
ABSTRACT
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive malignancy characterized by a dismal five-year survival rate of less than 10%. Neutrophils are key components of the innate immune system, playing a pivotal role in the PDAC immune microenvironment.
Areas covered
This review provides a comprehensive survey of the pivotal involvement of neutrophils in the tumorigenesis and progression of PDAC. Furthermore, it synthesizes preclinical and clinical explorations aimed at targeting neutrophils within the milieu of PDAC, subsequently proposing a conceptual framework to propel further inquiry focused on enhancing the therapeutic efficacy of PDAC through neutrophil-targeted strategies. PubMed and Web of Science databases were utilized for researching neutrophils in pancreatic cancer publications prior to 2024.
Expert opinion
Neutrophils play roles in promoting tumor growth and metastasis in PDAC and are associated with poor prognosis. However, the heterogeneity and plasticity of neutrophils and their complex relationships with other immune cells and extracellular matrix also provide new insights for immunotherapy targeting neutrophils to achieve a better prognosis for PDAC.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) comprises the majority of pancreatic cancer cases and is associated with a significant mortality rate, demonstrating an overall 5-year survival rate of approximately 9% [Citation1]. At present, surgical resection stands as the sole established curative intervention for PDAC. Nonetheless, upon diagnosis, only a mere 20% of patients meet the criteria for surgical intervention [Citation2]. Given the increasing incidence of PDAC and the limited treatment options available, there exists an urgent imperative for the development of innovative strategies to combat this formidable malady.
Tumor immunotherapy is a cutting-edge field of cancer research that aims to make tumor cells more recognizable by the immune system, enhance the anti-tumor immune response, and stop tumor growth and spread. PDAC is a type of tumor that does not trigger a strong immune reaction. Compared to immunotherapy-responsive cancers, PDAC exhibits a distinct immune profile and tumor microenvironment (TME) rich in immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs), tumor-associated neutrophils (TANs), regulatory T cells (Tregs), and collagen-producing carcinoma-associated fibroblasts (CAFs), promoting fibrosis [Citation3,Citation4]. The thick stroma of PDAC contains many components, such as extracellular matrix, stellate cells, fibroblasts, myofibroblasts, immune cells, cytokines, and growth factors, that help the tumor progress and spread through complex interactions [Citation5]. Notably, neutrophils, which are important innate immune cells, have a key role in the pancreatic cancer immune microenvironment.
This comprehensive review delineates the pivotal role of neutrophils in the genesis and progression of PDAC, summarizes preclinical and clinical studies on targeting neutrophils in the context of PDAC, and subsequently posits a theoretical framework for advanced investigations into targeting neutrophils to enhance therapeutic efficacy in PDAC. The PubMed and Web of Science databases were systematically searched for studies examining the role of neutrophils in pancreatic cancer published prior to 2024, using the keywords ‘Pancreatic Cancer’ and ‘neutrophil.’
2. Neutrophils in pancreatic cancer tumorigenesis and progression
2.1. Stepwise pathogenesis of PDAC
The origin of PDAC is attributed to a variety of noninvasive precancerous lesions with distinct histological features. The majority of PDAC are believed to arise from pancreatic intraepithelial neoplasia (PanIN), characterized by microscopic lesions with a diameter of less than 5 millimeters that affect the pancreatic ducts [Citation6]. PanIN can be morphologically classified as low-grade or high-grade dysplasia, which is very common in the human body but does not usually develop into pancreatic cancer [Citation7,Citation8]. In general, low-grade dysplasia is more common, and high-grade dysplasia has a higher risk of developing pancreatic cancer [Citation9]. KRAS, CDKN2A, TP53 and SMAD4 are the four most commonly mutated driver genes in pancreatic cancer, and the progression of PanIN from low to high grade is associated with mutations in these four genes [Citation10].
In approximately 90% of pancreatic cancer patients, the occurrence of grade 1 and 2 PanIN is associated with point mutations in the KRAS gene, specifically codon 12, resulting in the activation of the RAS-RAF and PI3K-AKT pathways, intracellular signaling pathways that regulate the cell cycle [Citation11]. Grade 2 disease is associated with the inactivation of two cyclin-dependent kinase inhibitors, CDKN2A (and its coding protein p16) and CDKN1A (and its coding protein p21), which promote the transition of the cell cycle from G1 to S phase [Citation11]. Finally, grade 3 and 4 PanIN often represent malignant changes in the tumor. Mutations in the key tumor suppressor gene TP53 are observed in 50–70% of patients with PDAC, as well as SMAD4 inactivation mutations observed in 60–90% of these patients during this transition [Citation12]. Inactivation of SMAD4 and its coding protein SMAD4 blocks typical signaling downstream of transforming growth factor-β (TGF-β) receptors, while inactivation of TP53 and its coding protein p53 promotes the progression of cell cycle, survival, and apoptosis.
2.2. Evolving inflammatory cell infiltration during the stepwise pathogenesis of PDAC
The inflammatory response plays a significant role in the pathological changes of pancreatic cancer. Chronic pancreatitis is generally considered a risk factor for the onset of PDAC. Patients with chronic pancreatitis have a higher risk of developing PDAC, and the cumulative risk of developing PDAC in patients with hereditary pancreatitis is 40% over a lifetime [Citation13]. Chronically inflamed pancreatic tissue harbors early genetic alterations that promote cytokine production, initiating a multi-step process encompassing tumor formation, growth, metastasis, and suppression of anti-tumor immune responses [Citation14,Citation15]. This immunosuppressive microenvironment is further underscored by the prominent influx of myeloid cells even in the lowest grade of pre-invasive lesions. Generally, the pathological changes in the tumorigenesis of pancreatic cancer involve a variety of complex inflammatory cell infiltrates, with neutrophils typically being the first effector cells recruited to sites of acute inflammation. Additionally, compared to normal pancreatic tissue, the infiltration of neutrophils, macrophages, Tregs, CAFs with suppressive activity, and MDSCs is significantly increased in PanINs and PDAC tissue, while the infiltration of immune effector cells such as CD8+ cytotoxic T and natural killer (NK) cells is typically reduced [Citation16–18].
2.3. KRAS-driven neutrophil and G-MDSC infiltration
KRAS is the most commonly mutated driver gene in PDAC (70% to 95%) [Citation19] and is closely related to the infiltration of diverse immune cells, including neutrophils and granulocytic MDSCs (G-MDSCs). G-MDSCs are characterized as neutrophils that exhibit immunosuppressive attributes. Studies have indicated that KRAS mutations are closely associated with tumor-induced inflammation and tumorigenesis through several induced inflammatory cytokines and chemokines, such as granulocyte colony stimulating factor (G-CSF), granulocyte-macrophage CSF (GM-CSF) [Citation20], and interleukin 6 (IL-6) [Citation21], which play important roles in recruiting and activating neutrophils. G-CSF and GM-CSF are upregulated in PDAC cells, promoting the survival and recruitment of G-MDSCs and CD11b+Ly6G+ neutrophils in vivo [Citation22]. The Janus kinase (JAK)- Signal transducer and activator of transcription 3 (STAT3) and NF-κB pathways are involved in the production of KRAS-induced cytokines/chemokines. Mutant KRAS activity has been proved to upregulate the expression of IL-6, which induces STAT3/SOCS3 activity in a paracrine or autocrine manner. On the contrary, the genetic deficiency of IL-6 or STAT3 inhibits PanIN progression and reduces the development of PDAC in mouse models [Citation23]. Furthermore, studies have shown that in KRAS-driven PDAC mouse models, the percentage of pro-tumor macrophages and MDSCs within tumors significantly decreases following IL-6 deficiency [Citation24]. Additionally, CXC motif chemokine receptor 2 (CXCR2) is a receptor for a group of chemokines that enhances the recruitment of granulocytes to inflammatory sites and promotes angiogenesis by recruiting MDSCs and immunosuppressive neutrophils. In pancreatic cancer, mutated KRAS drives the release of CXC motif chemokine ligand 1 (CXCL1), CXCL2, and CXCL5, all of which are ligands of CXCR2 [Citation25]. Conversely, inhibition of RAS signaling can lead to decreased pancreatic myeloperoxidase (MPO) activity induced by taurocholic acid, indicating inhibited neutrophil recruitment [Citation26]. Research has also shown that the overall deficiency of CXCR2 in KrasG12D/+ p53R172H/+ transgenic mice significantly increases T cell infiltration in pancreatic cancer animal models and prevents metastasis [Citation27]. Moreover, the activation of the IL-17/IL-17 R pathway driven by KrasG12D leads to increased neutrophil recruitment and neutrophil extracellular traps (NETs) [Citation28,Citation29], which promote the development of pancreatic cancer. Similarly, there is evidence that IL-17 promotes tumor progression by enhancing the effector function of MDSCs [Citation30].
