

1. Introduction
Development of a new drug currently takes 10–12 years with costs of around 2 billion EUR. The two main reasons for failures comprise lack of efficacy and unforeseen xicity. For the latter, a standard process pursued to minimize the risk is the so-called toxicological read across. Briefly, toxicologists query the available literature and databases for compounds, which are structurally similar to their development candidate in order to retrieve information on potential hazards. In addition, computational models might be applied which are either trained for a single protein, such as hERG or P-glycoprotein, or for a respective in vivo endpoint (cholestasis, steatosis, drug-induced liver injury (DILI), ….). In both cases, a proper ‘description’ of the compound of interest is key for the predictive ability of the models. In the following editorial, we will highlight a few general approaches for compound description with a focus on bioactivity-based characterization of compounds.
2. Direct prediction versus knowledge-based approach
In principle, there are three general concepts how in silico models for prediction of in vivo effects might be derived:
2.1. Mechanism-agnostic concepts
In this setting, a data set of toxic and nontoxic compounds is derived, and then various machine-learning models are developed. Descriptors used for characterizing the compounds comprise all sorts of physicochemical descriptors as well as fingerprints and graphs. The list of models being put in the public domain is huge, and almost weekly new models are published using larger datasets and more complex modeling techniques.
2.2. Mechanism-based approaches
In this setting, distinct biological effects which are causally linked to the adverse outcome are modeled. A prominent example is inhibition of the hERG potassium channel and Torsade de Points. In this case, all sorts of tools and techniques are applied, ranging from conventional quantitative structure–activity relationship (QSAR)/machine learning (ML) based on physicochemical descriptors and/or fingerprints to pharmacophore models and protein structure-based approaches such as docking. Of course, this approach can be extended toward more than one target as e.g. shown by Dracheva et al. [Citation1] and by de Lomana et al. [Citation2]. Both groups developed a series of classification models for Molecular Initiating Events (MIEs) involved in thyroid homeostasis, whereby de Lomana also used multitask neural networks combining several end points as a possible way to improve the performance of models for which the experimental data available for model training were limited. Obiol et al. went even a step further and used a combination of docking simulations on two potassium channels, hERG and KCNQ1 and 3D-QSAR studies for blockers of the potassium currents IKr and IKs. The results have been used as input to electrophysiological models of the cardiomyocytes and the ventricular tissue, allowing the direct prediction of the drug effects on electrocardiogram simulations [Citation3].
2.3. Combined approaches
Going further, it is also possible to combine both approaches, as e.g. shown by Kotsampasakou et al. In this study, the authors compiled a dataset of compounds to predict if they might lead to cholestasis. In addition, a set of models for transporters expressed in the liver was developed. Subsequently, the cholestasis dataset was pushed through the transporter models, thus creating a sort of transporter interaction profile for the compounds. Finally, the predicted transporter interaction values were used as descriptors in addition to physicochemical descriptors for the compounds in order to create machine learning models for predicting cholestasis. Evaluation of this integrated model demonstrated a superior performance over the use of only physicochemical descriptors. Furthermore, descriptor importance analysis confirmed the predicted BSEP (bile salt export pump) inhibition as an important feature driving the model [Citation4].
3. Let’s talk about (biological) descriptors
However, any modeling attempt starts with the choice of the descriptors, i.e. how the compounds are encoded. This is the most crucial step in model building, as finally any model links differences in chemical structures to differences in biological activity. Software packages such as Mordred, Paddel, or alvaDesc allow the calculation of thousands of molecular descriptors. In addition to 2D- and 3D-molecular descriptors, also a set of different molecular fingerprints, such as ECFP4, are available. However, considering the complexity of Adverse Events such as DILI, steatosis, or DNT (developmental neurotoxicity), just to mention a few, a more biology-oriented description of compounds might be superior toward a pure chemical description. In the first instance, these could be measured bioactivity values for targets linked to the adverse event of interest. Nevertheless, although ChEMBL [Citation5] and PubChem [Citation6] provide millions of bioactivity values, for most of the targets less than 100 compounds are reported. Even in the case of established off-targets, such as those routinely tested by Roche [Citation7], publicly available experimental data are quite sparse.
Especially in the field of toxicity prediction also the Tox21/ToxCast initiatives need to be mentioned. These large-scale programs generated more than 150 million data points by screening thousands of compounds against 60 important targets and pathways. Combining Tox21 derived biological signatures with data from gene expression signatures, FDA reporting of adverse events (FAERS), and molecular descriptors provided DILI classification models with decent performance [Citation8].
