
ABSTRACT
Introduction
Anterior segment dysgeneses are congenital anomalies that predominantly involve the cornea, iris, anterior chamber, iridocorneal angle structures, and ciliary body, but may also have posterior segment findings. Genetic causes of these diseases have gradually been identified over the last 30 years. The clinical genetics combined with animal studies have given important insight into the pathogenesis of these diseases.
Areas Covered
An overview of anterior segment development will be followed by a review of the genetics and pathogenesis underlying primary congenital glaucoma, aniridia, Axenfeld-Rieger syndrome, Peters anomaly, sclerocornea, congenital ectropion uvea, and megalocornea/megalophthalmos.
Expert Opinion
Lack of genetic testing is a critical barrier for increasing our understanding of these diseases and ultimately improving outcomes. Genetic testing is important for patients and gives greater insight into genotype–phenotype correlations regarding treatments and prognosis. Nevertheless, there is a significant percentage of patients with no identified genetic cause. This demonstrates the great opportunity for gene discovery, which requires wider access to whole-genome sequencing and increased support for research efforts and funding. The increased knowledge of genetics and basic science will ultimately lead to the development of novel molecularly targeted treatments.
1. Introduction
Anterior segment dysgeneses (ASDs) encompass a heterogenous group of congenital eye diseases in which there are varying degrees of abnormalities involving the cornea, iris, anterior chamber, iridocorneal angle structures, and ciliary body [Citation1,Citation2]. There is still much to be learned regarding the underlying genetics and pathogenesis of these diseases. In some of the diseases which have autosomal dominant transmission, large pedigrees have facilitated the identification of causative genes. However, in many of these diseases autosomal recessive inheritance and sporadic cases make identification of new genes more challenging. As gene therapies as well as molecularly targeted treatments have become a reality, it is important to better understand their genetics and pathogenesis. This will open up the possibility of novel vision-saving treatments for children with these rare, blinding conditions.
While the clinical findings and management of primary congenital glaucoma (PCG), aniridia, Axenfeld-Rieger syndrome (ARS), iridogoniodysgenesis, Peters anomaly, sclerocornea, congenital ectropion uvea (CEU), anterior megalophthalmos and megalocornea were detailed in the first part of this review series, this segment will focus on the genetics and pathogenesis of ASD. A brief overview of eye development is first presented to lay the groundwork [Citation2,Citation3]. Subsequently, our current knowledge regarding the genetics and diseases basis will be detailed.
2. Ocular development
Ocular development involves complex interactions between the neural epithelial-derived optic cup, the surrounding neural crest-derived periocular mesenchyme, and the overlying surface ectoderm () [Citation4]. The optic vesicles that form as outpouchings of the anterior diencephalon interact with the overlying surface ectoderm to induce formation of the bilayered optic cup [Citation5–7]. The pigmented outer layer gives rise to the retinal pigment epithelium, while the non-pigmented inner layer forms the photoreceptors [Citation8,Citation9]. Further, the ciliary body epithelium (both pigmented and non-pigmented), posterior epithelium of the iris, and the pupillary sphincter and dilator muscles are derived from the hinge region [Citation4]. The optic fissure spans the length of the cup on the inferomedial aspect and transmits the hyaloid vasculature which supports lens and anterior segment development [Citation10]. Concurrent with optic cup formation, neural crest cells are specified at the neural plate border during neural tube closure and migrate into the pericoular mesenchyme [Citation11–16]. Together with dorsal-ventral and anterior-posterior patterning of the retina, neural crest cells regulate optic fissure closure [Citation17–19].
Figure 1. Eye development.
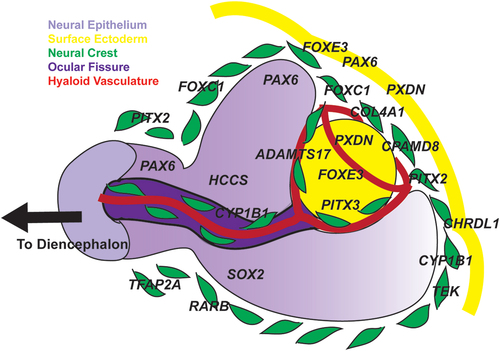
The hinge region of the optic cup induces the lens placode, which will then form the lens vesicle as it separates from the overlying surface ectoderm [Citation8,Citation20]. Neural crest cells migrate into the newly formed anterior segment via two routes: 1) between the surface ectoderm and the hinge of the optic cup and 2) through the optic fissure [Citation21,Citation22]. These neural crest cells have endothelial-like characteristics and form a layer over the iris and on the posterior surface of the cornea [Citation23]. Classic human fetal histology studies suggest that these cells migrate into the anterior segment in three waves. The first wave gives rise to the corneal stroma and endothelium while the second forms the iris stroma. The final wave then becomes the trabecular meshwork. In addition, the ciliary body stroma and the majority of the sclera are derived from neural crest cells.
The interactions between neural crest cells with surface epithelium in the cornea and the neural epithelial-derived optic cup in the iris and ciliary body are critical for differentiation and development of the anterior segment [Citation22,Citation24]. At the juxtaposition of these structures is the trabecular meshwork and Schlemm’s canal. During the first few post-natal months, the trabecular meshwork matures as extracellular matrix secreted by neural crest-derived cells is organized into beams and channels [Citation25]. Aqueous humor flows through the meshwork into Schlemm’s canal whose endothelial cells originate from adjacent blood vessels and share molecular profiles with both vascular and lymphatic endothelial cells [Citation26].
Genotype–phenotype correlations in combination with animal studies have helped to identify the signals involved in ocular morphogenesis and subsequently gain better understanding of the molecular pathogenesis of congenital eye diseases [Citation3].
3. Primary congenital glaucoma (PCG)
The prevalence of PCG varies around the globe with a rate of 1:10,000–20,000 in the United States, but is as high as 1:1,250–2,500 in some Eastern European and Arabic populations [Citation27,Citation28]. The basis for this wide range is largely attributed to the prevalence of autosomal recessive variants in the CYP1B1 gene, which are the most commonly identified genetic cause of PCG. While the majority of cases in the United States are sporadic without family history, both the founder effect and consanguinity have propagated certain CYP1B1 variants in some populations and geographic regions [Citation28,Citation29]. Consequently, while less than 20% of PCG cases in the United States are attributed to CYP1B1 variants, the rate is between 60% and 100% in Saudia Arabia and the Slovakia Roma populations [Citation27,Citation28,Citation30].
The CYP1B1 gene was originally identified as the GLC3A locus on 2p22-p21 [Citation31,Citation32]. CYP1B1 is a cytochrome p450 heme-thiolate monooxygenase enzyme consisting of a hinge region, four conserved helices, and the conserved core structures that bind the target organic substrate, a complement reductase enzyme, and a heme molecule [Citation25]. Over 150 disease-causing variants have been localized throughout the coding region [Citation33]. The Glycine to Glutamic acid (G61E) variant is commonly reported, especially within the Middle East, and is within the hinge region affecting protein folding and activity [Citation25]. This variant is associated with a milder form of PCG which is highly responsive to goniotomy or trabeculotomy surgery [Citation34]. The R390 site is also frequently mutated to either result in an Arginine to Histidine (R390H) or Arginine to Cysteine (R390C) change that disrupts the heme-protein complex [Citation25]. In contrast to the G61E variant, the R390H variant causes a more severe PCG phenotype with worse corneal edema at presentation and less responsiveness to angle surgery [Citation35].
In zebrafish, cyp1b1 is expressed in the neural epithelial-derived optic cup and regulates timing of neural crest migration through the optic fissure [Citation21]. In post-natal mice, Cyp1b1 is expressed in the trabecular meshwork, and knockout mice show normal trabecular meshwork at 1 week of age, but trabecular meshwork collapse and atrophy with subsequent increased IOP by 3 months of age [Citation25,Citation36]. The CYP1B1 enzyme oxidizes organic substrates, including environmental toxins, estradiol, and retinol, but its specific physiologic target within the developing eye and trabecular meshwork remains unknown [Citation37,Citation38]. However, Cyp1b1 deficient mouse trabecular meshwork cells show increased oxidative stress such that antioxidant injections prevented trabecular meshwork atrophy in knockout mice [Citation25]. Nevertheless, this information has not been verified in humans, and thus has yet to show applicability for treatments.
Four other gene loci, GLC3B on 1p36.2-p36.1, GLC3C on 14p24.3, GLC3D on 14q24.2-p24.3, and GLC3E on 9p21 have also been associated with PCG [Citation39–41]. Specific genes at the GLC3B and GLC3C loci have yet to be identified [Citation39,Citation41]. The LTBP2 gene which encodes latent transforming growth factor beta-binding protein 2 has been identified as GLC3D and variants have been predominantly identified in Pakistani, Indian, and Roma/Gypsy families [Citation40]. However, there is debate as to whether the ocular disease associated with LTBP2 variants is true PCG, as the majority of the reported phenotypes display ectopia lentis, microspherophakia, and often a Marfanoid habitus in addition to infantile-onset glaucoma [Citation42]. The GLC3E locus correlates with the TIE2 or TEK gene, which encodes the tunica internal endothelial cell kinase that is part of the angiopoietin (ANGPT)/TEK signaling pathway [Citation26,Citation43]. This pathway has traditionally been associated with vascular development such that knockout mice models are embryonic lethal due to cardiac abnormalities and lack of vasculogenesis [Citation44]. The TEK protein, which is the receptor for the ANGPT ligands is expressed within endothelial cells in Schlemms canal. Conditional knockout mice that overcome early embryonic lethality demonstrate increased IOP due to a hypoplastic trabecular meshwork and Schlemm’s canal [Citation45,Citation46]. In addition, variants in ANGPT1 as well as in the SEVP1 (Sushi, Von Willebrand factor type A, epidermal growth factor, and pentraxin domain containing-1) gene, which modifies TEK expression have also been identified in individuals with PCG [Citation47,Citation48]. Correlation of mice models with human disease histology is limited, and due to low penetrance and rarity of PCG, there is minimal genotype–phenotype data, especially regarding the response to angle surgery in individuals who carry TEK or ANGPT1 variants [Citation48].