2.4. Microbiome in driving neutrophil infiltration
The research indicates that the interactions of the host microbiota can influence cellular bioactivity, and regulate inflammation, immunity, and cancer progression [Citation31–33]. This mechanism within pancreatic cancer is referred to as the tumor-microbiota-immune-pancreatic cancer axis [Citation34]. Neutrophils, being the principal phagocytic cells in the body, are pivotal to the body’s defense against pathogenic microorganisms, including bacteria, fungi, viruses, and parasites. The primary mechanisms through which neutrophils function are phagocytosis, degranulation, and the release of NETs. NETs, comprised of chromatin and mitochondrial DNA fibers along with antimicrobial enzymes and histones, are discharged by neutrophils to ensnare and eliminate pathogens. Nevertheless, the presence of NETs in tumors can lead to tumor recurrence, heightened tumor migration and invasiveness, and the promotion of tumor cell proliferation [Citation35]. Research has suggested that P. gingivalis can stimulate the secretion of neutrophil chemoattractants (CXCL1 and CXCL2) in the tumor microenvironment of PDAC, thereby fostering the accumulation of TANs in the tumor microenvironment, although this mechanism is not yet fully elucidated [Citation36]. Conversely, the enrichment of pro-tumor subtype of TANs and the progression of PDAC can be blocked by CXCR2 inhibitors.
2.5. The role of neutrophils in the stepwise evolution of the PDAC TME
Neutrophils play a critical role in bridging inflammation and tumor progression. While neutrophils recruited during the inflammatory process exhibit effective anti-inflammatory and immune functions, excessive recruitment and anti-inflammatory responses can result in extended healing periods, exacerbation of tissue damage, and the progression of acute inflammation into chronic conditions. It is worth noting that chronic pancreatitis significantly predisposes individuals to pancreatic cancer. The accumulation of neutrophils in pathological lesions can lead to increased tissue damage and a heightened risk of carcinogenesis due to the release of proteases and reactive oxygen species (ROS) by the neutrophils themselves [Citation37]. Moreover, mutations in the KRAS gene in PanINs are notably associated with an elevated risk of developing pancreatic cancer. As previously stated, mutations in the RAS gene can independently facilitate heightened activation and recruitment of neutrophils, further augmenting the risk of pancreatic cancer development.
In addition to their involvement in the tumorigenesis of pancreatic cancer, neutrophils are also implicated in the progression of PDAC (). Evidence from studies suggests that in a mouse model of pancreatic cancer, the release of matrix metalloproteinase-9 (MMP-9) by neutrophils amplifies the activity of vascular endothelial growth factor (VEGF), thereby fostering angiogenesis [Citation37]. Additionally, neutrophils can secrete pro-inflammatory cytokines such as IL-17, as well as CXC and CC chemokines, within the tumor microenvironment [Citation38–40]. These pro-inflammatory cytokines, particularly IL-17, can induces stem cell features of PanINs [Citation41], or inducing the upregulation of CXCR2 ligands, which in turn promote neutrophil mobilization and indirectly facilitate tumor progression, while simultaneously excluding cytotoxic CD8+ T lymphocytes from the TME [Citation29]. Furthermore, PDAC tumor cells can release various factors, such as members of the CXC family chemokines (CXCL1, CXCL2, CXCL5, and CXCL8), growth factors (GM-CSF and G-CSF), IL-1, and CD200, which recruit neutrophils [Citation42].
Figure 1. The role of tumor-associated neutrophils in PDAC. PDAC cells secrete chemokines (such as IL-8, CXCR1, CXCR2, CXCR4, etc.) and cytokines (such as G-CSF, GM-CSF, etc.), which recruit neutrophils from the circulation system and direct them to the tumor microenvironment, where they polarized into pro-tumor TAN under the influence of TGF-β produced by tumor cells and other immune cells. These TANs not only secrete MMP-9 and VEGF to stimulate angiogenesis, but also induce tissue damage and tumorigenesis of normal ductal epithelial cells by generating ROS and proteases or inducing NETs. In addition, such TANs also express PD-L1, which interacts with PD-1 on T cells to facilitate checkpoint mediated immune escape. CXCR1, CXC motif chemokine receptor 1; CXCR2, CXC motif chemokine receptor 2; CXCR4, CXC motif chemokine receptor 4; G-CSF, granulocyte colony stimulating factor; GM-CSF, granulocyte-macrophage colony stimulating factor; IL-8, interleukin 8; IL-17, interleukin 17; MMP-9, matrix metalloproteinase-9; NETs, neutrophil extracellular traps; PanIN, pancreatic intraepithelial neoplasia; PD-1, programmed cell death; PD-L1, programmed death-ligand 1; PDAC, pancreatic ductal adenocarcinoma; ROS, reactive oxygen species; TAN, tumor-associated neutrophil; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor.
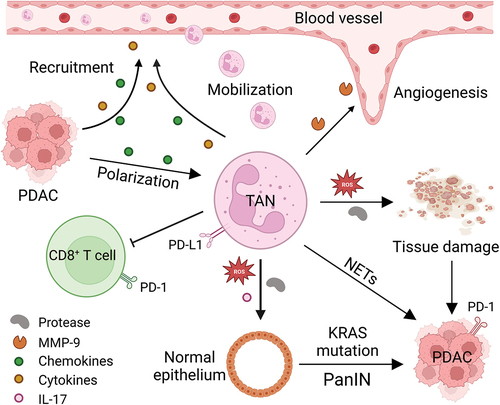
Neutrophils also participate in promoting the metastasis of pancreatic cancer. NETs can entrap tumor cells in the circulation and encourage the adhesion of tumor cells to hepatic sinusoids by binding to intercellular adhesion molecule-1 via Mac-1 [Citation43,Citation44]. Moreover, neutrophil elastase (NE) within NETs can degrade VE-cadherin, leading to the disruption of endothelial cell connections, thereby enhancing cancer cell motility, increasing vascular permeability, and promoting epithelial-mesenchymal transition [Citation45,Citation46]. The presence of cancer-associated fibroblasts in liver micro-metastases may also be attributed to NETs, thereby supporting a pre-metastatic niche [Citation47].
3. Neutrophil plasticity and reprogramming
Neutrophils are a highly plastic cell population presenting with heterogeneous phenotypes in response to various environmental stimuli [Citation48,Citation49]. It has been shown that neutrophils assume different functions based on their activation status to an immunostimulatory (N1-like) or immunosuppressive (N2-like) subtype similar to the nomenclature used for macrophages [Citation50]. N1 neutrophils exhibit normal density, multi-segmented nuclei, and demonstrate cytotoxicity against cancer cells. Conversely, the predominant subtype, N2 neutrophils, possess the capability to suppress T cell activation and function through the secretion of immunosuppressive molecules such as arginase 1 (ARG1) and prostaglandin E2. This immunosuppressive activity underscores their significant role in modulating immune responses [Citation48]. Importantly, the interconversion between these two phenotypes can occur under specific physiological circumstances. In a TGF-β rich environment, neutrophils typically have a N2 type phenotype, which is generally thought to promote tumor progression, while in the presence of interferon (IFN) β or inhibition of TGF-β signaling, neutrophils switch to N1 type, which is associated with antitumor characteristics [Citation51]. The adoption of either phenotype categorizes the neutrophil as a TAN.
3.1. Tumor killings by neutrophil
Neutrophils kill tumor cells directly through cytotoxic effects, with the most well-known example being the generation of ROS such as superoxide and hydrogen peroxide [Citation52]. However, the mechanism and role of ROS in the TME are not yet fully understood. Research has indicated that the ROS-mediated cytotoxic effect depends on the expression of TRPM2 in tumor cells, which is a hydrogen peroxide-dependent Ca2+ channel that, upon activation, leads to the entry of calcium ions into the cells, causing cell death [Citation53]. Additionally, TANs can also suppress metastatic seeding in the lungs through the generation of hydrogen peroxide [Citation54]. Apart from ROS, neutrophils also secrete reactive nitrogen species, such as nitric oxide and peroxynitrite, but their effects on human cancer cells have not been thoroughly studied.
Neutrophils also exert anti-tumor effects by coordinating the recruitment and function of other immune cells within the TME. Research has proved that neutrophils are capable of presenting antigens to T cells to stimulate adaptive immune responses and the production of IFN-γ [Citation55]. Neutrophils can also interact with T cells through NETs, which can directly activate T cells by reducing their activation threshold [Citation56]. Moreover, studies have shown that IFN-γ-stimulated neutrophils can recruit and activate NK cells by secreting IL-18 [Citation57]. Furthermore, research has demonstrated the important role of neutrophils in eliminating tumor antigen escape variants when treated with melanoma-specific CD4+ T cells in combination with O×40co-stimulation therapy for melanoma [Citation58].
Neutrophils also mediate antibody-dependent cell cytotoxicity. Studies have shown that this effect utilizes antibody-mediated myodegeneration, including phagocytosis of neutrophils and direct destruction of the cancer cell membrane [Citation59]. Moreover, the inflammatory molecules secreted by neutrophils can reduce tumor burden. Studies have found an increase in tumor necrosis factor-α (TNF-α) expression in mouse N1 cells [Citation50], which has been shown to promote tumor cell death by triggering the release of reactive oxygen species by neutrophils [Citation60]. Additionally, TNF-α has been shown to upregulate the receptor tyrosine kinase MET expression in various cancer types and promote the anti-tumor activity of neutrophils [Citation61].
