
ABSTRACT
The neonatal Fc receptor (FcRn) promotes antibody recycling through rescue from normal lysosomal degradation. The binding interaction is pH-dependent with high affinity at low pH, but not under physiological pH conditions. Here, we combined rational design and saturation mutagenesis to generate novel antibody variants with prolonged half-life and acceptable development profiles. First, a panel of saturation point mutations was created at 11 key FcRn-interacting sites on the Fc region of an antibody. Multiple variants with slower FcRn dissociation kinetics than the wildtype (WT) antibody at pH 6.0 were successfully identified. The mutations were further combined and characterized for pH-dependent FcRn binding properties, thermal stability and the FcγRIIIa and rheumatoid factor binding. The most promising variants, YD (M252Y/T256D), DQ (T256D/T307Q) and DW (T256D/T307W), exhibited significantly improved binding to FcRn at pH 6.0 and retained similar binding properties as WT at pH 7.4. The pharmacokinetics in human FcRn transgenic mice and cynomolgus monkeys demonstrated that these properties translated to significantly prolonged plasma elimination half-life compared to the WT control. The novel variants exhibited thermal stability and binding to FcγRIIIa in the range comparable to clinically validated YTE and LS variants, and showed no enhanced binding to rheumatoid factor compared to the WT control. These engineered Fc mutants are promising new variants that are widely applicable to therapeutic antibodies, to extend their circulation half-life with obvious benefits of increased efficacy, and reduced dose and administration frequency.
Introduction
The interaction between antibodies and the neonatal Fc receptor (FcRn) is a key determinant in maintaining and prolonging antibody plasma half-life. Protection and recycling of monoclonal antibodies (mAbs) mediated by FcRn is well documented as one of the most important mechanisms in modulating mAb clearance through pH-dependent interactions in the endosomal compartment.Citation1 FcRn is a heterodimer of a major histocompatibility complex (MHC) class-I-like α-domain and a β2-microglobulin (β2-m) subunit ()Citation2 that recognizes regions on the Fc distinct from the other Fc receptors.Citation3 Antibody binding to FcRn is highly pH-dependent and occurs with high affinity at low pH (pH <6.5),4 but not at physiological pH (pH ~7.4),Citation5 due to pH titratable histidine residues.Citation6 This interaction is sufficient to rescue antibodiesCitation7 from lysosomal degradation and recycle them to the cell surface, where the increased pH compared to acidified endosomes weakens the interaction and allows for antibody release. While this receptor is expressed in various tissues, this FcRn-based recycling mechanism is thought to mainly occur in the vascular endothelium, kidneys and the blood brain barrier.Citation8,Citation9 However, the expression of FcRn on bone marrow-derived cells has also been shown to contribute to antibody half-life extension through a similar mechanism.Citation10 Engineering therapeutic antibodies to enhance the interaction with FcRn would provide increased efficacy and reduced dose and administration frequency as a direct result of a prolonged plasma elimination half-life.
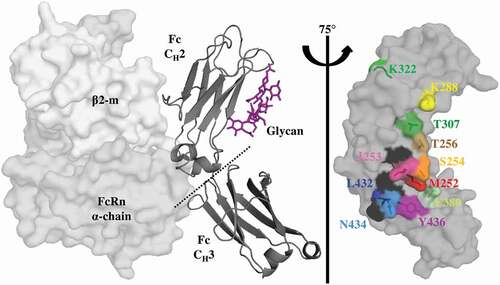
Figure 1. Key Residues at the Fc-FcRn Interface. Crystal structure (pdb: 4n0u) showing one Fc monomer and its glycosylation (dark gray ribbon), in complex with the α-domain (gray) and β2-m (light gray) hFcRn subunits. FcRn interacts with the Fc region at the CH2-CH3 interface with a pH-dependent affinity as a result of key surface histidine residues (black). The glycosylation of the CH2 domain is not required for FcRn binding. A saturation library of key residues was created at the Fc-FcRn interface and each position investigated in this study is shown as sticks and uniquely colored.