In the future, genetic information may help to direct surgical management (e.g. identify which patients will respond to angle surgery versus those who will need angle bypass surgery), yet the current limitations in genetic testing (e.g. insurance coverage, length of time for results) prevents applicability for immediate and urgent surgical interventions. Despite advancements in the field, the yield on genetic testing in PCG within European countries and the US remains low (<40%) [Citation28,Citation49]. Since most cases are sporadic without family history, the identification of new genes remains a slow, arduous, but important process for improving our understanding of angle development and disease pathogenesis.
4. Aniridia
The majority of cases of aniridia are due to autosomal dominant variants in the PAX6 gene on 11p13 [Citation50]. PAX6 encodes a transcription factor that in the neural epithelium demarcates domains of the central nervous system and within the eye is required for retina and optic nerve differentiation as well as cornea and lens formation [Citation20,Citation51]. In the mouse neural epithelial-derived optic vesicle, Pax6 in conjunction with Pax2, Six6, Gsx2, Pax5, and Lhx2 regulates invagination to form the bilayered optic cup and then further retinal differentiation [Citation20,Citation52]. This role in the optic cup accounts for posterior segment findings including optic nerve abnormalities (hypoplasia, dysplasia, morning glory disc) and foveal hypoplasia. Pax6 is also expressed at the interface between the lens placode and the hinge region of the optic cup through the interplay of Sox2, Oct-1, and Foxe3 in the surface ectoderm and inhibition by TGFβ and Wnt signaling in the periocular neural crest [Citation53–55]. In this capacity, Pax6 is critical for the separation of the lens vesicle and migration of ocular neural crest cells into the developing anterior segment.
The PAX6 protein has a paired domain and a homeodomain that are connected via a linker region and both of these bind to DNA through their respective consensus sequences. In addition, there are numerous regulatory regions within the gene as well as downstream in the introns of the adjacent ELP4 gene [Citation56]. More than 500 variants, including substitutions (nonsense and missense), deletions, insertions, and duplications in the PAX6 gene have been reported [Citation50,Citation57–60]. The majority are within the gene and are spread throughout the entire coding sequence. However, variants are clustered within the regions coding for the paired and homeodomain DNA binding sites [Citation61]. Truncating nonsense substitutions and deletion or insertion variants resulting in a premature termination codon are most commonly identified and are typically associated with classic aniridia. Four nonsense substitutions involving cytosine to thymine changes (c.607C>T, c.718C>T, c.781C>T, and c.949C>T) within methylated CpG islands account for one-fifth of reported PAX6 (NM_000280.3) variants [Citation56]. Variants that alter splice sites are also often associated with aniridia as are deletions within regulatory domains in untranslated regions [Citation56,Citation62,Citation63]. Further, deletions downstream of PAX6 in the 11p13 region involving genes including ELP4, IMMP1L, DNAJC24, DCDC1, and DCDC5 have been commonly associated with aniridia as well neurodevelopmental disorders such as developmental delay and autism spectrum disorders [Citation64]. Missense substitutions can involve the DNA binding domains (paired or homeodomain) resulting in altered transactivation of targeted genes. As a result, these phenotypes can be different and include microphthalmia, coloboma, morning glory disc anomaly, Peters anomaly, ASD, and foveal hypoplasia [Citation50,Citation57,Citation59,Citation65–67]. Further, large multi-generational families have demonstrated phenotypic variation with some affected individuals showing aniridia and others displaying other anterior segment (e.g. Peters anomaly) and optic nerve manifestations (e.g. morning glory disc) [Citation63,Citation68].
Further, it is important to note that a proportion of individuals with sporadic aniridia have WAGR syndrome, which is due to a contiguous gene deletion involving at least a portion, if not all of the PAX6 and WT1 genes. While the most common ophthalmic finding is aniridia, Peters anomaly and microphthalmia have also been reported in WAGR syndrome [Citation69]. Further, due to involvement of the WT1 gene, affected individuals commonly have genitourinary anomalies, developmental delay and are at high risk for developing a Wilms tumor [Citation69]. Taken together, any PAX6 variant whether it result in a premature termination codon, amino acid substitution, absence of an allele, or abnormal gene regulation can lead to a variety of ocular phenotypes.
In addition to PAX6 variants, other genetic causes of aniridia have been identified. Variants in PITX2 and FOXC1, while more commonly associated with Axenfeld-Rieger syndrome (discussed below) can cause partial to complete aniridia [Citation70–72]. Interestingly, the aniridia associated with FOXC1 variants shows a high rate of infantile-onset glaucoma (7 of 9 patients). This is in contrast to PAX6 variants in which glaucoma affects approximately 50% of patients, but is less often diagnosed in the first year of life [Citation70,Citation71,Citation73]. In Gillespie syndrome there is iris sphincter aplasia leading to partial aniridia, which is phenotypically distinct from PAX6-related aniridia [Citation72]. Further, Gillespie syndrome is also characterized by developmental delay and cerebellar ataxia and has in some, but not all cases, been linked to ITPR1 variants [Citation74,Citation75]. Specific ACTA2 variants (pArg179*) also display a similar partial aniridia phenotype, but is systemically distinct due to the presence of cardiac abnormalities including patent ductus arterioles and aortopulmonary septal defect [Citation72,Citation76]. In addition, there remains a small percentage (estimated to be 5%) of cases of aniridia in which the genetic cause remains to be identified [Citation72]. Continued genetic testing in affected individuals will improve our understanding of genes associated with aniridia and overall eye development.
5. Axenfeld-Rieger syndrome (ARS) and other anterior segment dysgeneses (ASDs)
Autosomal dominant variants in either the PITX2 or the FOXC1 gene are identified in approximately half of ARS cases as well in other individuals with ASD that do not fit under other clinical diagnoses [Citation77–79]. The PITX2 gene is localized to the 4q25 chromosome and is labeled as Type I ARS while the FOXC1 gene is on the 6p25 chromosome and correlates to Type III ARS [Citation80,Citation81]. The PITX2 gene and FOXC1 gene encode homeobox and forkhead transcription factors, respectively. Both genes are expressed within neural crest cells that are first within the periocular mesenchyme and then subsequently migrate into the anterior segment both through the optic fissure as well as between the neural epithelial-derived optic cup and the overlying surface ectoderm [Citation18,Citation82]. Mice and zebrafish knockout studies have shown that complete absence of Pitx2 or Foxc1, inhibits neural crest migration from the edge of the neural tube, resulting in a more severe phenotype that is embryonic lethal [Citation18,Citation83]. In the heterozygous state, the animal models survive to adulthood and recapitulate the ocular and systemic manifestations [Citation84–86]. The common cranial neural crest origin of many of the anterior segment structures, craniofacial bones and connective tissues, and teeth demonstrates the pathogenic connection between these abnormalities in ARS [Citation87]. Further the cardiac outflow tracts and autonomic innervation of the gastrointestinal tract are also neural crest derived and mice and zebrafish models show that Foxc1 and Pitx2 are required in their development [Citation83,Citation85].
Over 250 families with heterozygous variants in PITX2 have been reported [Citation88]. Nonsense substitution or frameshift deletion or insertion variants resulting in gene truncation are most commonly identified, followed by missense substitutions and then large deletions either within the coding or regulatory regions. Almost 90% of the missense substitutions have been localized to the DNA binding homeodomain of the PITX2 gene. More than 300 families with FOXC1 variants have also been reported with a close to one-third split between missense substitutions, premature truncation variants, and large deletions. Similar to PITX2, more than 75% of the missense substitutions are found within the forkhead DNA binding domain [Citation88]. Various studies have shown differences in the prevalence and type of ocular and systemic findings associated with PITX2 and FOXC1 variants. Iris abnormalities are more common in PITX2 variants while glaucoma was diagnosed earlier in some studies of FOXC1 variants [Citation88,Citation89]. Craniofacial dysmorphism, umbilical abnormalities, and dental anomalies were also more often associated with PITX2 variants. In contrast, hearing loss, cardiovascular defects, skeletal anomalies, and central nervous system abnormalities were more frequently seen with FOXC1 variants [Citation88,Citation90]. However, there is phenotypic variation in ocular and systemic findings within families that carry the same variant.