3.2. Immunosuppressive effects of neutrophils
Neutrophils play an immunosuppressive role in cancers by impeding the recruitment of other immune cells into the TME. Notably, neutrophils can release ROS and ARG1 into the TME which degrades extracellular ARG, a pivotal protein for T cell activation and proliferation [Citation62]. Cancer cell-secreted IL-8 has been demonstrated to enhance ARG1 release into the TME further exacerbating this immunosuppressive effect [Citation63]. Additionally, studies have shown that membrane-related Proteinase 3, a serine protease, expressed by neutrophils can hinder T cell proliferation in in vitro experiments [Citation64]. Additionally, by secreting CCL17, neutrophils are proficient in recruiting Treg cells [Citation65], resulting in the suppression of other inflammatory T-cell populations. Furthermore, neutrophils can instigate apoptosis of CD8+ T cells within the tumor environment through TNF-α and nitric oxide-dependent mechanisms [Citation66]. Programmed death-ligand 1 (PD-L1) and its ligand, programmed cell death-1 (PD-1), constitute a classic immune checkpoint. PD-L1, as a co-inhibitory molecule, binds with PD-1 on T cells and inhibits their proliferation, survival, and activity [Citation67]. In gastric cancer, tumor-derived GM-CSF activates neutrophils and induces PD-L1 expression on them via the JAK-STAT3 signaling pathway. This suppresses T cell-mediated immunity and facilitates gastric cancer progression [Citation68]. Similarly, in hepatocellular carcinoma, neutrophils expressing PD-L1 accumulate in the peritumoral area of patients and effectively impair T cell proliferation and activation [Citation69]. In addition to curtailing T cell immune activity, neutrophils also inhibit NK cell activity by releasing MPO and hydrogen peroxide [Citation70] and diminish IL-18 secretion, responsible for NK cell activation, through membrane-expressed CXCR4 [Citation71].
3.3. Neutrophils are pro-metastatic
Neutrophils have the potential to facilitate the metastasis of tumor tissue through various mechanisms. Firstly, mature neutrophils harbor a substantial quantity of enzymes such as MPO, NE, and MMPs, including neutrophil collagenase (MMP-8) and gelatinase B (MMP-9). These enzymes are implicated in promoting the invasion and migration of tumor cells. For instance, MMPs can enhance cancer migration and invasion by stabilizing integrin proteins [Citation72], while NE and MMP-9 can prompt the proliferation of dormant metastatic cancer cells, thereby fostering the development of metastatic tumors [Citation73]. Furthermore, NE and MPO can modulate the production of NETs, which, when released into the TME, can instigate cancer cell migration and invasion. Moreover, after the successful extravasation of tumor cells, their survival and proliferation in a newly established conducive microenvironment in distant organs are imperative for the formation of metastases. This microenvironment, known as the pre-metastatic niche, has been demonstrated to be significantly influenced by neutrophils. Neutrophils infiltrate the metastatic site preceding the arrival of tumor cells, promoting tumor metastasis while not impacting the growth of the primary tumor. In colorectal cancer, tissue inhibitor of MMP-1 facilitates liver metastasis by creating a pre-metastatic niche. This phenomenon is reliant on the upregulation of stromal cell-derived factor 1, which attracts CXCR4-dependent neutrophils to the liver [Citation74]. Furthermore, research has demonstrated that conditioned medium from pre-metastatic lung neutrophils, enriched in leukotrienes, proliferates tumor spheroids in vitro and enhances the likelihood of initiating cancer cell metastasis in vivo [Citation75].
3.4. NETosis
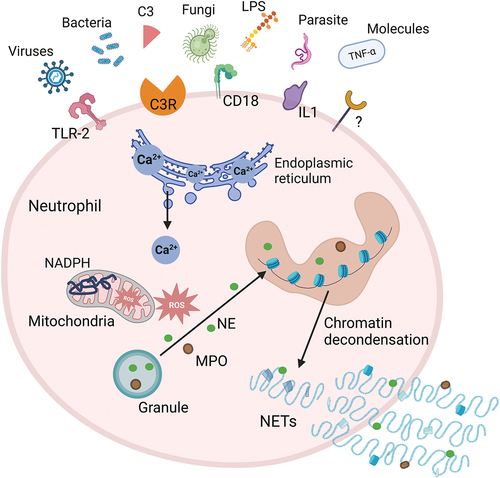
Neutrophil extracellular traps are extracellular structures consisting of decondensed chromatin, citrullinated histones such as histone H3, and proteins commonly found in azurophilic granules, including NE, histone protease G, and MPO. The majority of DNA in NETs originates from the neutrophil’s nuclear chromatin, with a smaller fraction possibly derived from mitochondrial DNA. Upon disruption of the nuclear and plasma membranes, neutrophils undergo cell death within 3–8 hours due to the release of nuclear DNA, a process referred to as NETosis () [Citation76,Citation77]. The specific signals that trigger NETosis are not yet fully elucidated, however, the release of NETs is instigated after the activation of surface receptors, subsequent perturbations in intracellular calcium levels, activation of kinase signaling cascades, and the production of ROS. These primary signaling events ultimately lead to morphological alterations, particularly heightened cell spreading and changes in cellular morphology [Citation78]. The main biological role of NETs is to trap and neutralize microorganisms, thereby preventing their further reproduction. However, dysregulation of NETs formation can lead to several types of pathology, including immune-related and inflammatory diseases, as well as cancer [Citation77,Citation79].
Figure 2. Mechanisms of NETosis. Exogenous microorganisms (such as bacteria, fungi, viruses, etc.) and immune complexes activate neutrophils by stimulating receptors on the cell membrane, such as TLR2, C3R, IL-1, CD18. Activated neutrophils induce the release of Ca2+ from the endoplasmic reticulum. Intracellular Ca2+ efflux activates protein kinase signaling to stimulate NADPH oxidase to produce ROS, which trigger a MPO pathway. MPO-mediated oxidative activation of NE is required for NE to degrade the actin cytoskeleton in the cytoplasm. MPO and NE subsequently translocate to the nucleus, causing nuclear membrane disruption and chromatin decondensation. The generated NETs are directly released into the cytoplasm, and the rupture of the plasma membrane leads to the release of NETs to the extracellular space and neutrophil death. C3, complement 3; C3R, complement 3 receptor; IL1, interleukin 1; LPS, lipopolysaccharide; MPO, myeloperoxidase; NADPH, nicotinamide adenine dinucleotide phosphate; NE, neutrophil elastase; NETs, neutrophil extracellular traps; ROS, reactive oxygen species; TLR-2, toll-like receptor 2; TNF-α, tumor necrosis factor-α.
3.5. Neutrophil reprogramming
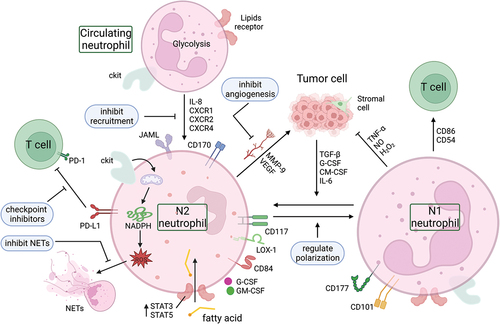
Normal neutrophils are usually glycolytic with minimal mitochondrial metabolism. However, in response to the low glucose availability in the TME, immature neutrophil subsets switch to mitochondrial oxidative metabolism, relying on mitochondrial fatty acid oxidation to support nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-dependent ROS generation. This process inhibits T cell responses through c-Kit signaling [Citation80]. Furthermore, the high lipid concentration in the TME, upregulation of tumor-derived cytokines (such as G-CSF and GM-CSF), and upregulation of lipid transport receptors mediated by STAT3 or STAT5 facilitate the uptake of exogenous fatty acids by G-MDSCs, leading to fatty acid oxidation, which in turn suppresses immune responses of T cells [Citation81]. While TME metabolic reprogramming and oncometabolites’ impact on immune cells have garnered extensive research, particularly in anti-tumor immunotherapy, the metabolic reprogramming of neutrophils remains largely unexplored [Citation82].
Besides metabolic reprogramming, neutrophils also experience functional reprogramming in the TME. Specific surface markers that have been suggested to identify TAN subpopulations include CD101 and CD177 [Citation83,Citation84], which are associated with tumor regression, and CD117, PD-L1, CD170, lectin-like low-density lipoprotein receptor-1 (LOX-1), CD84, and junction adhesion molecule like Gene (JAML) [Citation48], which are associated with T cell immunosuppression and tumor progression (). Under the influence of TGF-β, G-CSF, GM-CSF and IL-6 secreted by cancer cells and stromal cells, neutrophils can switch from anti-tumor (N1) phenotype to pro-tumor phenotype (N2). Moreover, TANs or normal neutrophils have been demonstrated to be reprogrammed by various methods to become anti-tumor neutrophils to kill cancer cells. For instance, PPM1D/Wip1 is a negative regulator of the tumor suppressor p53, which is overexpressed in several human solid cancers. Knocking out Ppm1d or chemically inhibiting Wip1 in human or mouse neutrophils enhances the anticancer phenotype, increases p53-dependent expression of co-stimulatory ligands and proliferation of co-cultured cytotoxic T cells [Citation85]. Functional reprogramming for neutrophils has significant implications for targeted therapy, which will be elaborated in the next section.