Fc engineering to enhance the FcRn binding affinity has been extensively pursued using high-throughput mutagenesis approaches.Citation3,Citation11,Citation12 The M252Y/S254T/T256E (YTE, Eu Numbering),Citation4 T307A/E380A/N434A (AAA),Citation13 Xtend Technology (M428L/N434S, LS)Citation14 and the NHance platformCitation15 have consistently shown a 2- to 11-fold enhancement in FcRn binding affinity at pH 6.0. In addition, other variants retained substantial FcRn binding at pH 7.4,Citation16 which negated any improvement in affinity pH 6.0 and use for serum half-life extension.Citation11,Citation17 The altered pH-dependent profiles are common in the literature and may provide guidelines for selecting antibody variants with enhanced serum half-life. Other studiesCitation3,Citation11,Citation14 have proposed variants with enhanced FcRn binding profiles that have yet to be investigated in vivo.
While significant improvements in the FcRn binding affinity have been achieved, mutations within the Fc regions typically result in unintended alterations to other aspects of antibody function and developability. Variants with large increases in FcRn affinity at pH 6.0 may show wildtype (WT)-like or severely reduced serum half-lives in animal models,Citation18 or are not universally applicable but suitable only to certain IgG Fv specificities.Citation19,Citation20 The most commonly used variants that have successfully demonstrated prolonged antibody circulation half-life in vivo and in clinical trials are M252Y/S254T/T256E (YTE)Citation4 and M428L/N434S (LS).Citation14,Citation21 However, the YTE variant suffers from decreased binding to FcγRIIIaCitation11 and C1q, reduced thermal stability and increased aggregation propensity, affecting antibody effector function and purification yields, respectively. The LS variant, among others,Citation12 exhibits enhanced binding to the rheumatoid factor (RF) autoantibody, suggestive of possible immunogenicity concerns in Fc-engineered therapeutic antibodies.
Here, rational design and saturation mutagenesis at key FcRn binding positions were used to successfully engineer novel Fc variants with improved pharmacokinetics (PK) properties in human FcRn transgenic mice and non-human primates (NHP), and acceptable developability features.
Results
Screening of saturation point mutation variants
In order to evaluate the saturation point mutations, a biolayer interferometry (BLI)-based assay was developed to screen for improved rat FcRn (rFcRn) off rates at pH 6.0. rFcRn was used for the 10-fold enhanced antibody affinityCitation22 and off rate compared to human FcRn (hFcRn) and allowed for the robust identification of lead mutations with improved FcRn off rates. The hFcRn off rate (3.9x10−1 s−1, ) was below the detection limit of the instrument. In addition, human and rat FcRn share a high sequence homology at the positions involved in the human IgG1 binding site (~91%),Citation23 and variants were desired for translational improvement in binding across FcRn of multiple species, such as those observed with AAA,Citation3 LSCitation21 and YTE (, Sup. Figure 1). Eleven positions in the Fc region of a model IgG1, mAb1, were selected based on their proximity or direct contribution to the FcRn interface ().
Table 1. In Vitro Characterization Parameters of the WT, Benchmark and Single Mutations. All data was obtained using the experimental techniques at the top of each column. FcRn affinity chromatography, DSF and FcγRIIIa binding were performed in triplicate (n = 3). FcRn binding kinetics to human and rat FcRn were obtained in quadruplicate and fit independently. *Mutations used in combination variants. Units: Biacore pH 6.0 hFcRn On Rate (x104 M−1 s−1), Off Rate (x10−1 s−1) and KD,app (x109 M); Biacore pH 6.0 rFcRn KD,app (x109 M); Biacore pH 7.4 hFcRn and rFcRn Steady State RU (RU), DSF Tm (°C); FcγRIIIa Binding (Steady State) .