There are many cases of ARS and ASD which do not have either FOXC1 or PITX2 variants. Other genes associated with ARS and ASD include CPAMD8, COL4A1, CYP1B1, PRDM5, and ADAMTS17. [Citation79,Citation88,Citation91–94] The CPAMD8 gene on 19p13.11 encodes the C3 and PZP Like Alpha-2 Macroglobulin Domain Containing 8 protein, a member of the protease inhibitor I39 family. Autosomal recessive variants in the CPAMD8 gene have been associated with iris stromal hypoplasia with corectopia, posterior embryotoxon, ectopia lentis, ectropion uvea, and congenital cataract [Citation93,Citation95]. One histological sample of an enucleated eye showed apoptosis of the trabecular meshwork cells and disorganized extracellular matrix within the iridocorneal angle [Citation95]. In zebrafish, cpamd8 is expressed within neural crest cells such that mutants show microphthalmia with abnormal iris stroma and collagen extracellular matrix, pharyngeal arch malformation, and pericardial effusion [Citation95]. In humans, CPAMD8 is found early within the lens and retina and then later in development in the iris and cornea [Citation93]. In contrast to zebrafish, CPAMD8 has not been detected within human embryonic neural crest or periocular mesenchyme [Citation95]. Nevertheless, while the role of this family of protease inhibitors is to mediate innate immunity by eliminating pathogens, the molecular function during development has yet to be determined. COL4A1 on 13q34 encodes for the pro-alpha 1 chain required for type IV collagen, and as a major component of basement membranes is ubiquitously expressed [Citation96]. Anterior segment findings are present in some patients with autosomal dominant COL4A1 variants and include cataract, iris hypoplasia, posterior embryotoxon, corneal opacification, microcornea, microphthalmia, and coloboma [Citation92]. In addition, posterior findings including optic nerve hypoplasia, retinal vascular tortuosity, and retinopathy have been described [Citation97]. Importantly, COL4A1 variants are more often associated with significant and potentially life threatening systemic findings such as cerebrovascular lesions including hemorrhagic stroke, kidney failure, and heart arrhythmias [Citation97–99]. Identification of COL4A1 variants may warrant referrals to genetics, neurology, nephrology and cardiology. The PRDM5 gene is on chromosome 4q27 and encodes a PR domain zinc finger protein transcription factor with numerous cellular functions including regulating extracellular matrix maintenance, cell migration and apoptosis. A few variants in the open reading frame of PRDM5 have been described in an autosomal recessive form of Brittle Cornea syndrome [Citation100,Citation101]. There is one report of a family with a missense substitution variant within the zinc finger region that resulted in autosomal dominant transmission of ophthalmic findings including pseudopolycoria, corectopia, and goniodysgenesis and systemic associations such as teeth abnormalities, redundant periumbilical skin, hearing loss, and facial dysmorphism [Citation94]. Notably, COL4A1 is a target of PRDM5 in cultured dermal fibroblasts, however, it is unknown whether this is the mechanism that underlies the pathogenesis of ARS in this family [Citation102]. The ADAMTS17 gene on 15q26.3 encodes for a disintegrin and metalloproteinase enzyme that regulates structure and function of the extracellular matrix [Citation103]. Variants in ADAMTS17 are typically associated with ectopia lentis and microspherophakia, either as isolated ophthalmic findings or as part of Weill-Marchesani Syndrome 4, which also includes systemic findings such as brachydactyly, short stature and ichthyosis [Citation104,Citation105]. There is one report of a patient with a homozygous nonsense substitution variant in ADAMTS17 who had corectopia, pseudopolycoria, iridocorneal adhesions, and ectopia lentis [Citation79]. Further, common in Weill-Marchesani Syndrome 4, this patient also had short stature. The ADAMTS17 protease binds to Fibrillin-1 microfibrils, regulating both Fibronectin and Fibrillin-1 deposition within the extracellular matrix [Citation104]. Fibrillin-1, which is encoded by the FBN1 gene, is abundant within the lens zonules, and FBN1 variants are associated with ectopia lentis in Marfan syndrome. However, the role of ADAMTS17 in anterior segment development is unknown. Taken together, ARS and ASD associated with variants in CPAMD8, COL4A1, PRDM5, and ADAMTS17 are rare, and there is limited genotype–phenotype information correlations [Citation79,Citation88,Citation91–94]. Further, there continue to be patients who do not have a known genetic cause. Wider spread genetic testing is critical for identifying new genes and further understanding and characterizing these anterior segment diseases.
6. Peters anomaly
Embryologically, Peters anomaly is due to the abnormal or incomplete separation of the lens vesicle from the overlying surface ectoderm leading to impaired neural crest cell migration into anterior segment [Citation22]. In type 1 Peters anomaly, there are iridocorneal adhesions that are presumably due to abnormal neural crest cell migration and differentiation into iris and corneal structures. As a result, there is absence of the corneal endothelium underlying an avascular corneal opacity, and the presence of iridocorneal adhesions that can be peripheral to or in the area of the corneal defect [Citation106]. This differs from type 2 Peters anomaly in which the lens vesicle fails to separate from the surface ectoderm such that by ultrasound biomicroscopy the anterior capsule of the lens is indiscernible from the posterior cornea [Citation107]. This leads to a vascularized corneal opacity in the region of the keratolenticular adhesions, which may also be accompanied by peripheral iridocorneal adhesions. However, these phenotypes are not genetically distinct. Numerous genes have been associated with Peters anomaly, including many in which variants are commonly found in other congenital eye diseases as well as genes that otherwise have no other known ophthalmic connections.
PAX6 was the first gene associated with Peters Anomaly, and further studies have shown that PAX6 variants are most commonly identified in these patients [Citation108,Citation109]. The role of PAX6 in lens placode induction and vesicle formation likely underlies the connection between variants and Peters Anomaly [Citation8,Citation110]. As previously mentioned, ocular defects other than aniridia are more often seen with PAX6 missense substitution variants compared to nonsense substitutions, deletions, or insertions [Citation50,Citation57,Citation59,Citation65–67]. Numerous missense variants affecting the paired box domain or the homeodomain have been reported in cases of Peters Anomaly [Citation108,Citation109,Citation111,Citation112]. Additional molecular studies on the first PAX6 variant to be associated with Peters Anomaly, an Arginine to Glycine (R26G) substitution within the paired domain showed that the resultant protein had hypomorphic activity compared to wildtype [Citation108,Citation113]. However, whole gene deletions as well as splice site variants have also been reported in cases of Peters Anomaly further showing the heterogeneity of PAX6 phenotypes [Citation63,Citation109,Citation114].
FOXC1 and PITX2 regulate neural crest migration into the anterior segment and variants in these genes have also been identified in Peters Anomaly. Missense and nonsense substitution variants as well as duplications affecting the FOXC1 gene and missense substitution variants in PITX2 have been reported in individuals with Peters Anomaly [Citation109,Citation115–118]. Notably, some of these mutations had previously been found in patients with ARS or were identified in pedigrees where other family members had characteristic ARS findings [Citation109,Citation115]. A third gene associated with ARS, COL4A1 has also been found in a few cases of Peters Anomaly. These included a deletion-insertion resulting in an amino acid substitution in the 7S domain in a sporadic case and a splicing variant in four family members [Citation109,Citation119]. Additional genetic testing in patients with Peters Anomaly and ARS is likely to give important insight into phenotypic variations with these three genes.
The role of CYP1B1 in regulating neural crest cell migration into the anterior segment may be the basis for its association with Peters Anomaly, which phenotypically is much more severe than PCG [Citation21]. Six patients have been reported with CYP1B1 variants with bilateral Peters Anomaly, five of which also had glaucoma [Citation109,Citation120]. Variants included missense substitutions within the hinge region, helix bundles, and conserved core structures and nonsense and deletions that resulted in premature termination codons. Interestingly, most of the variants had previously been reported in cases of PCG [Citation109,Citation120].
The FOXE3, PITX3, and PXDN genes all play a role in lens development and variants have been found in Peters Anomaly, sclerocornea (described below) and congenital aphakia. The FOXE3 gene on 1p33 encodes a forkhead transcription factor and is a known downstream target of PAX6 within the embryonic lens vesicle [Citation53]. In three cases of Peters Anomaly associated with FOXE3, the variants were missense substitutions resulting in stop-loss with continued translation of the downstream coding region [Citation109,Citation121]. This is a common genetic finding in monoallelic autosomal dominant FOXE3 mutations, and is typically associated with more ‘mild’ ocular findings that are isolated to the anterior segment as compared to microphthalmia with retinal involvement [Citation122]. The PITX3 gene located on chromosome 10q24.32 encodes for a homeodomain transcription factor that has been shown to regulate FOXE3 expression by binding to the 5’upstream regulatory region [Citation123]. Missense, deletion, insertion, and duplication PITX3 variants have all been identified in cases of Peters Anomaly, but many of them are also associated with congenital cataracts and other forms of ASD such as ARS and sclerocornea [Citation109,Citation124,Citation125]. The PXDN gene on 2p25.3 encodes Peroxidasin, a peroxidase that facilitates sulfilimine bond formation in type IV collagen. Loss of protein function likely impairs basement membrane formation within the lens and cornea [Citation126]. In mice, Pxdn together with Foxe3 and Pax6 regulates cell proliferation and differentiation in the lens vesicle and is also expressed within the corneal endothelium and epithelium [Citation127]. There is one report of bi-allelic PXDN variants (an insertion and a missense substitution) in trans that are associated with type 2 Peters anomaly in one eye and anterior persistent fetal vasculature in the other eye. Furthermore, this individual’s identical twin with the same variants showed bilateral microphthalmia, congenital aphakia and sclerocornea [Citation128]. While there are some variants in these genes that are more often associated with Peters Anomaly, there is clinical variability that remains unexplained.
Additional genes have been identified in cases of Peters Anomaly including a few which typically are associated with the microphthalmia-anophthlamia-coloboma spectrum due to their roles in optic cup formation [Citation109]. SOX2 is located at 3q26.33 and encodes a transcription factor expressed within the anterior diencephalon from which the optic vesicles originate. In mice, hypomorphic Sox2 mutants show malformed optic stalks and cups and result in microphthalmic phenotypes [Citation129]. Further, animal models have demonstrated a role for Sox2 in neural retinal differentiation, which is also critical for optic cup maturation and optic fissure closure [Citation130]. Sox2 together with Pax6 regulates lens placode induction, and this may account for the Peters Anomaly phenotype seen with some SOX2 variants [Citation110]. Nonsense substitution, deletion and duplication SOX2 variants have been associated with Peters Anomaly, but it is important to note that these affected individuals often exhibited ipsilateral microphthalmia and/or contralateral anophthalmia, consistent with the previously reported SOX2 phenotypes [Citation109]. The RARB gene on chromosome 3p24.2 encodes the retinoic acid receptor beta protein, which mediates downstream signaling of retinoic acid, an essential morphogen that is derived from vitamin A. The enzymes required for the conversion of vitamin A to retinoic acid as well as those involved in retinoic acid degradation have a specific expression pattern in the optic cup, resulting in a retinoic acid gradient that mediates neural crest migration [Citation82,Citation131]. The neural crest cells within the periocular region in turn direct and support retinal differentiation and anterior-posterior and dorsal-ventral optic cup patterning which is critical for optic fissure closure [Citation82,Citation131]. RARB variants, typically missense substitutions within the ligand-binding domain are typically associated with Microphthalmia, syndromic 12 (MCOPS12) [Citation132]. Peters anomaly in conjunction with microphthalmia has been reported in a RARB missense variant also within the ligand-binding domain [Citation109]. The TFAP2A gene on 6p24.3 encodes a transcription factor that in Xenopus, mouse and zebrafish is expressed in neural crest cells. While Tfap2a is a key regulator in neural crest specification at the neural plate border, there is continued gene expression within migratory and post-migratory cranial neural crest cells [Citation133–136]. This is likely the basis for the craniofacial abnormalities and microphthalmia seen in brachio-oculo-facial syndrome which is typically associated with TFAP2A variants [Citation137,Citation138]. A splice site variant in TFAP2A has been identified in a patient with unilateral Peters anomaly [Citation112]. Following optic cup morphogenesis and optic fissure closure, Tfap2a is expressed in corneal and lens epithelium in zebrafish and mice [Citation139]. This may account for the rare cases of Peters Anomaly associated with TFAP2A variants. The HCCS gene, which is localized to chromosome Xp22.2, encodes the holocytochrome c-type synthase. This protein regulates apoptosis via mitochondrial oxidative phosphorylation. Variants are associated with the X-linked dominant syndrome, Microphthalmia with linear skin defects, that in addition to the microphthalmia and skin findings can affect the heart, nervous system, and genitourinary tract [Citation140,Citation141]. There are a couple of reports of HCCS nonsense substitution variants associated with Peters Anomaly as well as microphthalmia [Citation109,Citation112]. These abnormalities are presumed to be due to increased apoptosis during organ development, which in the case of Peters Anomaly may be related to the dysregulation of cell survival during lens vesicle separation.