Figure 3. Neutrophil reprogramming and potential targets. The metabolic mode of neutrophils in the circulation is mainly glycolysis. They can be recruited into the TME by chemokines (such as IL-8, CXCR1, CXCR2, CXCR4), where the low glucose and high lipid environment induces a metabolic reprogramming of neutrophils. The high lipid content in the TME, the upregulation of tumor-derived cytokines (G-CSF and GM-CSF), and the upregulation of lipid transport receptors mediated by STAT3 or STAT5 enhance the uptake of exogenous fatty acids by TANs, resulting in fatty acid oxidation that is immunosuppressive. In addition, there are two subtypes of neutrophils within the TME, the anti-tumor N1 type and the pro-tumor N2 type, and these two different phenotypes of TANs can be converted to each other. Blocking the PD-L1 signal in TANs with immune checkpoint inhibitors, inhibiting the formation and/or structure of NETs, inhibiting the angiogenesis derived from TANs, and regulating the recruitment and polarization of neutrophils in the TME are potential targets for reprogramming TANs in tumors. CXCR1, CXC motif chemokine receptor 1; CXCR2, CXC motif chemokine receptor 2; CXCR4, CXC motif chemokine receptor 4; G-CSF, granulocyte colony stimulating factor; GM-CSF, granulocyte-macrophage colony stimulating factor; IL-6, interleukin 6; IL-8, interleukin 8; JAML, junction adhesion molecule-like protein; LOX-1, lectin-like oxidized low-density lipoprotein (LDL) receptor-1; MMP-9, matrix metalloproteinase-9; NADPH, nicotinamide adenine dinucleotide phosphate; NETs, neutrophil extracellular traps; NO, nitric oxide; PD-1, programmed cell death; PD-L1, programmed death-ligand 1; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor.
4. Targets to regulate neutrophil
Neutrophil-targeted therapies have been proven effective in human cancers [Citation51]. The role of neutrophils in modulating the effects of cancer therapies and the dynamics of neutrophils during these treatment processes in the TME is a novel research topic. The mechanistic understanding of neutrophil-targeted therapy pathways will shed light on the development of novel therapeutic alternatives () [Citation86].
4.1. Targets to reprogram the granulocytic myeloid cells
IL-8 (CXCL-8) is a proinflammatory chemokine that signals via CXCR1 and CXCR2 to recruit polymorphonuclear MDSCs (PMN-MDSCs) and TANs into the TME [Citation87,Citation88]. In a platform clinical trial evaluating vaccine therapy combined with nivolumab as neoadjuvant treatments for resectable PDAC patients, we observed that higher plasma levels of IL-8 were associated with worse outcomes (NCT02451982, Unpublished data). Previous studies have shown that IL-8 can stimulate the proliferation, migration, and survival of pancreatic cancer cells, such as the AsPC-1-cell line, which secretes IL-8 [Citation89]. In a prior study, we investigated whether human anti-IL-8 antibody treatment could enhance the anti-tumor activity of PD-1 blockade in a humanized murine model of PDAC. This model had a reconstituted immune system with human T cells and a mix of CD14+ and CD16+ myeloid cells, which included all monocytic and granulocytic myeloid cells among the peripheral blood mononuclear cells. The findings represent a significant advancement in the field as they demonstrate, for the first time, the potential of IL-8 targeted antibodies to enhance the efficacy of PD-1 checkpoint blockade in combating tumors. Surprisingly, anti-IL-8 antibodies boosted anti-tumor activity through increased granulocytic myeloid cell infiltration and upregulation of both innate and type I cytokine response signals, suggesting a potential role in reprogramming these cells. Most importantly, this study indicated that peripherally derived myeloid cells, such as neutrophils, might have inherent antitumor activities, presumably mediated by the innate immune response [Citation90].
Besides IL-8 targeted therapy, which can augment granulocytic myeloid cells and elicit anti-tumor responses, another potential approach is to inhibit specific cytokines that mediate the recruitment of TANs in TME. Signaling by CXCR1 and CXCR2 are major mechanisms for recruiting neutrophils and MDSCs into the TME which then differentiate into TANs or PMN-MDSCs [Citation91]. CXCR1 is very selective for IL-8, whereas CXCR2 also binds other chemokines, including CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and IL-8. CXCR1 and CXCR2 antagonists have been evaluated as strategies to deplete the TME of immune suppressive N2 TANs and PMN-MDSCs [Citation92,Citation93]. For example, preclinical evaluation of ladarixin, a dual CXCR1/2 antagonist, demonstrated significantly improved activity in a mouse model of PDAC compared to either agent alone [Citation94].
Accumulating evidence suggests that the chemokine CXCL12 interacting with its major receptor CXCR4 plays a key role in controlling neutrophil homeostasis [Citation95]. We previously published a study investigating the combinational treatment of stromal hyaluronan degradation with PD-1 blockade and focal adhesion kinase inhibitor in a murine liver metastasis syngeneic model of PDAC. The results indicated that the triple therapy reduces granulocytes, including G-MDSCs, and decreases CXCR4+ myeloid cells, particularly the CXCR4+ granulocytes. Adding anti-CXCR4 antibody combined with focal adhesion kinase inhibitor and PD-1 blockade significantly decreased metastatic rates in the PDAC liver metastasis model [Citation96].
4.2. Targets to regulate the development and polarization of neutrophils
Lorlatinib is a third-generation, ATP-competitive small-molecule tyrosine kinase inhibitor that has already been approved for the treatment of patients with anaplastic lymphoma kinase positive cancers [Citation97,Citation98]. A study using pre-clinical mouse models of PDAC found that tumor cells activate non-receptor tyrosine kinase FES signaling in neutrophils which can be blocked by lorlatinib in vitro. This research also showed that lorlatinib indirectly inhibits the growth of PDAC at both primary and metastatic locations by regulating neutrophil generation and mobilization from the bone marrow, and by suppressing neutrophil-driven tumor growth within the TME [Citation99].
The polarization of neutrophils into a N2 is mediated by TGF-β, which can be blocked to modulate their function. For instance, TGF-β has been shown to induce the polarization of TAN in the genetically engineered mouse model of oncogenic KRAS-driven lung adenocarcinoma, as well as in lung cancer and mesothelioma transplantation models [Citation100]. Blocking TGF-β in the lung cancer model resulted in the recruitment and activation of N1-type neutrophils, which exhibited tumor-killing activity and suppressed tumor growth [Citation50]. Likewise, in murine models, the phenotypic transition of neutrophils toward an anti-tumor phenotype was facilitated by targeting angiotensin-converting enzyme and angiotensin II type 1 receptor, or nicotinamide phosphoribosyltransferase or CXCR4 [Citation71,Citation101]. Furthermore, in a genetically engineered mouse model of uterine cancer, the anti-tumor potential of neutrophils was activated by reversing high oxygen and hypoxia [Citation102].
4.3. Target neutrophil in the setting of treatment induced TAN influx
When considering neutrophil-targeted immunotherapy, the effect of immunotherapy on causing excessive neutrophil influx into the TME should be taken into account. For instance, targeting CCR2+ tumor associated macrophages (TAMs) induce a compensatory influx of TANs in PDAC patients, which highlights the possibility of immunoregulatory therapies to elicit dynamic myeloid responses in primary tumors, potentially limiting the expected therapeutic effects. In tumor-bearing animals, the simultaneous targeting of CXCR2+ TANs and CCR2+ TAMs has shown the capacity to inhibit compensatory myeloid cell influx and induce a more robust anti-tumor immune response [Citation103].
4.4. Target PD-L1 immune checkpoint signaling on neutrophil
PD-L1 is expressed on several types of immune cells including human neutrophils to induce immunosuppressive functions. In a mouse model of colon cancer, PD-L1+ TANs not only influence adaptive immune cells but also directly engage with PD-1+ NK cells, leading to the downregulation of the anti-tumor immune activity of NK cells. Conversely, PD-L1 inhibitors can restore the anti-tumor immune activity of NK cells [Citation104]. Furthermore, the heightened recruitment of neutrophils is concomitant with the creation of NETs, and studies have indicated that this process is also implicated in TAN-induced resistance to anti-PD-1 blockade [Citation105]. Another pre-clinical study demonstrated that inhibition of TANs with lorlatinib could improve the therapeutic effect of anti-PD-L1 inhibitors in PDAC, indicating that targeting TANs is a possible strategy to prevent PD-1/PD-L1-induced tumor immune evasion [Citation99].
4.5. Target NETs
NETs are implicated in angiogenesis, immunosuppression, and metastasis of cancer. Therefore, inhibiting NET formation or disrupting their structure have been suggested as novel therapeutic strategies for cancer [Citation106]. IL-17 attracts neutrophils and induces NET formation, which impairs the cytotoxicity of CD8+ T cells in the TME of PDAC [Citation29]. Recent research indicates that IL-17 May be derived from infiltrating immune cells and facilitate the progression of PanIN, and that chronic pancreatitis may promote tumorigenesis by increasing IL-17 production [Citation107]. In mice with IL-17 overexpression, antibodies against IL-17 or its receptor reduce PanIN and neutrophil infiltration. Such antibodies are under investigation in clinical trials for the treatment of autoimmune diseases such as psoriasis and rheumatoid arthritis (NCT01350804, NCT05320159), which may have potential applications in PDAC in the future. Moreover, a study showed that exenatide, a glucagon-like peptide-1 receptor agonist, can diminish NET formation by limiting ROS levels. The combination of exenatide and PD-1 blockade can augment anti-tumor CD8+ T cell responses in lung cancer and colon cancer [Citation108]. Serum DNA and citrullinated histone H3 represent indicators of NET formation. Recognizing the detrimental role of NETs in cancer progression, deoxyribonuclease (DNase) has emerged as a promising therapeutic candidate. Its ability to degrade circulating free DNA effectively dismantles the structural and functional integrity of NETs, as demonstrated in a preclinical model of pancreatic cancer. Notably, DNase I administration significantly reduced the accumulation of fibroblasts in liver metastases, suggesting its potential to attenuate NET-mediated cancer invasion and metastasis [Citation47,Citation109].