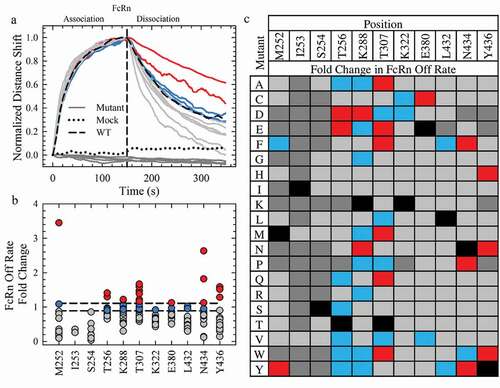
The normalized rFcRn binding Octet sensorgrams for a subset of the variants are shown in with the WT and mock controls. Several mutants completely disrupted the rFcRn binding, as little to no signal change was observed in the profiles (, dark gray). In the subset of mutations shown in , two variants had significantly reduced off rates (red), while the remaining variants had similar (blue) or faster (gray) off rates than WT. The FcRn binding off rates for all single point mutations were compiled in and and classified into one of four categories. Forty-six mutants (dark gray) showed little to no binding in a similar manner as the mock negative control (, dotted). Most of these mutations were localized to the M252, I253 and S254 loop, indicative of the importance of the CH2-CH3 interface to the FcRn interaction. Indeed, the only tolerable mutations at I253 are methionine and valine, and both significantly increase the off rate (), further supporting the importance of an isoleucine at this position.Citation3 Another 120 variants ( and , gray) destabilized the interaction, while 25 had a WT-like off rate ( and , blue). Eight of the 11 Fc positions examined in this study possessed a WT-like mutation that may provide useful engineering opportunities without compromising antibody half-life, such as improved thermal stability, reduced aggregation propensity or other developability properties. The following 18 mutations had a significantly reduced off rate compared to WT in the screening with rFcRn (, red): M252Y, T256D/E, K288D/N, T307A/E/F/M/Q/W, E380C, N434F/P/Y and Y436H/N/W. The M252Y, N434F and N434Y mutations possessed off rates greater than 1.8-fold slower than the WT antibody ().
Figure 2. Saturation Library Screening for Improved FcRn Off Rates. a. Normalization of the rFcRn binding kinetics at pH 6.0 of a subset of mutants (16) from the Octet screen. The mock and WT controls are represented by a black dotted and solid line, respectively. All mutants, represented by solid lines, fall into one of four categories: no FcRn binding (dark gray), faster (gray), WT-like (blue) or slower (red) rFcRn off rates compared to WT. The color scheme is identical for a, b and c. b. Scatterplot of the fold change in rFcRn off rate compared to WT. c. Fold change in the Octet FcRn binding off rates of the saturation library. WT positions are denoted by the solid black squares.
Titration of FcRn binding at pH 6.0
In order to examine the alteration of FcRn binding by these 18 mutations, concentration-dependent binding to FcRn was performed at pH 6.0 with human and rat FcRn using a surface plasmon resonance (Biacore) based-assay (Sup. Figure 2). Several mutations showed preferential binding to rFcRn compared to hFcRn, including those at K288 and Y436 (), while E380C and N434P did not show any improvement in FcRn binding in this assay. As a result of the species-specific effects of some variants, only variants with improved binding to both human and rat FcRn were considered for further study (; Sup. Figure 2; Table 1). In this assay, the human and rat FcRn binding affinity of the WT antibody were 2,380 ± 470 and 207 ± 43 nM, respectively, similar to affinities previously reported,Citation4,Citation22 while LS and YTE showed a 10- and 7-fold enhanced binding affinity, respectively (Sup. Figure 1, Table 1). A majority of the lead variants, except for N434F/Y, had slower on rates than the WT or the benchmarks, resulting in overall weaker binding affinities (). A subset improved the binding off rates compared to WT for both FcRn species by 1.2–7.7-fold, but all showed faster off rates and weaker affinities than the benchmark YTE and LS variants (, ). These results indicated that single mutations had limited improvement, and that combinations may further improve the FcRn binding properties.
