase: results from the ENESTfreedom study. Leukemia. 2017;31(7):1525–1531.
- Mahon F-X, Réa D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–1035.
- Ross DM, Branford S, Seymour JF, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122(4):515–522.
- Clark RE, Polydoros F, Apperley JF, et al. De-escalation of tyrosine kinase inhibitor therapy before complete treatment discontinuation in patients with chronic myeloid leukaemia (DESTINY): a non-randomised, phase 2 trial. Lancet Haematology. 2019;6(7):e375–e383.
- Rea D, Ame S, Berger M, et al. Discontinuation of tyrosine kinase inhibitors in chronic myeloid leukemia: recommendations for clinical practice from the French chronic myeloid leukemia study group. Cancer. 2018;124(14):2956–2963.
- Cayssials E, Torregrosa-Diaz J, Gallego-Hernanz P, et al. Low-dose tyrosine kinase inhibitors before treatment discontinuation do not impair treatment-free remission in chronic myeloid leukemia patients: results of a retrospective study. Cancer. 2020;126(15):3438–3447. doi:10.1002/cncr.32940.
- Fassoni AC, Baldow C, Roeder I, et al. Reduced tyrosine kinase inhibitor dose is predicted to be as effective as standard dose in chronic myeloid leukemia: a simulation study based on phase III trial data. Haematologica. 2018;103(11):1825–1834. doi:10.3324/haematol.2018.194522.
- Lesko LJ, Schmidt S. Individualization of drug therapy: history, present state, and opportunities for the future. Clin Pharmacol Ther. 2012;92(4):458–466.
- García-Ferrer M, Wojnicz A, Mejía G, et al. Utility of therapeutic drug monitoring of imatinib, nilotinib, and dasatinib in chronic myeloid leukemia: a systematic review and meta-analysis. Clin Ther. 2019;41:2558–2570.e7.e7. doi:10.1016/j.clinthera.2019.10.009.
- Takahashi N, Wakita H, Miura M, et al. Correlation between imatinib pharmacokinetics and clinical response in Japanese patients with chronic-phase chronic myeloid leukemia. Clin Pharmacol Ther. 2010;88(6):809–813.
- Marin D, Bazeos A, Mahon FX, et al. Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J Clin Oncol. 2010;28(14):2381–2388.
- Davies A, Giannoudis A, Zhang JE, et al. Dual glutathione-S-transferase-θ1 and -μ1 gene deletions determine imatinib failure in chronic myeloid leukemia. Clin Pharmacol Ther. 2014;96(6):694–703.
- Qiu HB, Zhuang W, Wu T, et al. Imatinib-induced ophthalmological side-effects in GIST patients are associated with the variations of EGFR, SLC22A1, SLC22A5 and ABCB1. Pharmacogenomics J 2018;18(3):460–466.
- Yamakawa Y, Hamada A, Nakashima R, et al. Association of genetic polymorphisms in the influx transporter SLCO1B3 and the efflux transporter ABCB1 with imatinib pharmacokinetics in patients with chronic myeloid leukemia. Ther Drug Monit. 2011;33(2):244–250.
- Petain A, Kattygnarath D, Azard J, et al. Population pharmacokinetics and pharmacogenetics of imatinib in children and adults. Clin Cancer Res. 2008;14(21):7102.
- Delbaldo C, Chatelut E, Ré M, et al. Pharmacokinetic-pharmacodynamic relationships of imatinib and its main metabolite in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2006;12(20 Pt 1):6073–6078.
- Widmer N, Decosterd LA, Csajka C, et al. Population pharmacokinetics of imatinib and the role of alpha1-acid glycoprotein. Br J Clin Pharmacol. 2006;62(1):97–112.
- Judson I, Ma P, Peng B, et al. Imatinib pharmacokinetics in patients with gastrointestinal stromal tumour: a retrospective population pharmacokinetic study over time. EORTC soft tissue and bone sarcoma group. Cancer Chemother Pharmacol. 2005;55(4):379–386.
- Adeagbo BA, Olugbade TA, Durosinmi MA, et al. Population pharmacokinetics of imatinib in nigerians with chronic myeloid leukemia: clinical implications for dosing and resistance. The Journal of Clinical Pharmacology. 2017;57(12):1554–1563.
- Martinez-Lage M, Torres-Ruiz R, Puig-Serra P, et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat. Commun. 2020;11:1–14. doi:10.1038/s41467-019-13993-7.
- Garcia-Tunon I, Alonso-Perez V, Vuelta E, et al. Splice donor site sgRNAs enhance CRISPR/Cas9-mediated knockout efficiency. PLoS ONE. 2019;14:e0216674. doi:10.1371/journal.pone.0216674.
- Luo Z, Gao M, Huang N, et al. Efficient disruption of BCR-ABL gene by CRISPR RNA-guided FokI nucleases depresses the oncogenesis of chronic myeloid leukemia cells. J Exp Clin Cancer Res. 2019;38:224, doi:10.1186/s13046-019-1229-5.
- Huang N, Huang Z, Gao M, et al. Induction of apoptosis in imatinib sensitive and resistant chronic myeloid leukemia cells by efficient disruption of BCR-ABL oncogene with zinc finger nucleases. J Exp Clin Cancer Res. 2018;37:62, doi:10.1186/s13046-018-0732-4.
- García-Tuñón I, Hernández-Sánchez M, Ordoñez JL, et al. The CRISPR/Cas9 system efficiently reverts the tumorigenic ability of BCR/ABL in vitroand in a xenograft model of chronic myeloid leukemia. Oncotarget. 2017;8(16):26027–26040. doi:10.18632/oncotarget.15215.
- Vuelta E, García-Tuñón I, Hernández-Carabias P, et al. Future approaches for treating chronic myeloid leukemia: CRISPR therapy. Biology. 2021;10(2).
- Kevin Z, Mohammad Y, Sawsan M. Optimizing clinical workflow using precision medicine and advanced data analytics. Processes. 2023;11(3).
- Al-Dewik NI, Younes SN, Essa MM, et al. Making biomarkers relevant to healthcare innovation and precision medicine. Processes. 2022;10(6):1107. doi:10.3390/pr10061107.
- AL-Dewik NI, Qoronfleh MW. Genomics and precision medicine: molecular diagnostics innovations shaping the future of healthcare in Qatar. Advances in Public Health. 2019;2019:1–11. doi:10.1155/2019/3807032.