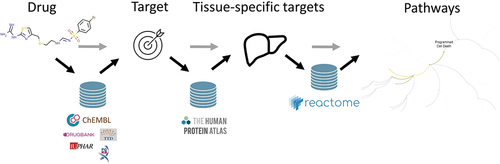
Of course, as outlined above, experimental data might be enriched by predicted values from respective target-based models. These might be individually derived ones or model sets available in the public domains, such as those described by Bosc et al., Lane et al., or Mayr et al., just to mention a few. The drawback of these approaches relates to the fact that mostly conformal prediction is used, which does not provide a prediction for every compound. Alternatively, one might derive predicted compound–target interaction profiles by using target prediction tools such as SEA, HitPick, TargetPred, or SwissTarget (for a comprehensive list and comparison of different methods see Ren et al. [Citation9]). When given the structure of a compound, these tools provide a list of targets the compound might interact with plus respective probability values. Unfortunately, in comparison to individual target-based models, using these tools will not provide a fingerprint with a fixed length, which would be necessary when used as input for machine learning. However, these target prediction tools were of value when approaching the next level of complexity – biological pathways. In their paper Path4Drug, Füzi et al. introduced the concept of compound/pathway interaction fingerprints and used them as descriptors for machine learning models for prediction of DILI. Briefly, for every compound in the dataset, bioactivity values are retrieved from ChEMBL. Subsequently, the respective target profile is enriched with predicted targets and the complete list of targets is used as input for an interactome database such as STRING. For this, a further enriched list of protein enrichment analyses in a pathway database is performed. This finally leads to a list of pathways linked to every compound () [Citation10]. This compound/pathway interaction fingerprint has a fixed length determined by the number of pathways in the pathway database and can be used as a biological fingerprint. Combining these fingerprints with chemical fingerprints not only provided DILI classification models with decent performance, but also allowed to identify major pathways involved in DILI via feature importance analysis of the model [Citation11].
Figure 1. Workflow for generating compound/pathway interaction fingerprints.
4. Expert opinion and outlook
A proper description of chemical entities is key for a successful development of predictive in silico models for complex Adverse Events. While there is a plethora of chemical descriptors available, methods for deriving biologically informed descriptors need considerable attention. One of the major problems is the sparsity of public databases, which, in addition, show a heavy bias toward active compounds [Citation12]. However, methods such as proteochemometric modeling, transfer learning, and data imputation will allow to derive proper predictions of large sets of compounds for whole protein families such as SLC transporters. Furthermore, with the increasing availability of large AI-based models also filling a substantial part of ChEMBL with predicted bioactivity values comes into reach. This could serve for generation of bioactivity fingerprints with a length of thousands of bins, each bin representing one protein. Finally, protocols for docking of ultra-large scale compound libraries in combination with the increasing reliability of AlphaFold structures [Citation13] open the path for docking-derived descriptors for model building. First attempts have recently been presented. The next few years will show if the large-scale AI models currently being launched will fulfil all the promises they claim and indeed revolutionize drug discovery and also toxicity prediction.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Dracheva E, Norinder U, Rydén P, et al. In silico identification of potential thyroid hormone system disruptors among chemicals in human serum and chemicals with a high exposure index. Environ Sci Technol. 2022;56(12):8363–8372. doi: 10.1021/acs.est.1c07762
- Garcia De Lomana M, Weber AG, Birk B, et al. In silico models to predict the perturbation of molecular initiating events related to thyroid hormone homeostasis. Chem Res Toxicol. 2021;34(2):396–411. doi: 10.1021/acs.chemrestox.0c00304
- Obiol-Pardo C, Gomis-Tena J, Sanz F, et al. A multiscale simulation system for the prediction of drug-induced cardiotoxicity. J Chem Inf Model. 2011;51(2):483–492. doi: 10.1021/ci100423z
- Kotsampasakou E, Ecker GF. Predicting drug-induced cholestasis with the help of hepatic transporters—an in silico modeling approach. J Chem Inf Model. 2017;57(3):608–615. doi: 10.1021/acs.jcim.6b00518
- Zdrazil B, Felix E, Hunter F, et al. The ChEMBL database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 2024;52(D1):D1180–92. doi: 10.1093/nar/gkad1004
- Kim S, Chen J, Cheng T, et al. PubChem 2023 update. Nucleic Acids Res. 2023;51(D1):D1373–80. doi: 10.1093/nar/gkac956
- Bendels S, Bissantz C, Fasching B, et al. Safety screening in early drug discovery: an optimized assay panel. J Pharmacol Toxicol Methods. 2019;99:106609. doi: 10.1016/j.vascn.2019.106609
- Adeluwa T, McGregor BA, Guo K, et al. Predicting drug-induced liver injury using machine learning on a diverse set of predictors. Front Pharmacol. 2021;12:648805. doi: 10.3389/fphar.2021.648805
- Ren X, Yan CX, Zhai R, et al. Comprehensive survey of target prediction web servers for traditional Chinese Medicine. Heliyon. 2023;9(8):e19151. doi: 10.1016/j.heliyon.2023.e19151
- Füzi B, Gurinova J, Hermjakob H, et al. Path4Drug: data science workflow for identification of tissue-specific biological pathways modulated by toxic drugs. Front Pharmacol. 2021;12:708296. doi: 10.3389/fphar.2021.708296
- Füzi B, Mathai N, Kirchmair J, et al. Toxicity prediction using target, interactome, and pathway profiles as descriptors. Toxicol Lett. 2023;381:20–26. doi: 10.1016/j.toxlet.2023.04.005
- Smajić A, Rami I, Sosnin S, et al. Identifying differences in the performance of machine learning models for off-targets trained on publicly available and proprietary data sets. Chem Res Toxicol. 2023;36(8):1300–1312. doi: 10.1021/acs.chemrestox.3c00042
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. doi: 10.1038/s41586-021-03819-2