Other genes not typically associated with ASD or microphthalmia have also been identified in cases of Peters Anomaly. The NDP gene on Xp11.3 is usually linked to congenital retinal vascular diseases such as Norrie Disease and familial exudative vitreoretinopathy [Citation142–144]. The NDP gene product, Norrin is predominantly within the extracellular matrix and regulates Wnt signaling during angiogenesis [Citation145]. Peters Anomaly either with or without retinal vascular abnormalities has been associated with NDP variants that affect the C-terrminus of the protein; however, the exact molecular basis for the anterior segment findings has yet to be determined [Citation112,Citation143]. The SLC4A11 gene located on chromosome 20p13 encodes a transmembrane channel that regulates oxidative stress via transporting peroxide and hydrogen. Variants are typically associated with both autosomal recessive and autosomal dominant corneal dystrophies, which is likely due to disruption of oxidative stress response within the corneal endothelium [Citation146,Citation147]. There is one report of a patient with compound heterozygous variants (nonsense substitution and missense substitution) with bilateral Peters anomaly and microphthalmia, but the pathogenesis of this finding is unknown [Citation112]. The FLNA gene on Xq28 encodes Filamen A, a protein that interacts with the intracellular actin cytoskeleton to regulate cell migration. Variants are not typically associated with ophthalmic findings, but there is one report of a missense variant in a patient with Peters Anomaly as well as widespread systemic abnormalities such that the individual did not survive the perinatal time frame [Citation112]. In addition, copy number variations in 22q11.2, 8q21, and 13q31–133, have been identified in patients with Peters Anomaly, although specific genes within these regions have not been identified [Citation109,Citation148].
While there may be systemic findings associated with the gene variants discussed above, the most common syndromic form is Peters Plus Syndrome, which is characterized by short stature, cleft palate and lip, brachydactyly, developmental delay, and characteristic craniofacial features [Citation109]. Peters Plus syndrome is attributed to autosome recessive variants in the B3GLCT gene [Citation149]. The B3GLCT gene on 13q12.3 encodes a ubiquitous glucosyltransferase enzyme that is involved in glycosylation of proteins expressed throughout the body. However, the specific role in eye development is unknown [Citation150].
Despite the number of genes that have been associated with Peters Anomaly, genetic causes are identified in less than half of affected individuals [Citation109,Citation112]. Bilateral cases and those with a family history of congenital eye anomalies have higher yield. Nevertheless, genetic testing should be encouraged in all affected individuals in order to better understand the disease pathogenesis.
7. Sclerocornea
Variants in FOXE3, PITX3 and PXDN genes have been associated with sclerocornea. Over 120 individuals with 20 different FOXE3 variants have been reported with both autosomal dominant and autosomal recessive inheritance patterns [Citation122]. Missense substitutions, which only affected the DNA binding forkhead domain, were more common than nonsense substitutions or frameshift deletions or insertions that were found throughout the gene. In addition to sclerocornea, individuals with biallelic (compound heterozygous or homozygous) FOXE3 variants often also had microphthalmia and congenital aphakia [Citation122]. However, due to the sclerocornea the congenital aphakia may have only been diagnosed intraoperatively or with additional imaging such as ultrasound biomicroscopy or optical coherence tomography. While mouse models show phenotypic similarities between Pitx3 and Foxe3 knockouts, PITX3 variants are more commonly associated with congenital cataracts, and there are only a few reports of sclerocornea [Citation151–153]. Compared to FOXE3, there have been fewer reports of biallelic (compound heterozygous or homozygous) PXDN variants associated with sclerocornea and ASD. Missense and nonsense substitutions and frameshift deletions and insertions have been reported, and while corneal involvement is almost universal, microphthalmia, congenital cataract, and glaucoma are variable phenotypes [Citation128,Citation154]. In both FOXE3 and PXDN associated sclerocornea, some individuals also show systemic findings including developmental delay, ventricular septal defect, renal pelvic dilatation, polycystic ovary syndrome, and Arnold-Chiari malformation [Citation122].
In addition, variants in PAX6 and PITX2 have been associated with sclerocornea, especially when found with other anterior segment anomalies [Citation79,Citation155,Citation156]. As a result, the disease pathogenesis is attributed to abnormal lens formation, disruption of neural crest cell migration into the anterior segment and absence of the limbal anlage that demarcates the embryonic cornea from sclera. Lens vesicle formation followed by differentiation is likely critical to demarcating the limbal anlage and accounts for the connection with FOXE3 and PITX3 variants. The anlage signals the steepening of the corneal tissues relative to the sclera and also directs differentiation of neural crest cells into corneal versus scleral fates; however, the exact mechanisms by which PXDN, PAX6, and PITX2 regulate the limbal anlage have yet to be described.
8. Congenital ectropion uvea (CEU)
An argument has been made that bilateral and unilateral cases of CEU are different entities, although in both forms the neural crest-derived endothelial-like cell layer that covers the iris fails to regress. These cells pull the iris pigment epithelium through the pupil, suppress normal iris surface architecture, and cover the angle structures. In bilateral cases, a report from India shows an association with the R390H CYP1B1 variant; however, this has not been confirmed in other populations [Citation35]. There have also been reports of ectropion uvea associated with PAX6 variants, but these cases typically have other ocular findings including partial aniridia, cataracts, limbal stem cell deficiency, and foveal hypoplasia [Citation157,Citation158]. Neurofibromatosis type 1 (NF1), which is due to autosomal dominant variants in the NF1 gene, has been associated with unilateral CEU [Citation159]. The NF1 gene on 17q11.2 encodes the tumor suppressor protein, Neurofibromin that negatively regulates the Ras signaling transduction pathway [Citation160]. While NF1 variants most commonly cause neurofibromas, optic pathway gliomas, and skin findings including Café-au-lait macules and inguinal freckling, CEU is a more rare finding [Citation159,Citation161]. Although the exact molecular pathogenesis of CEU in this setting has not been studied, it likely arises from the common neural crest origin of peripheral neurons and the endothelial-like cell layer covering the developing iris. However, no genetic causes have been identified in non-syndromic isolated cases of unilateral CEU. This may reflect a less severe form due to somatic variants that have yet to be identified due to paucity of available tissue/specimens for genetic testing.
9. Anterior megalophthalmos and megalocornea
Anterior megalophthalmos and megalocornea are related diseases, and the terms are often used interchangeably despite the difference in textbook definitions of whether lens diaphragm and ciliary body are (megalophthalmos) or are not (megalocornea) involved. Nevertheless, cases of megalocornea have been reported with enlarged ciliary body rings in conjunction with zonular abnormalities and iridodonesis especially in the context of cataract surgery [Citation162]. The denotation between megalophthalmos and megalocornea may stem from the identification of CHRDL1 variants in X-linked megalocornea type 1 (MGC1) [Citation163,Citation164]. Multiple disease-causing variants including missense and nonsense substitutions and frameshift deletions and insertions have been reported [Citation165]. The CHRDL1 gene on Xq23 encodes for Chordin-like 1 or ventroptin, an inhibitor of bone morphogenic protein-4 (BMP-4), a member of the transforming growth factor beta (TGF-ß) superfamily that is expressed in the embryonic and adult cornea [Citation166]. BMP-4 and Wnt/ß-catenin signaling act antagonistically to regulate limbal stem cell differentiation and corneal epithelial stratification in induced pluripotent stem cells and adult cornea injury models [Citation167]. Although the Xenopus chrdl1-/- animal recapitulates MGC1 findings, the exact molecular mechanisms that stimulate corneal and anterior segment overgrowth during development have yet to be identified [Citation166].
An autosomal dominant nonsense substitution variant in the SIN3A gene has been identified in siblings with anterior megalophathlmos and Wittveen-Kolk syndrome [Citation168]. The SIN3A gene encodes for a paired amphipathic helix protein that via the histone deacetylase complex regulates transcription throughout the body accounting for a wide range of findings including the characteristic facial features and developmental delay [Citation169]. In addition, connective tissue disorders including Marfan syndrome, osteogenesis imperfecta, and Alport syndrome have been associated, albeit rarely, with anterior megalophthalmos [Citation170]. However, the pathogenesis and specific effects on the anterior segment of the eye in these diseases have not been elucidated.
10. Expert opinion
The genetics of the diseases encompassed under the ASD-heading lags behind that of other eye pathologies such as retinal dystrophies. This has inhibited our understanding of disease pathogenesis and importantly the development of novel therapies. Wide access to genetic testing is a critical barrier that needs to be addressed to improve diagnosis and outcomes. Genetic testing, especially in isolated ocular diseases may not be considered or encouraged by ophthalmologists. This may be for a number of reasons including lack of knowledge of the disease and genetics, the added time this discussion takes in clinic, and poor access to genetic counselors and geneticists with ophthalmic experience. Additionally, insurance companies have a history of denying payment for genetic testing especially if there are no systemic abnormalities or if the results of the test are likely to have minimal effect on the current treatment plan. Nevertheless, genetic testing gives essential information to patients and families on prognosis and heritability of the disease in future generations. Further, genetic testing improves our understanding of disease pathogenesis that could lead to molecularly targeted treatments.