5. Clinical development of treatments targeting neutrophils in PDAC
Strong evidence has shown that TANs play a remarkable role in the development of various cancers. Based on a deeper understanding of the components within the TME of PDAC, neutrophils, especially TANs, as one of the predominant infiltrating immune cells, have emerged as a potential therapeutic target [Citation42]. Currently, many preclinical studies using different mouse models have demonstrated promising results in targeting neutrophils in pancreatic cancer. Notably, some of these interventions have progressed to the clinical testing stage in PDAC patients ().
Table 1. Clinical trials targeting neutrophils in treating PDAC.
As mentioned earlier, IL-8 has proved to be a good target for neutrophil-targeted therapy in pre-clinical PDAC studies [Citation90]. In our platform clinical trial for resectable PDAC patients (NCT02451982), we prospectively collected paired tumor specimens before and after neoadjuvant treatment with GVAX and nivolumab. By applying multi-omic analyses, we found that higher densities of TANs within the TME after GVAX plus nivolumab combinational treatment portend poorer overall survival. Mechanistic analyses revealed that nivolumab modulates CD4+ T cell chemotaxis signaling pathways, potentially linked to the degranulation of CD11b+ neutrophils [Citation114]. These findings illuminate immune regulation by PD-1 in PDAC, paving the way for the development of more efficacious therapeutic combinations, including regulators of TANs. Therefore, we added an anti-IL-8 antibody (BMS-986253) to the same ongoing platform trial (NCT02451982) as a next step in improving effector T cell responses.
SX-682 is an orally bioavailable, potent allosteric inhibitor of CXCR1 and CXCR2 that able to inhibit tumor MDSCs trafficking and enhance NK cell immunotherapy in rodent models of head and neck cancer [Citation110]. SX-682 is currently being tested in clinical trials in combination with immune checkpoint inhibitors such as nivolumab [Citation115] (NCT04477343) and tislelizumab (NCT05604560) in patients with metastatic and resectable PDAC, respectively. AZD5069 is a highly selective small molecule antagonist of CXCR2 receptors that has been shown to deplete TAN density in patients with metastatic castration resistant prostate cancer [Citation111]. It is currently tested combined with durvalumab, an anti-PD-L1 monoclonal antibody in metastatic PDAC patients, however, no outcomes have been reported at present (NCT02583477).
AMD3100 is the only marketed CXCL12/CXCR4 antagonist and was approved by the FDA. A completed dose escalation study (NCT02179970) evaluated the immune responses induced by AMD3100 in patients with advanced pancreatic cancer. The results demonstrated that CXCR4 impaired the function of the chemokine receptors that mediate the intratumoral accumulation of immune cells, and treatment with AMD3100 for 1 week significantly reduced the levels of circulating tumor DNA and circulating CXCL8 in patients with colorectal cancer and PDAC [Citation112]. In addition, a phase 2 clinical trial is currently evaluating the safety and clinical activity of AMD3100 in combination with cemiplimab (PD-1 blockade) in patients with metastatic pancreatic cancer (NCT04177810) [Citation116].
BL-8040 is a small synthetic peptide that binds to CXCR4, which demonstrates higher affinity and longer receptor occupancy when compared to other CXCR4 inhibitors such as AMD3100 [Citation113,Citation117,Citation118]. A phase IIa, open-label, two-cohort study (NCT02826486) assessed the safety, efficacy and immunobiological effects of BL-8040 with pembrolizumab and chemotherapy in metastatic PDAC. The results demonstrated that BL-8040 can increase CD8+ effector T cell tumor infiltration, decrease MDSCs, and further decrease circulating Tregs, which combined with the clinical data suggest that the co-inhibition of CXCR4 and PD-1 immune checkpoint may favor the outcome of chemotherapy in PDAC patients [Citation119].
At present, considerable strides have been made by researchers in the domain of targeting neutrophils, with several pharmaceutical agents progressing to the clinical trial phase. However, regrettably, the unique immunosuppressive TME inherent to PDAC presents formidable challenges. Factors such as the development of interstitial fibrosis, expansion of MDSCs, compromised cellular infiltration, as well as the presence of physical and chemical barriers, alongside diminished cytokine secretion collectively contribute to the ‘cold’ tumor phenotype characteristic of pancreatic cancer. This renders immunotherapeutic interventions less efficacious. Nevertheless, the intricate interplay between neutrophils and other immune effectors is well recognized. Modulating the mobilization and differentiation of neutrophils holds promise in influencing the functionality of surrounding immune cells. Such interventions have the potential to recalibrate the response of cancer cells to immunotherapy, thereby augmenting therapeutic outcomes.
6. Conclusion
Various strategies targeting immune suppressor cells within the TME to overcome or mitigate immune suppression have been investigated. In conclusion, immunotherapy targeting neutrophils in PDAC is an emerging field with considerable potential. However, the current body of research on neutrophils in this domain is relatively scarce compared to that on macrophages and T cells. Nevertheless, the unique heterogeneity and adaptability of neutrophils offer opportunities for their selective modulation, which may have implications for the future treatment of PDAC.
7. Expert opinion
Neutrophils are considered one of the major immune cells in the TME of PDAC. In recent years, more and more studies have revealed the important role of neutrophils in PDAC tumorigenesis, progression, and metastasis, attracting research attention to neutrophils in PDAC. This article reviews the dual anti-tumor and pro-tumor effects of neutrophils in PDAC tumorigenesis and progression and summarizes the current progress of cancer therapies targeting neutrophils. Finally, the status on clinical trials testing these experimental therapies that target TANs is updated. In summary, this review summarizes the latest research progress on neutrophils in PDAC, which has important clinical significance for promoting the translation of TAN-targeting therapeutics from preclinical research to clinical research.
Neutrophils have unique plasticity and heterogeneity, which enable them to exert both anti-tumor and pro-tumor effects in tumors. In PDAC, neutrophils are presented predominantly with the N2 subtype of TANs, which exhibit immunosuppressive effects; therefore, inhibition of the recruitment, mobilization, and polarization of these tumor-promoting neutrophil would potentially achieve the therapeutic benefit. However, prior published mechanistic and preclinical studies demonstrated that neutrophils have antitumor tumor capabilities including tumor-killing and tumor antigen presenting potentials. According to our group’s published preclinical study, neutrophils in PDAC can be reprogrammed by anti-IL-8 antibodies, suggesting that reprogramming neutrophils instead of depleting neutrophils would be an ideal therapeutic strategy. Future research needs to identify and develop more specific and effective reprogramming targets to support the anti-tumor effects of neutrophils. NETs are an important form of inflammatory cell death of neutrophils, and tumor cells can exploit NETosis to suppress effector T cell functions and enhance the tumor’s ability to grow, invade, and metastasize. Therefore, inhibiting the formation of NETs or destroying their structure is one of the potential therapeutic strategies reprogramming neutrophils.
It should be noted that simply targeting neutrophils would unlikely yield sufficient antitumor activities. On another hand, in PDAC, immune checkpoint inhibitors as a monotherapy is in general ineffective. Therefore, the combination immunotherapy shall be the strategy for developing the therapies targeting neutrophils. For PDAC and other immunogenically ‘cold’ tumors, by targeting TANs or NETosis, antitumor effector T cells may be unleashed. For immunogenically ‘hot’ tumors, targeting TANs or NETosis may overcome the refractory mechanisms for immune checkpoint inhibitor treatments. Therapies that targeting neutrophils may be synergized with standard of care chemotherapy and/or radiation therapy to activate the innate immune response, leading to a better T cell priming in PDACs.
In summary, there are still many aspects of neutrophil research in PDAC that remain unclear. Studies targeting neutrophils represent an emerging and promising research field in immunology. To fully elucidate the potential therapeutic applications of neutrophil-based interventions, further research in this area is of paramount importance. Given these findings, it is evident that researchers are striving to enhance our multifaceted understanding of the role of neutrophils in cancer immunology. This has the potential to significantly improve the efficacy and specificity of future therapeutic approaches for various cancer patients.
Article highlights
Neutrophils participate in the tumorigenesis, progression, and metastasis of PDAC.
KRAS gene mutations and microbiome drive the infiltration of neutrophils in the tumor microenvironment of PDAC.
The plasticity of neutrophils within the TME allows them to be dynamically reprogrammed to exhibit either pro-tumor or anti-tumor functions.
Potential strategies for neutrophil-based therapy include reprogramming the granulocytic myeloid cells, modulating the recruitment and polarization of neutrophils, applying immune checkpoint inhibitors, and inhibiting the formation and/or structure of NETs.
The main targets of clinical trials on neutrophils in PDAC are IL-8, CXCR1/2, and CXCR4.