Through genetic testing, variants in known genes can be identified and classified. The second step is to better curate the genetic information with phenotype including ophthalmic findings, response to treatments, and visual outcomes. Improved genotype–phenotype correlations yields valuable insight for response to treatment and overall visual prognosis. Global registries can help especially in rare diseases with data sharing and bringing together thousands of patients across different populations. However, restrictions in sharing de-identified data at academic institutions can be onerous and prevent participation.
Our current knowledge of the genetics of these diseases is far from complete, as signified by the significant percentage of patients in which genetic testing on standard panels yields negative results. This has led to a push for whole-genome sequencing, not only for the affected individual, but also ideally includes the biological parents. Interpretation and analysis of the mountain of data generated from whole-genome sequencing requires research effort and funding to search for new causative genes. This has been a slow, but essential pipeline that needs continued support from clinicians, researchers, and funding institutions in order to gain a more complete genetic picture of these diseases.
Taking a lesson from retinal dystrophies, which are similarly as rare as ASDs, understanding the genetics and basic science has led to one FDA approved gene therapy and many others currently in clinical trials. As a result, genetic testing is much more common and accepted for retinal dystrophies now that there are potential treatments. By improving our knowledge of disease pathogenesis of ASDs, there is a huge potential for molecularly targeted treatments. The prevention of glaucoma with reversal of angle abnormalities especially in PCG, aniridia, ARS, and CEU as well as the repair of corneal defects in aniridia, Peters anomaly, and sclerocornea are attractive, but lofty goals. While stem cell replacement and gene therapy to either replace defective genes or the downstream targets in the case of transcription factors are attractive, other approaches such antioxidant treatment in CYP1B1 variants, enzyme replacement in CPAMD8, ADAMTS17 or PXDN variants or collagen supplementation with COL4A1 variants could be explored. Much work has yet to be done in this field, but the future is wide open for better treatments resulting in fewer surgeries and better visual outcomes in individuals affected with ASD.
Article highlights
Anterior segment dysgeneses are a heterogeneous group of congenital eye anomalies involving the cornea, iris, anterior chamber, iridocorneal angle, and ciliary body.
The pathogenesis of these diseases are due to disruption of eye development including optic cup formation, neural crest migration, lens formation, and differentiation into anterior segment structures.
There are overlapping as well as distinct genetic causes of diseases such as primary congenital glaucoma congenital aniridia, Axenfeld-Rieger syndrome, Peters Anomaly, sclerocornea, congenital ectropion uvea, megalophthalmos, and megalocornea.
Continued genetic testing in patients with anterior segment dysgenesis is important and will give greater insight into genotype–phenotype correlations regarding treatments and prognosis.
Additional work on gene discovery in cases of anterior segment dysgenesis will improve our understanding of disease pathogenesis and may lead to molecularly targeted treatments in the future.
Declaration of interests
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Additional information
Funding
References
- Reis LM, Semina EV. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol. 2011 Sep;22(5):314–24. doi: 10.1097/ICU.0b013e328349412b
- Ito YA, Walter MA. Genomics and anterior segment dysgenesis: a review. Clin Exp Ophthalmol. 2014 Jan;42(1):13–24. doi: 10.1111/ceo.12152
- Ma AS, Grigg JR, Jamieson RV. Phenotype-genotype correlations and emerging pathways in ocular anterior segme Brenda L. Bohnsack1nt dysgenesis. Hum Genet. 2019 Sep;138(8–9):899–915. doi: 10.1007/s00439-018-1935-7
- Miesfeld JB, Brown NL. Eye organogenesis: a hierarchical view of ocular development. Curr Top Dev Biol. 2019;132:351–393.
- Chow RL, Lang RA. Early eye development in vertebrates. Annu Rev Cell Dev Biol. 2001;17(1):255–96. doi: 10.1146/annurev.cellbio.17.1.255
- Zagozewski JL, Zhang Q, Eisenstat DD. Genetic regulation of vertebrate eye development. Clin Genet. 2014 Nov;86(5):453–60. doi: 10.1111/cge.12493
- Abitbol MM. Networks of genes governing the development of optic and otic vesicles: implications for eye and ear development. Invest Ophthalmol Vis Sci. 2015 Feb 5;56(2):892. doi: 10.1167/iovs.15-16420
- Fuhrmann S. Eye morphogenesis and patterning of the optic vesicle. Curr Top Dev Biol. 2010;93:61–84.
- Cardozo MJ, Sánchez-Bustamante E, Bovolenta P. Optic cup morphogenesis across species and related inborn human eye defects. Development. 2023 Jan 15;150(2). doi: 10.1242/dev.200399
- Lingam G, Sen AC, Lingam V, et al. Ocular coloboma-a comprehensive review for the clinician. Eye (Lond). 2021 Aug;35(8):2086–2109.
- Beebe DC, Coats JM. The lens organizes the anterior segment: specification of neural crest cell differentiation in the avian eye. Dev Biol. 2000;220(2):424–431. doi: 10.1006/dbio.2000.9638
- Creuzet S, Vincent C, Couly G. Neural crest derivatives in ocular and periocular structures. Int J Dev Biol. 2005;49(2–3):161–171. doi: 10.1387/ijdb.041937sc
- Sauka-Spengler T, Bronner-Fraser M. Development and evolution of the migratory neural crest: a gene regulatory perspective. Curr Opin Genet Dev. 2006;16(4):360–366. doi: 10.1016/j.gde.2006.06.006
- Theveneau E, Mayor R. Neural crest delamination and migration: from epithelium-to-mesenchyme transition to collective cell migration. Dev Biol. 2012;366(1):34–54. doi: 10.1016/j.ydbio.2011.12.041
- Simões-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142(2):2420257. doi: 10.1242/dev.105445
- Schille C, Schambony A. Signaling pathways and tissue interactions in neural plate border formation. Neurogenesis (Austin). 2017;4(1):e1292783. doi: 10.1080/23262133.2017.1292783
- Kulesa PM, Bailey CM, Kasemeier-Kulesa JC, et al. Cranial neural crest migration: new rules for an old road. Dev Biol. 2010;344(2):543–554. doi: 10.1016/j.ydbio.2010.04.010
- Chawla B, Schley E, Williams AL, et al. Retinoic Acid and pitx2 regulate early neural crest survival and migration in craniofacial and ocular development. Birth Defects Res B Dev Reprod Toxicol. 2016;107(3):126–135. doi: 10.1002/bdrb.21177
- Eason J, Williams AL, Chawla B, et al. Differences in neural crest sensitivity to ethanol account for the infrequency of anterior segment defects in the eye cmpared with craniofacial anomalies in a zebrafish model of fetal alcohol syndrome. Birth Defects Res. 2017;109(15). doi: 10.1002/bdr2.1069
- Canto-Soler MV, Adler R. Optic cup and lens development requires Pax6 expression in the early optic vesicle during a narrow time window. Dev Biol. 2006;294(1):119–132. doi: 10.1016/j.ydbio.2006.02.033
- Williams AL, Eason J, Chawla B, et al. Cyp1b1 regulates ocular fissure closure through a retinoic acid–independent pathway. Invest Ophthalmol Vis Sci. 2017;58(2):1084–1097. doi: 10.1167/iovs.16-20235
- Williams AL, Bohnsack BL. The ocular neural crest: specification, migration, and then what? Front Cell Dev Biol. 2020;8:595896. doi: 10.3389/fcell.2020.595896
- McMenamin PG. A morphological study of the inner surface of the anterior chamber angle in pre and postnatal human eyes. Curr Eye Res. 1989 Jul;8(7):727–39. doi: 10.3109/02713688909025808
- Seo S, Chen L, Liu W, et al. Foxc1 and Foxc2 in the neural crest are required for ocular anterior segment development. Invest Ophthalmol Vis Sci. 2017;58(3):1368–1377. doi: 10.1167/iovs.16-21217
- Zhao Y, Wang S, Sorenson CM, et al. Cyp1b2 mediates periostin regulation of trabecular meshwork development by suppression of oxidative stress. Mol Cell Bill. 2013;33(21):4225–4240. doi: 10.1128/MCB.00856-13
- Souma T, Tompson SW, Thomson BR, et al. Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest. 2016 Jul 1;126(7):2575–87. doi: 10.1172/JCI85830
- Haddad A, Ait Boujmia OK, El Maaloum L, et al. Meta-analysis of CYP1B1 gene mutations in primary congenital glaucoma patients. Eur J Ophthalmol. 2021 Nov;31(6):2796–2807. doi: 10.1177/11206721211016308
- Shah M, Bouhenni R, Benmerzouga I. Geographical variability in CYP1B1 mutations in primary congenital glaucoma. J Clin Med. 2022 Apr 6;11(7):2048. doi: 10.3390/jcm11072048
- Plásilová M, Stoilov I, Sarfarazi M, et al. Identification of a single ancestral CYP1B1 mutation in Slovak Gypsies (roms) affected with primary congenital glaucoma. J Med Genet. 1999 Apr;36(4):290–4.
- Wiggins RE, Tomey KF. The results of glaucoma surgery in aniridia. Arch Ophthalmol. 1992;110(4):503–505. doi: 10.1001/archopht.1992.01080160081036
- Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum Mol Genet. 1997 Apr;6(4):641–647. doi: 10.1093/hmg/6.4.641
- Stoilov I, Akarsu AN, Alozie I, et al. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P450 1B1. Am J Hum Genet. 1998;62(3):573–584. doi: 10.1086/301764
- Chouiter L, Nadifi S. Analysis of CYP1B1 gene mutations in patients with primary congenital glaucoma. J Pediatr Genet. 2017 Dec;6(4):205–214. doi: 10.1055/s-0037-1602695
- Zhao Y, Sorenson CM, Sheibani N. Cytochrome P450 1B1 and primary congenital glaucoma. J Oophthalmic Vis Res. 2015;10(1):60–67. doi: 10.4103/2008-322X.156116
- Kaushik S, Choudhary S, Kaur A, et al. Neonatal-onset congenital ectropion uveae may be caused by a distinct CYP1B1 pathologic variant. Am J Ophthalmol. 2022 Jul;239:54–65.