Abbreviation
ARG1, arginase 1; CAFs, carcinoma-associated fibroblasts; C3, complement 3; C3R, complement 3 receptor; CXCL1, CXC motif chemokine ligand 1; CXCR2, CXC motif chemokine receptor 2; DNase, Deoxyribonuclease; ECM, extracellular matrix; G-CSF, granulocyte colony stimulating factor; G-MDSCs, granulocytic MDSCs; GM-CSF, granulocyte-macrophage CSF; IFN-β, interferon β; IL, interleukin; JAML, junction adhesion molecule-like protein; LPS, lipopolysaccharide; LOX-1, lectin-like oxidized low-density lipoprotein (LDL) receptor-1; MMP-9, matrix metalloproteinase-9; MPO, myeloperoxidase; MDSCs, myeloid-derived suppressor cells; NADPH, nicotinamide adenine dinucleotide phosphate; NETs, neutrophil extracellular traps; NK, natural killer; NO, nitric oxide; PanIN, pancreatic intraepithelial neoplasia; PDAC, pancreatic ductal adenocarcinoma; PD-1, programmed cell death-1; PD-L1, programmed death-ligand 1; PMN-MDSCs, polymorphonuclear MDSCs; ROS, reactive oxygen species; TAN, tumor-associated neutrophils; TAMs, tumor associated macrophages; TGF-β, transforming growth factor-β; TLR-2, toll-like receptor 2; TME, tumor microenvironment; TNF-α, tumor necrosis factor-α; Treg, regular T; VEGF, vascular endothelial growth factor.
Declaration of interest
L Zheng receives grant support from Bristol-Meyer Squibb, Merck, AstraZeneca, iTeos, Amgen, NovaRock, Inxmed, Halozyme and Abmeta. L Zheng is a paid consultant/Advisory Board Member at Biosion, Alphamab, NovaRock, Ambrx, Akrevia/Xilio, QED, Novagenesis, Snow Lake Capitals, Amberstone, Pfizer, Tavotek, and Mingruizhiyao. L Zheng holds shares at Alphamab, Amberstone, Mingruizhiyao, and Cellaration. The other authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Authors’ contributions
Concept was conceived by L Zheng and K Li. The strategy and the overall study were designed by K Li and L Zheng Original draft manuscript was written by Y Jin and E Christenson. Manuscript was reviewed and revised was by K Li and L Zheng. Supervision was made by L Zheng.
Additional information
Funding
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019 [article]. Ca A Cancer J Cli. 2019 Jan;69(1):7–34. doi: 10.3322/caac.21551
- McGuigan A, Kelly P, Turkington RC, et al. Pancreatic cancer: a review of clinical diagnosis, epidemiology, treatment and outcomes [review]. WJG. 2018 Nov 21;24(43):4846–4861. doi: 10.3748/wjg.v24.i43.4846
- Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition [letter]. N Engl J Med. 2017 Dec 21;377(25):2500–2501. doi: 10.1056/NEJMc1713444
- Laklai H, Miroshnikova YA, Pickup MW, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression [article]. Nature Med. 2016 May;22:(5):497–505.
- Li K-Y, Yuan J-L, Trafton D, et al. Pancreatic ductal adenocarcinoma immune microenvironment and immunotherapy prospects. Chron Dis Transl Med. 2020;6(1):6–17. doi: 10.1016/j.cdtm.2020.01.002
- Basturk O, Hong S-M, Wood LD, et al. A revised classification system and recommendations from the Baltimore consensus meeting for neoplastic precursor lesions in the pancreas [article]. Am J Surg Pathol. 2015 Dec;39:(12):1730–1741.
- Rezaee N, Barbon C, Zaki A, et al. Intraductal papillary mucinous neoplasm (IPMN) with high-grade dysplasia is a risk factor for the subsequent development of pancreatic ductal adenocarcinoma [article]. HPB. 2016 Mar;18:(3):236–246.
- Oyama H, Tada M, Takagi K, et al. Long-term risk of malignancy in branch-duct intraductal papillary mucinous neoplasms [article]. Gastroenterology. 2020 Jan;158:(1):226–237.
- Matsuda Y, Furukawa T, Yachida S, et al. The prevalence and clinicopathological characteristics of high-grade pancreatic intraepithelial neoplasia: autopsy study evaluating the entire pancreatic parenchyma [article]. Pancreas. 2017 May;46:(5):658–664.
- Kamisawa T, Wood LD, Itoi T, et al. Pancreatic cancer [review]. Lancet. 2016;388(10039):73–85. doi: 10.1016/S0140-6736(16)00141-0
- Mizrahi JD, Surana R, Valle JW, et al. Pancreatic cancer [review]. Lancet. 2020 Jun 27;395(10242):2008–2020. doi: 10.1016/S0140-6736(20)30974-0
- Guo J, Xie K, Zheng S. Molecular biomarkers of pancreatic intraepithelial neoplasia and their implications in early diagnosis and therapeutic intervention of pancreatic cancer [review]. Int J Biol Sci. 2016;12(3):292–301. doi: 10.7150/ijbs.14995
- Vitone LJ, Greenhalf W, Howes NR, et al. Hereditary pancreatitis and secondary screening for early pancreatic cancer [review]. Rocz Akad Med Bialymst. 2005;50:73–84.
- Wachsmann MB, Pop LM, Vitetta ES. Pancreatic ductal adenocarcinoma: a review of immunologic aspects [review]. J Invest Med. 2012 Apr;60(4):643–663. doi: 10.2310/JIM.0b013e31824a4d79
- Jura N, Archer H, Bar-Sagi D. Chronic pancreatitis, pancreatic adenocarcinoma and the black box in-between [review]. Cell Res. 2005 Jan;15(1):72–77. doi: 10.1038/sj.cr.7290269
- Feig C, Gopinathan A, Neesse A, et al. The pancreas cancer microenvironment [article]. Clin Cancer Res. 2012 Aug 15;18(16):4266–4276. doi: 10.1158/1078-0432.CCR-11-3114
- Zheng L, Xue J, Jaffee EM, et al. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma [review]. Gastroenterology. 2013 May;144:(6):1230–1240.
- Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion [article]. Cancer Res. 2007 Oct 1;67(19):9518–9527. doi: 10.1158/0008-5472.CAN-07-0175
- Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer [review]. Nat Rev Gastroenterol Hepatol. 2020 Mar;17(3):153–168. doi: 10.1038/s41575-019-0245-4
- Tape CJ, Ling S, Dimitriadi M, et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation (vol 165, pg 910, 2016) [correction]. Cell. 2016 Jun 16;165(7):1818–1818. doi: 10.1016/j.cell.2016.05.079
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma [review]. N Engl J Med. 2014 Sep 11;371(11):1039–1049. doi: 10.1056/NEJMra1404198
- Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity [article]. Gut. 2014 Nov;63:(11):1769–1781.
- Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer [article]. Cancer Cell. 2011 Apr 12;19(4):456–469. doi: 10.1016/j.ccr.2011.03.009
- Zhang Y, Yan W, Collins MA, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance [article]. Cancer Res. 2013 Oct 15;73(20):6359–6374. doi: 10.1158/0008-5472.CAN-13-1558-T
- O’Hayer KM, Brady DC, Counter CM. ELR plus CXC chemokines and oncogenic ras-mediated tumorigenesis [article]. Carcinogenesis. 2009 Nov;30(11):1841–1847. doi: 10.1093/carcin/bgp198
- Yu C, Merza M, Luo L, et al. Inhibition of Ras signalling reduces neutrophil infiltration and tissue damage in severe acute pancreatitis [article]. Eur J Pharmacol. 2015 Jan 5;746:245–251. doi: 10.1016/j.ejphar.2014.11.020
- Steele CW, Karim SA, Leach JDG, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma [article]. Cancer Cell. 2016 Jun 13;29(6):832–845. doi: 10.1016/j.ccell.2016.04.014
- McAllister F, Leach SD. Targeting IL-17 for pancreatic cancer prevention [editorial material]. Oncotarget. 2014 Oct 30;5(20):9530–9531. doi: 10.18632/oncotarget.2618
- Zhang Y, Chandra V, Riquelme Sanchez E, et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer [article]. J Exp Med. 2020 Dec;217(12). doi: 10.1084/jem.20190354
- He D, Li H, Yusuf N, et al. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells [article]. J Immunol. 2010 Mar 1;184(5):2281–2288. doi: 10.4049/jimmunol.0902574
- Koh A, De Vadder F, Kovatcheva-Datchary P, et al. From dietary fiber to Host physiology: short-chain fatty acids as key bacterial metabolites [review]. Cell. 2016 Jun 2;165(6):1332–1345. doi: 10.1016/j.cell.2016.05.041
- Wang Y, Du J, Wu X, et al. Crosstalk between autophagy and microbiota in cancer progression [review]. Mol Cancer. 2021 Dec 11;20(1). doi: 10.1186/s12943-021-01461-0
- Tilg H, Zmora N, Adolph TE, et al. The intestinal microbiota fuelling metabolic inflammation [Review]. Nat Rev Immunol. 2020 Jan;20:(1):40–54.
- Sepich-Poore GD, Zitvogel L, Straussman R, et al. The microbiome and human cancer [review]. Science. 2021;371(6536):eabc4552. doi: 10.1126/science.abc4552
- Demkow U. Neutrophil extracellular traps (NETs) in cancer Invasion, evasion and metastasis [review]. Cancers (Basel). 2021 Sep;13(17):4495. doi: 10.3390/cancers13174495
- Tan Q, Ma X, Yang B, et al. Periodontitis pathogen porphyromonas gingivalis promotes pancreatic tumorigenesis via neutrophil elastase from tumor-associated neutrophils [article]. Gut Microbes. 2022 Dec 31;14(1). doi: 10.1080/19490976.2022.2073785
- Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis [article]. Proc Natl Acad Sci, USA. 2006 Aug 15;103(33):12493–12498. doi: 10.1073/pnas.0601807103
- Wu L, Saxena S, Awaji M, et al. Tumor-associated neutrophils in cancer: going pro [review]. Cancers (Basel). 2019 Apr;11:(4):564.
- Galdiero MR, Varricchi G, Loffredo S, et al. Potential involvement of neutrophils in human thyroid cancer [article]. PLOS ONE. 2018 Jun 28;13(6):e0199740. doi: 10.1371/journal.pone.0199740
- Li T-J, Jiang Y-M, Hu Y-F, et al. Interleukin-17-producing neutrophils link inflammatory stimuli to disease progression by promoting angiogenesis in gastric cancer [article]. Clin Cancer Res. 2017 Mar 15;23(6):1575–1585. doi: 10.1158/1078-0432.CCR-16-0617
- Zhang Y, Zoltan M, Riquelme E, et al. Immune cell production of interleukin 17 induces stem cell features of pancreatic intraepithelial neoplasia cells [article]. Gastroenterology. 2018 Jul;155:(1):210–223.e3.
- Jin L, Kim HS, Shi J. Neutrophil in the pancreatic tumor microenvironment [review]. Biomolecules. 2021 Aug;11(8):1170. doi: 10.3390/biom11081170
- Cools-Lartigue J, Spicer J, McDonald B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Investig. 2013 Aug;123(8):3446–3458. Article. doi: 10.1172/JCI67484
- Spicer JD, McDonald B, Cools-Lartigue JJ, et al. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells [article]. Cancer Res. 2012 Aug 15;72(16):3919–3927. doi: 10.1158/0008-5472.CAN-11-2393
- Pieterse E, Rother N, Garsen M, et al. Neutrophil extracellular traps drive endothelial-to-mesenchymal transition [article]. Arteriosclerosis Thrombosis Vasc Biol. 2017 Jul;37:(7):1371–1379.
- Yang L, Liu Q, Zhang X, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25 [Article]. Nature. 2020 Jul 2;583(7814):133–138. doi: 10.1038/s41586-020-2394-6
- Takesue S, Ohuchida K, Shinkawa T, et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancer‑associated fibroblasts. Int J Oncol. 2020 Feb;56:(2):596–605.
- Jaillon S, Ponzetta A, Di Mitri D, et al. Neutrophil diversity and plasticity in tumour progression and therapy [review]. Nat Rev Cancer. 2020 Sep;20:(9):485–503.
- Hedrick CC, Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted [review]. Nat Rev Immunol. 2022 Mar;22(3):173–187. doi: 10.1038/s41577-021-00571-6
- Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN [article]. Cancer Cell. 2009 Sep 8;16(3):183–194. doi: 10.1016/j.ccr.2009.06.017
- Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer [review]. Nat Rev Clin Oncol. 2019 Oct;16(10):601–620. doi: 10.1038/s41571-019-0222-4
- Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment [review]. Blood. 2019 May 16;133(20):2159–2167. doi: 10.1182/blood-2018-11-844548
- Gershkovitz M, Caspi Y, Fainsod-Levi T, et al. TRPM2 mediates neutrophil killing of disseminated tumor cells [article]. Cancer Res. 2018 May 15;78(10):2680–2690. doi: 10.1158/0008-5472.CAN-17-3614
- Granot Z, Henke E, Comen EA, et al. Tumor entrained neutrophils inhibit seeding in the premetastatic lung [article]. Cancer Cell. 2011 Sep 13;20(3):300–314. doi: 10.1016/j.ccr.2011.08.012
- Beauvillain C, Delneste Y, Scotet M, et al. Neutrophils efficiently cross-prime naive T cells in vivo [article]. Blood. 2007 Oct 15;110(8):2965–2973. doi: 10.1182/blood-2006-12-063826
- Tillack K, Breiden P, Martin R, et al. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses [article]. J Immunol. 2012 Apr 1;188(7):3150–3159. doi: 10.4049/jimmunol.1103414
- Sun R, Luo J, Li D, et al. Neutrophils with protumor potential could efficiently suppress tumor growth after cytokine priming and in presence of normal NK cells [article]. Oncotarget. 2014 Dec 30;5(24):12621–12634. doi: 10.18632/oncotarget.2181
- Hirschhorn D, Budhu S, Kraehenbuehl L, et al. T cell immunotherapies engage neutrophils to eliminate tumor antigen escape variants [article]. Cell. 2023 Mar 30;186(7):1432–1447.e17. doi: 10.1016/j.cell.2023.03.007
- Matlung HL, Babes L, Zhao XW, et al. Neutrophils kill antibody-opsonized cancer cells by trogoptosis [article]. Cell Rep. 2018 Jun 26;23(13):3946–3959. doi: 10.1016/j.celrep.2018.05.082
- Comen E, Wojnarowicz P, Seshan VE, et al. TNF is a key cytokine mediating neutrophil cytotoxic activity in breast cancer patients [article]. NPJ Breast Cancer. 2016;2(1):16009–16009. doi: 10.1038/npjbcancer.2016.9
- Finisguerra V, Di Conza G, Di Matteo M, et al. MET is required for the recruitment of anti-tumoural neutrophils [article]. Nature. 2015 Jun 18;522(7556):349–353. doi: 10.1038/nature14407
- Gungor N, Knaapen AM, Munnia A, et al. Genotoxic effects of neutrophils and hypochlorous acid [article]. Mutagenesis. 2010 Mar;25:(2):149–154.
- Rotondo R, Barisione G, Mastracci L, et al. IL-8 induces exocytosis of arginase 1 by neutrophil polymorphonuclears in nonsmall cell lung cancer [article]. Intl J Cancer. 2009 Aug 15;125(4):887–893. doi: 10.1002/ijc.24448
- Yang T-H, St John LS, Garber HR, et al. Membrane-associated proteinase 3 on granulocytes and acute myeloid leukemia inhibits T cell proliferation [article]. J Immunol. 2018 Sep 1;201(5):1389–1399. doi: 10.4049/jimmunol.1800324
- Mishalian I, Bayuh R, Eruslanov E, et al. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17—A new mechanism of impaired antitumor immunity. Intl J Cancer. 2014 Sep 1;135(5):1178–1186. doi: 10.1002/ijc.28770
- Michaeli J, Shaul ME, Mishalian I, et al. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFα and NO-dependent mechanism, promoting a tumor-supportive environment [article]. Oncoimmunology. 2017;6(11):e1356965. doi: 10.1080/2162402X.2017.1356965
- Kornepati AV, Vadlamudi RK, Curiel TJ. Programmed death ligand 1 signals in cancer cells. Nat Rev Cancer. 2022;22(3):174–189. doi: 10.1038/s41568-021-00431-4
- Wang T-T, Zhao Y-L, Peng L-S, et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway [article]. Gut. 2017 Nov;66:(11):1900–1911.
- He G, Zhang H, Zhou J, et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma [article]. J Exp Clin Cancer Res. 2015 Nov 18;34(1). doi: 10.1186/s13046-015-0256-0
- Segal BHH, Giridharan T, Suzuki S, et al. Neutrophil interactions with T cells, platelets, endothelial cells, and of course tumor cells [review]. Immunol Rev. 2023 Mar;314(1):13–35. doi: 10.1111/imr.13178
- Yang J, Kumar A, Vilgelm AE, et al. Loss of CXCR4 in myeloid cells enhances antitumor immunity and reduces melanoma growth through NK cell and FASL mechanisms [article]. Cancer Immunol Res. 2018 Oct;6:(10):1186–1198.
- Das A, Monteiro M, Barai A, et al. MMP proteolytic activity regulates cancer invasiveness by modulating integrins [article]. Sci Rep. 2017 Oct 27;7(1). doi: 10.1038/s41598-017-14340-w
- Albrengues J, Shields MA, Ng D, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361(6409):eaao4227. Article: doi: 10.1126/science.aao4227
- Seubert B, Gruenwald B, Kobuch J, et al. Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice [article]. Hepatology. 2015 Jan;61:(1):238–248.
- Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells [article]. Nature. 2015 Dec 17;528(7582):413–417. doi: 10.1038/nature16140
- Poli V, Zanoni I. Neutrophil intrinsic and extrinsic regulation of NETosis in health and disease [review]. Trends Microbiol. 2023 Mar;31(3):280–293. doi: 10.1016/j.tim.2022.10.002
- Papayannopoulos V. Neutrophil extracellular traps in immunity and disease [review]. Nat Rev Immunol. 2018 Feb;18(2):134–147. doi: 10.1038/nri.2017.105
- Thiam HR, Wong SL, Wagner DD, et al. Cellular mechanisms of NETosis [review]. Annu Rev Cell Dev Biol. 2020;36(1):191–218. doi: 10.1146/annurev-cellbio-020520-111016
- Lood C, Blanco LP, Purmalek MM, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease [article]. Nature Med. 2016 Feb;22:(2):146–153.