- Teixeira LB, Zhao Y, Dubielzig RR, et al. Ultrastructural abnormalities of the trabecular meshwork extracellular matrix in Cyp1b1-deficient mice. Vet Pathol. 2015 Mar;52(2):397–403.
- Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005 Sep 28;227(2):115–24. doi: 10.1016/j.canlet.2004.10.007
- Yin H-C, Tseng H-P, Chung H-Y, et al. Influence of TCDD on zebrafish CYP1B1 transcription during development. Toxicol Sci. 2008;103(1):158–168. doi: 10.1093/toxsci/kfn035
- Akarsu AN, Turacli ME, Aktan SG, et al. A second locus (GLC3B) for primary congenital glaucoma (buphthalmos) maps to the 1p36 region. Hum Mol Genet. 1996 Aug;5(8):1199–1203.
- Ali M, McKibbin M, Booth A, et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am J Hum Genet. 2009 May;84(5):664–71.
- Chen X, Chen Y, Wang L, et al. Confirmation and further mapping of the GLC3C locus in primary congenital glaucoma. Front Biosci (Landmark Ed). 2011 Jun 1;16(6):2052–2059. doi: 10.2741/3838
- Désir J, Sznajer Y, Depasse F, et al. LTBP2 null mutations in an autosomal recessive ocular syndrome with megalocornea, spherophakia, and secondary glaucoma. Eur J Hum Genet. 2010 Jul;18(7):761–7.
- Qiao Y, Chen Y, Tan C, et al. Screening and functional analysis of TEK mutations in Chinese children with primary congenital glaucoma. Front Genet. 2021;12:764509. doi: 10.3389/fgene.2021.764509
- Sato TN, Qin Y, Kozak CA, et al. Tie-1 and tie-2 define another class of putative receptor tyrosine kinase genes expressed in early embryonic vascular system. Proc Natl Acad Sci USA. 1993 Oct 15;90(20):9355–8. doi: 10.1073/pnas.90.20.9355
- Thomson BR, Heinen S, Jeansson M, et al. A lymphatic defect causes ocular hypertension and glaucoma in mice. J Clin Invest. 2014 Oct;124(10):4320–4.
- Kim J, Park DY, Bae H, et al. Impaired angiopoietin/Tie2 signaling compromises Schlemm’s canal integrity and induces glaucoma. J Clin Invest. 2017 Oct 2;127(10):3877–3896. doi: 10.1172/JCI94668
- Thomson BR, Souma T, Tompson SW, et al. Angiopoietin-1 is required for Schlemm’s canal development in mice and humans. J Clin Invest. 2017 Dec 1;127(12):4421–4436. doi: 10.1172/JCI95545
- Young TL, Whisenhunt KN, Jin J, et al. SVEP1 as a genetic modifier of TEK-Related primary congenital glaucoma. Invest Ophthalmol Vis Sci. 2020 Oct 1;61(12):6. doi: 10.1167/iovs.61.12.6
- Yu-Wai-Man C, Arno G, Brookes J, et al. Primary congenital glaucoma including next-generation sequencing-based approaches: clinical utility gene card. Eur J Hum Genet. 2018 Nov;26(11):1713–1718.
- Glaser T, Jepeal L, Edwards JG, et al. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nerve defects. Nat Genet. 1994;7(4):463–471. doi: 10.1038/ng0894-463
- Davis N, Yoffe C, Raviv S, et al. Pax6 dosage requirements in iris and ciliary body differentiation. Dev Biol. 2009;333(1):132–142. doi: 10.1016/j.ydbio.2009.06.023
- Reza HM, Takahashi Y, Yasuda K. Stage-dependent expression of Pax6 in optic vesicle/cup regulates patterning genes through signaling molecules. Differentiation. 2007;75(8):726–736. doi: 10.1111/j.1432-0436.2007.00168.x
- Blixt A, Landgren H, Johansson BR, et al. Foxe3 is required for morphogenesis and differentiation of the anterior segment of the eye and is sensitive to Pax6 gene dosage. Dev Biol. 2007;302(1):218–229. doi: 10.1016/j.ydbio.2006.09.021
- Grocott T, Johnson S, Bailey AP, et al. Neural crest cells organize the eye via TGF-β and canonical wnt signalling. Nat Commun. 2011;2(1):265. doi: 10.1038/ncomms1269
- Kamachi Y, Uchikawa M, Tanouchi A, et al. Pax6 and SOX2 form a co-DNA binding partner complex that regulates initiation of lens development. Genes Dev. 2001;15(10):1272–1286. doi: 10.1101/gad.887101
- Lima Cunha D, Arno G, Corton M, et al. The spectrum of PAX6 mutations and genotype-phenotype correlations in the eye. Genes (Basel). 2019 Dec 17;10(12):1050. doi: 10.3390/genes10121050
- Prosser J, van Heyningen V. PAX6 mutations reviewed. Hum Mutat. 1998;11(2):93–108. doi: 10.1002/(SICI)1098-1004(1998)11:2<93:AID-HUMU1>3.0.CO;2-M
- Tzoulaki I, White IMS, Hanson IM. PAX6mutations: genotype-phenotype correlations. BMC Genet. 2005;6(1):27. doi: 10.1186/1471-2156-6-27
- Cvekl A, Callaerts P. Pax6: 25th anniversary and more to learn. Exp Eye Res. 2017;156:10–21. doi: 10.1016/j.exer.2016.04.017
- Daruich A, Duncan M, Robert MP, et al. Congenital aniridia beyond black eyes: from phenotype and novel genetic mechanisms to innovative therapeutic approaches. Prog Retin Eye Res. 2023 Jul;95:101133.
- Tzoulaki I, White IM, Hanson IM. PAX6 mutations: genotype-phenotype correlations. BMC Genet. 2005 May 26;6(1):27. doi: 10.1186/1471-2156-6-27
- Plaisancié J, Tarilonte M, Ramos P, et al. Implication of non-coding PAX6 mutations in aniridia. Hum Genet. 2018 Oct;137(10):831–846.
- Wang GM, Prasov L, Al-Hasani H, et al. Phenotypic variation in a four-generation family with aniridia carrying a novel PAX6 mutation. J Ophthalmol. 2018;2018:1–10. doi: 10.1155/2018/5978293
- Wawrocka A, Krawczynski MR. The genetics of aniridia - simple things become complicated. J Appl Genet. 2018;59(2):151–159. doi: 10.1007/s13353-017-0426-1
- Brown A, McKie M, van Heyningen V, et al. The human PAX6 mutation database. Nuc Acid Res. 1998;26:259–264. doi: 10.1093/nar/26.1.259
- Dubey SK, Mahalaxmi N, Vijayalakshmi P, et al. Mutational analysis and genotype-phenotype correlations in southern Indian patients with sporadic and familial aniridia. Mol Vis. 2015;27(21):88–97.
- Hingorani M, Williamson KA, Moore AT, et al. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Invest Ophthalmol Vis Sci. 2009;50(6):2581–2590. doi: 10.1167/iovs.08-2827
- Azuma N, Yamaguchi Y, Handa H, et al. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet. 2003 Jun;72(6):1565–70.
- Duffy KA, Trout KL, Gunckle JM, et al. Results from the WAGR syndrome patient registry: characterization of WAGR spectrum and recommendations for care management. Front Pediatr. 2021;9:733018. doi: 10.3389/fped.2021.733018
- Ito YA, Footz TK, Berry FB, et al. Severe molecular defects of a novel FOXC1 W152G mutation result in aniridia. Invest Ophthalmol Vis Sci. 2009 Aug;50(8):3573–9.
- Ansari M, Rainger J, Hanson IM, et al. Genetic analysis of ‘PAX6-negative’ individuals with aniridia or Gillespie syndrome. PLoS One. 2016;11(4):e0153757. doi: 10.1371/journal.pone.0153757
- Hall HN, Williamson KA, FitzPatrick DR. The genetic architecture of aniridia and Gillespie syndrome. Hum Genet. 2019 Sep;138(8–9):881–898. doi: 10.1007/s00439-018-1934-8
- Sadagopan KA, Liu GT, Capasso JE, et al. Anirdia-like phenotype caused by 6p25 dosage aberrations. Am J Med Genet A. 2015 Mar;167(3):524–8.
- Gerber S, Alzayady KJ, Burglen L, et al. Recessive and dominant De novo ITPR1 mutations cause Gillespie syndrome. Am J Hum Genet. 2016 May 5;98(5):971–980. doi: 10.1016/j.ajhg.2016.03.004
- McEntagart M, Williamson KA, Rainger JK, et al. A restricted repertoire of De novo mutations in ITPR1 cause gillespie syndrome with evidence for dominant-negative effect. Am J Hum Genet. 2016 May 5;98(5):981–992. doi: 10.1016/j.ajhg.2016.03.018
- Regalado ES, Mellor-Crummey L, De Backer J, et al. Clinical history and management recommendations of the smooth muscle dysfunction syndrome due to ACTA2 arginine 179 alterations. Genet Med. 2018 Oct;20(10):1206–1215.
- Strungaru MH, Dinu I, Walter MA. Genotype-phenotype correlations in axenfeld-rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci. 2007;48(1):228–237. doi: 10.1167/iovs.06-0472
- Leis LM, Tyler RC, Volkmann Kloss BA, et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet. 2012;20(12):1224–1233. doi: 10.1038/ejhg.2012.80
- Ma A, Yousoof S, Grigg JR, et al. Revealing hidden genetic diagnoses in the ocular anterior segment disorders. Genet Med. 2020 Oct;22(10):1623–1632.
- Kulak SC, Kozlowski K, Semina EV, et al. Mutation in the RIEG1 gene in patients with iridogoniodysgenesis syndrome. Hum Mol Genet. 1998;7(7):1113–1117. doi: 10.1093/hmg/7.7.1113
- Suzuki K, Nakamura M, Amano E, et al. Case of chromosome 6p25 terminal deletion associated with Axenfeld–Rieger syndrome and persistent hyperplastic primary vitreous. Am J Med Genet A. 2006;140(5):503–508. doi: 10.1002/ajmg.a.31085
- Bohnsack BL, Kasprick D, Kish PE, et al. A zebrafish model of Axenfeld-Rieger syndrome reveals thatpitx2regulation by retinoic acid is essential for ocular and craniofacial development. Invest Ophthalmol Vis Sci. 2012;53(1):7–22. doi: 10.1167/iovs.11-8494
- Kitamura K, Miura H, Miyagawa-Tomita S, et al. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development. 1999;126(24):5749–5758. doi: 10.1242/dev.126.24.5749
- Evans AL, Gage PJ. Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum Mol Genet. 2005;14(22):3347–3359. doi: 10.1093/hmg/ddi365
- French CR, Seshadri S, Destefano AL, et al. Mutation of FOXC1 and PITX2 induces cerebral small-vessel disease. J Clin Invest. 2014;124(11):4877–4881. doi: 10.1172/JCI75109
- Chen L, Gage PJ. Heterozygous Pitx2 null mice accurately recapitulate the ocular features of Axenfeld-Rieger Syndrome and congenital glaucoma. Invest Ophthalmol Vis Sci. 2016;57(11):5023–5030. doi: 10.1167/iovs.16-19700
- Minoux M, Rijli FM. Molecular mechanisms of cranial neural crest cell migration and patterning in craniofacial development. Development. 2010 Aug;137(16):2605–21. doi: 10.1242/dev.040048
- Zhou L, Wang X, An J, et al. Genotype-phenotype association of PITX2 and FOXC1 in axenfeld-rieger syndrome. Exp Eye Res. 2023 Jan;226:109307. doi: 10.1016/j.exer.2022.109307
- Prem Senthil M, Knight LSW, Taranath D, et al. Comparison of anterior segment abnormalities in individuals with FOXC1 and PITX2 variants. Cornea. 2022 Aug 1;41(8):1009–1015. doi: 10.1097/ICO.0000000000003020
- French CR. Mechanistic insights into axenfeld-rieger syndrome from zebrafish foxc1 and pitx2 mutants. Int J Mol Sci. 2021;22(18):10001. doi: 10.3390/ijms221810001
- Chavarria-Soley G, Michels-Rautenstrauss K, Caliebe A, et al. Novel CYP1B1 and known PAX6 mutations in anterior segment dysgenesis (ASD). J Glaucoma. 2006 Dec;15(6):499–504.
- Sibon I, Coupry I, Menegon P, et al. COL4A1 mutation in axenfeld-rieger anomaly with leukoencephalopathy and stroke. Ann Neurol. 2007 Aug;62(2):177–184.
- Cheong SS, Hentschel L, Davidson AE, et al. Mutations in CPAMD8 cause a unique form of autosomal-recessive anterior segment dysgenesis. Am J Hum Genet. 2016 Dec 1;99(6):1338–1352. doi: 10.1016/j.ajhg.2016.09.022
- Micheal S, Siddiqui SN, Zafar SN, et al. Whole exome sequencing identifies a heterozygous missense variant in the PRDM5 gene in a family with axenfeld-rieger syndrome. Neurogenetics. 2016 Jan;17(1):17–23.
- Bonet-Fernández JM, Aroca-Aguilar JD, Corton M, et al. CPAMD8 loss-of-function underlies non-dominant congenital glaucoma with variable anterior segment dysgenesis and abnormal extracellular matrix. Hum Genet. 2020 Oct;139(10):1209–1231.
- Hudson BG, Reeders ST, Tryggvason K. Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of goodpasture and Alport syndromes and diffuse leiomyomatosis. J Biol Chem. 1993 Dec 15;268(35):26033–6. doi: 10.1016/S0021-9258(19)74270-7
- Jeanne M, Gould DB. Genotype-phenotype correlations in pathology caused by collagen type IV alpha 1 and 2 mutations. Matrix Biol. 2017 Jan;57-58:29–44. doi: 10.1016/j.matbio.2016.10.003
- Volonghi I, Pezzini A, Del Zotto E, et al. Role of COL4A1 in basement-membrane integrity and cerebral small-vessel disease. The COL4A1 stroke syndrome. Curr Med Chem. 2010;17(13):1317–24. doi: 10.2174/092986710790936293
- Søndergaard CB, Nielsen JE, Hansen CK, et al. Hereditary cerebral small vessel disease and stroke. Clin Neurol Neurosur. 2017 Apr;155:45–57.
- Micheal S, Khan MI, Islam F, et al. Identification of mutations in the PRDM5 gene in brittle cornea syndrome. Cornea. 2016 Jun;35(6):853–9.
- Sklar BA, Pisuchpen P, Bareket M, et al. Identification and management of a novel PRDM5 gene pathologic variant in a family with brittle cornea syndrome. Cornea. 2023 Sep 15;42(12):1572–1577. doi: 10.1097/ICO.0000000000003372
- Burkitt Wright EMM, Spencer HL, Daly SB, et al. Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance. Am J Hum Genet. 2011 Jun 10;88(6):767–777. doi: 10.1016/j.ajhg.2011.05.007
- Stanley S, Balic Z, Hubmacher D. Acromelic dysplasias: how rare musculoskeletal disorders reveal biological functions of extracellular matrix proteins. Ann N Y Acad Sci. 2021 Apr;1490(1):57–76. doi: 10.1111/nyas.14465
- Karoulias SZ, Beyens A, Balic Z, et al. A novel ADAMTS17 variant that causes Weill-Marchesani syndrome 4 alters fibrillin-1 and collagen type I deposition in the extracellular matrix. Matrix Biol. 2020 Jun;88:1–18.
- Guo D, Liu L, Yang F, et al. Characteristics and genotype-phenotype correlations in ADAMTS17 mutation-related weill-marchesani syndrome. Exp Eye Res. 2023 Sep;234:109606.
- Bhandari R, Ferri S, Whittaker B, et al. Peters anomaly: review of the literature. Cornea. 2011 Aug;30(8):939–44.
- Nischal KK. Genetics of congenital corneal opacification–impact on diagnosis and treatment. Cornea. 2015 Oct;34(Suppl 10):S24–34. doi: 10.1097/ICO.0000000000000552
- Hanson IM, Fletcher JM, Jordan T, et al. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat Genet. 1994 Feb;6(2):168–73.
- Chesneau B, Aubert-Mucca M, Fremont F, et al. First evidence of SOX2 mutations in Peters’ anomaly: lessons from molecular screening of 95 patients. Clin Genet. 2022 May;101(5–6):494–506.
- Smith AN, Miller LA, Radice G, et al. Stage-dependent modes of Pax6-Sox2 epistasis regulate lens development and eye morphogenesis. Development. 2009 Sep;136(17):2977–85.
- Zhang X, Tong Y, Xu W, et al. Two novel mutations of the PAX6 gene causing different phenotype in a cohort of Chinese patients. Eye (Lond). 2011 Dec;25(12):1581–9.
- Weh E, Reis LM, Happ HC, et al. Whole exome sequence analysis of Peters anomaly. Hum Genet. 2014 Dec;133(12):1497–511.
- Tang HK, Chao LY, Saunders GF. Functional analysis of paired box missense mutations in the PAX6 gene. Hum Mol Genet. 1997 Mar;6(3):381–6. doi: 10.1093/hmg/6.3.381
- Nanjo Y, Kawasaki S, Mori K, et al. A novel mutation in the alternative splice region of the PAX6 gene in a patient with Peters’ anomaly. Br J Ophthalmol. 2004 May;88(5):720–721.
- Honkanen RA, Nishimura DY, Swiderski RE, et al. A family with axenfeld-rieger syndrome and Peters Anomaly caused by a point mutation (Phe112Ser) in the FOXC1 gene. Am J Ophthalmol. 2003 Mar;135(3):368–375.
- Weisschuh N, Wolf C, Wissinger B, et al. A novel mutation in the FOXC1 gene in a family with Axenfeld-Rieger syndrome and Peters’ anomaly. Clin Genet. 2008 Nov;74(5):476–480.
- Hassed SJ, Li S, Xu W, et al. A novel mutation in PITX2 in a patient with Axenfeld-Rieger Syndrome. Mol Syndromol. 2017 Mar;8(2):107–109.
- Li Y, Zhang J, Dai Y, et al. Novel mutations in COL6A3 that associated with Peters’ anomaly caused abnormal intracellular protein retention and decreased cellular resistance to oxidative stress. Front Cell Dev Biol. 2020;8:531986. doi: 10.3389/fcell.2020.531986
- Faber H, Puk O, Holz A, et al. Identification of a new genetic mutation associated with Peters Anomaly. Cornea. 2021 Mar 1;40(3):373–376. doi: 10.1097/ICO.0000000000002611
- Vincent A, Billingsley G, Priston M, et al. Further support of the role of CYP1B1 in patients with Peters anomaly. Mol Vis. 2006 May 16;12:506–10.
- Doucette L, Green J, Fernandez B, et al. A novel, non-stop mutation in FOXE3 causes an autosomal dominant form of variable anterior segment dysgenesis including Peters anomaly. Eur J Hum Genet. 2011 Mar;19(3):293–9.
- Plaisancié J, Ragge NK, Dollfus H, et al. FOXE3 mutations: genotype-phenotype correlations. Clin Genet. 2018 Apr;93(4):837–845. doi: 10.1111/cge.13177
- Ahmad N, Aslam M, Muenster D, et al. Pitx3 directly regulates Foxe3 during early lens development. Int J Dev Biol. 2013;57(9–10):741–51. doi: 10.1387/ijdb.130193jg
- Summers KM, Withers SJ, Gole GA, et al. Anterior segment mesenchymal dysgenesis in a large Australian family is associated with the recurrent 17 bp duplication in PITX3. Mol Vis. 2008;14:2010–5.
- Zazo Seco C, Plaisancié J, Lupasco T, et al. Identification of PITX3 mutations in individuals with various ocular developmental defects. Ophthalmic Genet. 2018 Jun;39(3):314–320.