- Rice CM, Davies LC, Subleski JJ, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression [article]. Nat Commun. 2018 Nov 30;9(1). doi: 10.1038/s41467-018-07505-2
- Al-Khami AA, Zheng L, Del Valle L, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells [article]. Oncoimmunology. 2017;6(10):e1344804. doi: 10.1080/2162402X.2017.1344804
- Lian X, Yang K, Li R, et al. Immunometabolic rewiring in tumorigenesis and anti-tumor immunotherapy [review]. Mol Cancer. 2022 Jan 21;21(1). doi: 10.1186/s12943-021-01486-5
- Evrard M, Kwok IWH, Chong SZ, et al. Developmental analysis of bone marrow neutrophils reveals populations specialized in expansion, trafficking, and effector functions [article]. Immunity. 2018 Feb 20;48(2):364–379. doi: 10.1016/j.immuni.2018.02.002
- Zhou G, Peng K, Song Y, et al. CD177+ neutrophils suppress epithelial cell tumourigenesis in colitis-associated cancer and predict good prognosis in colorectal cancer [article]. Carcinogenesis. 2018 Feb;39:(2):272–282.
- Uyanik B, Goloudina AR, Akbarali A, et al. Inhibition of the DNA damage response phosphatase PPM1D reprograms neutrophils to enhance anti-tumor immune responses [article]. Nat Commun. 2021 Jun 15;12(1). doi: 10.1038/s41467-021-23330-6
- Jiang W, Li X, Xiang C, et al. Neutrophils in pancreatic cancer: potential therapeutic targets [review]. Front Oncol. 2022 Oct 17; 12;12. doi: 10.3389/fonc.2022.1025805
- Waugh DJJ, Wilson C. The interleukin-8 pathway in cancer [article]. Clin Cancer Res. 2008 Nov 1;14(21):6735–6741. doi: 10.1158/1078-0432.CCR-07-4843
- Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases [review]. Theranostics. 2017;7(6):1543–1588. doi: 10.7150/thno.15625
- Fu S, Chen X, Lin H-J, et al. Inhibition of interleukin 8/C‑X-C chemokine receptor�1,/2 signaling reduces malignant features in human pancreatic cancer cells. Int J Oncol. 2018 Jul;53:(1):349–357.
- Li P, Rozich N, Wang J, et al. Anti-IL-8 antibody activates myeloid cells and potentiates the anti-tumor activity of anti-PD-1 antibody in the humanized pancreatic cancer murine model [article]. Cancer Lett. 2022 Jul 28;539:215722. doi: 10.1016/j.canlet.2022.215722
- Yuen KC, Liu L-F, Gupta V, et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade [article]. Nature Med. 2020 May;26:(5):693–698.
- Che J, Song R, Chen B, et al. Targeting CXCR1/2: the medicinal potential as cancer immunotherapy agents, antagonists research highlights and challenges ahead [review]. Eur J Med Chem. 2020 Jan 1;185:111853. doi: 10.1016/j.ejmech.2019.111853
- Han Z-J, Li Y-B, Yang L-X, et al. Roles of the CXCL8-CXCR1/2 axis in the tumor microenvironment and immunotherapy [review]. Molecules. 2022 Jan;27:(1):137.
- Piro G, Carbone C, Agostini A, et al. CXCR1/2 dual-inhibitor ladarixin reduces tumour burden and promotes immunotherapy response in pancreatic cancer [article]. Br J Cancer. 2023 Jan 19;128(2):331–341. doi: 10.1038/s41416-022-02028-6
- De Filippo K, Rankin SM. CXCR 4, the master regulator of neutrophil trafficking in homeostasis and disease. Eur J Clin Invest. 2018 Nov; 48;48(S2). doi: 10.1111/eci.12949
- Blair AB, Wang J, Davelaar J, et al. Dual stromal targeting sensitizes pancreatic adenocarcinoma for anti-programmed cell death protein 1 therapy [article]. Gastroenterology. 2022 Nov;163:(5):1267–1280.
- Zou HY, Friboulet L, Kodack DP, et al. PF-06463922, an ALK/ROS1 Inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models [article]. Cancer Cell. 2015 Jul 13;28(1):70–81. doi: 10.1016/j.ccell.2015.05.010
- Shaw AT, Felip E, Bauer TM, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial [article]. Lancet Oncol. 2017 Dec;18:(12):1590–1599.
- Nielsen SR, Strobech JE, Horton ER, et al. Suppression of tumor-associated neutrophils by lorlatinib attenuates pancreatic cancer growth and improves treatment with immune checkpoint blockade [article]. Nat Commun. 2021 Jun 7;12(1). doi: 10.1038/s41467-021-23731-7
- Shaul ME, Levy L, Sun J, et al. Tumor-associated neutrophils display a distinct N1 profile following TGFβ modulation: a transcriptomics analysis of pro- vs. antitumor TANs [article]. Oncoimmunology. 2016;5(11):e1232221. doi: 10.1080/2162402X.2016.1232221
- Pylaeva E, Harati MD, Spyra I, et al. NAMPT signaling is critical for the proangiogenic activity of tumor-associated neutrophils [article]. Intl J Cancer. 2019 Jan 1;144(1):136–149. doi: 10.1002/ijc.31808
- Mahiddine K, Blaisdell A, Ma S, et al. Relief of tumor hypoxia unleashes the tumoricidal potential of neutrophils [article]. J Clin Investig. 2020 Jan;130:(1):389–403.
- Nywening TM, Belt BA, Cullinan DR, et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma [article]. Gut. 2018 Jun;67:(6):1112–1123.
- Sun R, Xiong Y, Liu H, et al. Tumor-associated neutrophils suppress antitumor immunity of NK cells through the PD-L1/PD-1 axis [article]. Transl Oncol. 2020 Oct;13:(10):100825.
- Kwantwi LB. Overcoming anti-PD-1/PD-L1 immune checkpoint blockade resistance: the role of macrophage, neutrophils and mast cells in the tumor microenvironment [review]. Clin Exp Med. 2023 Nov;23(7):3077–3091. doi: 10.1007/s10238-023-01059-4
- Poto R, Cristinziano L, Modestino L, et al. Neutrophil extracellular traps, angiogenesis and cancer [article]. Biomedicines. 2022 Feb;10:(2):431.
- McAllister F, Bailey JM, Alsina J, et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia [article]. Cancer Cell. 2014 May 12;25(5):621–637. doi: 10.1016/j.ccr.2014.03.014
- Chen D, Li Q, Liang H, et al. Exenatide enhanced the antitumor efficacy on PD-1 blockade by the attenuation of neutrophil extracellular traps [article]. Biochem Biophys Res Commun. 2022 Sep 3;619:97–103. doi: 10.1016/j.bbrc.2022.06.052
- Alekseeva LA, Sen’kova AV, Zenkova MA, et al. Targeting circulating SINEs and LINEs with DNase I provides metastases inhibition in experimental tumor models [article]. Mol Ther Nucleic Acids. 2020 Jun 5;20:50–61. doi: 10.1016/j.omtn.2020.01.035
- Greene S, Robbins Y, Mydlarz WK, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 Inhibitor, enhances NK-Cell immunotherapy in head and neck cancer models [article]. Clin Cancer Res. 2020 Mar;26:(6):1420–1431.
- Guo C, Sharp A, Vogl U, et al. A phase (Ph) I/II trial of the CXCR2 antagonist AZD5069 in combination with enzalutamide (ENZA) in patients (pts) with metastatic castration resistant prostate cancer (mCRPC) [meeting abstract]. Ann Oncol. 2022 Sep;33:(7):S745–S745.
- Biasci D, Smoragiewicz M, Connell CM, et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response [article]. Proc Natl Acad Sci USA. 2020 Nov 17;117(46):28960–28970. doi: 10.1073/pnas.2013644117
- Peled A, Abraham M, Avivi I, et al. The high-affinity CXCR4 antagonist BKT140 is safe and induces a robust mobilization of human CD34+ cells in patients with multiple myeloma [article]. Clin Cancer Res. 2014 Jan 15;20(2):469–479. doi: 10.1158/1078-0432.CCR-13-1302
- Li K, Tandurella JA, Gai J, et al. Multi-omic analyses of changes in the tumor microenvironment of pancreatic adenocarcinoma following neoadjuvant treatment with anti-PD-1 therapy [article]. Cancer Cell. 2022 Nov 14;40(11):1374–1391.e7. doi: 10.1016/j.ccell.2022.10.001
- Dunne RF, Ullman NA, Belt BA, et al. A phase I study to evaluate the safety and tolerability of SX-682 in combination with PD-1 inhibitor as maintenance therapy for unresectable pancreatic adenocarcinoma [meeting abstract]. JCO. 2022 Feb 1;40(4):TPS631–TPS631. doi: 10.1200/JCO.2022.40.4_suppl.TPS631
- Shin SM, Hernandez A, Coyne E, et al. Abstract 2270: combination of CXCR4 antagonist and anti-PD1 therapy results in significant mobilization and increased infiltration of myeloid cells into the metastatic liver microenvironment of PDAC. Cancer Res. 2023;83(7_Supplement):2270–2270. doi: 10.1158/1538-7445.AM2023-2270
- Abraham M, Beider K, Wald H, et al. The CXCR4 antagonist 4F-benzoyl-TN14003 stimulates the recovery of the bone marrow after transplantation [article]. Leukemia. 2009 Aug;23:(8):1378–1388.
- Abraham M, Biyder K, Begin M, et al. Enhanced unique pattern of hematopoietic cell mobilization induced by the CXCR4 antagonist 4F-benzoyl-TN14003 [article]. Stem Cells (Dayton, Ohio). 2007 Sep;25:(9):2158–2166.
- Bockorny B, Semenisty V, Macarulla T, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial [article]. Nat Med. 2020 Jun;26:(6):878–885.