- Kim HK, Ham KA, Lee SW, et al. Biallelic deletion of Pxdn in mice leads to Anophthalmia and severe eye malformation. Int J Mol Sci. 2019 Dec 5;20(24):6144. doi: 10.3390/ijms20246144
- Yan X, Sabrautzki S, Horsch M, et al. Peroxidasin is essential for eye development in the mouse. Hum Mol Genet. 2014 Nov 1;23(21):5597–614. doi: 10.1093/hmg/ddu274
- Zhu AY, Costain G, Cytrynbaum C, et al. Novel heterozygous variants in PXDN cause different anterior segment dysgenesis phenotypes in monozygotic twins. Ophthalmic Genet. 2021 Oct;42(5):624–630.
- Langer L, Taranova O, Sulik K, et al. SOX2 hypomorphism disrupts development of the prechordal floor and optic cup. Mech Dev. 2012 Mar;129(1–4):1–12.
- Heavner WE, Andoniadou CL, Pevny LH. Establishment of the neurogenic boundary of the mouse retina requires cooperation of SOX2 and WNT signaling. Neural Dev. 2014 Dec 9;9(1):27. doi: 10.1186/1749-8104-9-27
- Williams AL, Bohnsack BL. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural cret in craniofacial and ocular development. Genesis. 2019;57(7–8):e23308. doi: 10.1002/dvg.23308
- Caron V, Chassaing N, Ragge N, et al. Clinical and functional heterogeneity associated with the disruption of retinoic acid receptor beta. Genet Med. 2023 Aug;25(8):100856.
- Luo T, Zhang Y, Khadka D, et al. Regulatory targets for transcription factor AP2 in Xenopus embryos. Dev Growth Differ. 2005;47(6):403–413. doi: 10.1111/j.1440-169X.2005.00809.x
- Hoffman TL, Javier AL, Campeau SA, et al. Tfap2 transcription factors in zebrafish neural crest development and ectodermal evolution. J Exp Zool B Mol Dev Evol. 2007;308(5):679–691. doi: 10.1002/jez.b.21189
- Li W, Cornell RA. Redundant activities of Tfap2a and Tfap2c ae required for neural crest induction and development of other non-neural ectoderm derivatives in zebrafish embryos. Dev Biol. 2007;304(1):338–354. doi: 10.1016/j.ydbio.2006.12.042
- Van Otterloo E, Li H, Jones KL, et al. AP-2α and AP-2β cooperatively orchestrate homeobox gene expression during branchial arch patterning. Development. 2017;145(2):dev157438. doi: 10.1242/dev.157438
- Milunsky JM, Maher TM, Zhao G, et al. TFAP2A mutations result in branchio-oculo-facial syndrome. Am J Hum Genet. 2008;82(5):1171–1177. doi: 10.1016/j.ajhg.2008.03.005
- Milunsky JM, Maher TM, Zhao G, et al. Genotype–phenotype analysis of the branchio-oculo-facial syndrome. Am J Med Genet A. 2011;155(1):22–32. doi: 10.1002/ajmg.a.33783
- Gestri G, Osborne RJ, Wyatt AW, et al. Reduced TFAP2A function causes variable optic fissure closure and retinal defects and sensitizes eye development to mutations in other morphogenetic regulators. Hum Genet. 2009;126(6):791–803. doi: 10.1007/s00439-009-0730-x
- Schaefer L, Ballabio A, Zoghbi HY. Cloning and characterization of a putative human holocytochrome c-type synthetase gene (HCCS) isolated from the critical region for microphthalmia with linear skin defects (MLS). Genomics. 1996 Jun 1;34(2):166–72. doi: 10.1006/geno.1996.0261
- van Rahden VA, Rau I, Fuchs S, et al. Clinical spectrum of females with HCCS mutation: from no clinical signs to a neonatal lethal form of the microphthalmia with linear skin defects (MLS) syndrome. Orphanet J Rare Dis. 2014 Apr 15;9:53. doi: 10.1186/1750-1172-9-53
- Battinelli EM, Boyd Y, Craig IW, et al. Characterization and mapping of the mouse NDP (Norrie disease) locus (NDP). Mamm Genome. 1996 Feb;7(2):93–7.
- Nikopoulos K, Venselaar H, Collin RW, et al. Overview of the mutation spectrum in familial exudative vitreoretinopathy and Norrie disease with identification of 21 novel variants in FZD4, LRP5, and NDP. Hum Mutat. 2010 Jun;31(6):656–666.
- Wawrzynski J, Patel A, Badran A, et al. Spectrum of mutations in NDP resulting in ocular disease; a systematic review. Front Genet. 2022;13:884722. doi: 10.3389/fgene.2022.884722
- Ohlmann A, Tamm ER. Norrin: molecular and functional properties of an angiogenic and neuroprotective growth factor. Prog Retin Eye Res. 2012 May;31(3):243–57. doi: 10.1016/j.preteyeres.2012.02.002
- Vithana EN, Morgan P, Sundaresan P, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet. 2006 Jul;38(7):755–7.
- Patel SP, Parker MD. SLC4A11 and the pathophysiology of congenital hereditary endothelial dystrophy. Biomed Res Int. 2015;2015:475392. doi: 10.1155/2015/475392
- Happ H, Schilter KF, Weh E, et al. 8q21.11 microdeletion in two patients with syndromic peters anomaly. Am J Med Genet A. 2016 Sep;170(9):2471–5.
- Zhang A, Venkat A, Taujale R, et al. Peters plus syndrome mutations affect the function and stability of human β1,3-glucosyltransferase. J Biol Chem. 2021 Jul;297(1):100843.
- Lesnik Oberstein SA, Kriek M, White SJ, et al. Peters plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet. 2006;79(3):562–566. doi: 10.1086/507567
- Medina-Martinez O, Shah R, Jamrich M. Pitx3 controls multiple aspects of lens development. Dev Dyn. 2009 Sep;238(9):2193–201. doi: 10.1002/dvdy.21924
- Aldahmesh MA, Khan AO, Mohamed J, et al. Novel recessive BFSP2 and PITX3 mutations: insights into mutational mechanisms from consanguineous populations. Genet Med. 2011 Nov;13(11):978–81.
- Pohlmann D, Rossel M, Salchow DJ, et al. Outcome of a penetrating keratoplasty in a 3-month-old child with sclerocornea. GMS Ophthalmol Cases. 2020;10:Doc35. doi: 10.3205/oc000162
- Choi A, Lao R, Ling-Fung Tang P, et al. Novel mutations in PXDN cause microphthalmia and anterior segment dysgenesis. Eur J Hum Genet. 2015 Mar;23(3):337–41. doi: 10.1038/ejhg.2014.119
- Vincent MC, Pujo AL, Olivier D, et al. Screening for PAX6 gene mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects. Eur J Hum Genet. 2003 Feb;11(2):163–9.
- Qin Y, Gao P, Yu S, et al. A large deletion spanning PITX2 and PANCR in a Chinese family with axenfeld-rieger syndrome. Mol Vis. 2020;26:670–678.
- Skeens HM, Brooks BP, Holland EJ. Congenital aniridia variant: minimally abnormal irides with severe limbal stem cell deficiency. Ophthalmol. 2011 Jul;118(7):1260–4. doi: 10.1016/j.ophtha.2010.11.021
- Nieves-Moreno M, Noval S, Peralta J, et al. Expanding the phenotypic spectrum of PAX6 mutations: from congenital cataracts to nystagmus. Genes (Basel). 2021 May 9;12(5):707. doi: 10.3390/genes12050707
- Edward DP, Morales J, Bouhenni RA, et al. Congenital ectropion uvea and mechanisms of glaucoma in neurofibromatosis type 1. Ophthalmol. 2012;119(7):1485–1494. doi: 10.1016/j.ophtha.2012.01.027
- Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017 Jun 29;170(1):17–33. doi: 10.1016/j.cell.2017.06.009
- Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1): diagnosis and management. Handb Clin Neurol. 2013;115:939–955.
- Messina M, Ross AR, Pocobelli G, et al. Cataract surgery with intraocular lens implantation in 3 brothers with megalocornea: long-term follow-up. J Cataract Refract Surg. 2018 Mar;44(3):399–402.
- Mackey DA, Buttery RG, Wise GM, et al. Description of X-linked megalocornea with identification of the gene locus. Arch Ophthalmol. 1991 Jun;109(6):829–833.
- Mangialavori D, Colao E, Carnevali A, et al. Novel mutation in the CHRDL1 gene detected in patients with Megalocornea. Cornea. 2015 Aug;34(8):976–9.
- Webb TR, Matarin M, Gardner JC, et al. X-linked megalocornea caused by mutations in CHRDL1 identifies an essential role for ventroptin in anterior segment development. Am J Hum Genet. 2012 Feb 10;90(2):247–59. doi: 10.1016/j.ajhg.2011.12.019
- Pfirrmann T, Emmerich D, Ruokonen P, et al. Molecular mechanism of CHRDL1-mediated X-linked megalocornea in humans and in Xenopus model. Hum Mol Genet. 2015 Jun 1;24(11):3119–32. doi: 10.1093/hmg/ddv063
- Zhang Y, Yeh LK, Zhang S, et al. Wnt/β-catenin signaling modulates corneal epithelium stratification via inhibition of Bmp4 during mouse development. Development. 2015 Oct 1;142(19):3383–93. doi: 10.1242/dev.125393
- Jacobson A, Bohnsack BL. Anterior megalophthalmos in sisters with Witteveen-Kolk syndrome. J AAPOS. 2022 Jun;26(3):148–150. doi: 10.1016/j.jaapos.2022.01.003
- Grzenda A, Lomberk G, Zhang JS, et al. Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta. 2009;1789(6–8):443–450. doi: 10.1016/j.bbagrm.2009.05.007
- Balasubramanian M, Dingemans AJM, Albaba S, et al. Comprehensive study of 28 individuals with SIN3A-related disorder underscoring the associated mild cognitive and distinctive facial phenotype. Eur J Hum Genet. 2021 Apr;29(4):625–636.