Figure 3. hFcRn Off Rates at pH 6.0. a. Normalized sensorgrams of the WT (dotted lines), LS (green solid lines), YTE (blue solid lines), and the lead single variants with improved hFcRn off rates compared to the WT (gray scale solid lines). b. Normalized sensorgrams of a representative variant of the single (dashed black lines), double (dark gray solid lines), triple (gray solid lines) and quadruple (black solid lines) combination variants. c. Binding affinity plots of the WT, LS (green circle), YTE (blue circle), single (white circles), double (gray circles), triple (dark gray circles) and quadruple (black circles) variants.
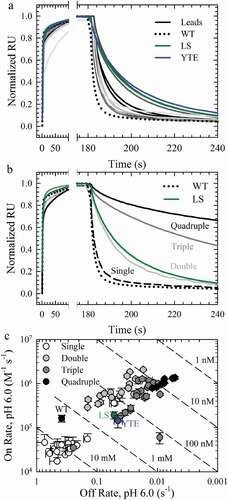
Mutations with the slowest off rates to human and rat FcRn (, asterisks) were used for the creation of combination variants and consisted of seven mutations at four positions: M252Y, T256D/E, T307Q/W and N434F/Y. These mutations were combined to obtain all possible combinations, 18 double, 20 triple and 8 quadruple variants, for additional in vitro characterization. In comparison to the single mutations, additional mutations continued to improve the FcRn off rates and binding affinities at pH 6.0 compared to the WT, benchmark and single lead variants in the FcRn binding assay to both human () and rat FcRn (Sup. Figure 3). Indeed, the quadruple variants (, Sup. Figure 3, black circles) possessed hFcRn binding affinities ~500-fold greater than WT. A plot of the on and off rates with hFcRn () revealed that two single, 15 double, 18 triple and eight quadruple variants provide enhanced binding affinity at pH 6.0 over the LS benchmark (, green).
The effect of combination mutations on the pH-dependence of the interaction with FcRn
FcRn affinity chromatography and steady-state binding measurements at pH 7.4 using Biacore were used to probe the effect of the combination variants on the pH-dependence of the FcRn interaction and was a critical criterion for lead combination selection. A common observation in the literature for variants with enhanced FcRn affinity is the simultaneous affinity improvement at physiological pH, which could negatively impact the PK of therapeutic antibodies. FcRn affinity chromatography employs a linear pH elution gradient to directly measure the perturbation of the pH-dependence by Fc-engineered mutations. The WT antibody eluted from the immobilized hFcRn at near physiological pH (pH 7.37 ± 0.05), as expected, while variants with weak FcRn binding, H435ACitation3 and H310A/H435Q, did not bind to the column regardless of the pH of the low pH buffer (Sup. Figure 4). The benchmark AAA, LS and YTE variants, with higher FcRn binding affinities at pH 6.0, each required a higher pH for column dissociation (). All of the combination variants and the seven lead single variants also required a higher elution pH than WT (Sup. Figure 4). The N434F/Y variants eluted at a greater pH than LS (), indicating that these mutations alone, and in combination, disrupted the pH-dependence of the FcRn interaction (). Representative chromatograms show a clear shift to higher elution pH with the increasing number of FcRn-binding mutations (, Sup. Figure 4), indicative of substantial FcRn binding affinity at physiological or higher pH. Indeed, a strong correlation (R2 = 0.94) between the elution pH and the hFcRn off rates at pH 6.0 () indicated that improvement in the FcRn binding properties at pH 6.0 directly contributed to the altered elution pH for these engineered FcRn variants.
Table 2. In Vitro Characterization Parameters of the Seven Combination Variants with Favorable FcRn Binding Properties. The inserted mutations are bold and underlined in the WT (M252/T256/T307/N434: MTTN) sequence (i.e. MDQN refers to T256D and T307Q mutations). All data was obtained using the experimental techniques at the top of each column. FcRn affinity chromatography, DSF and FcγRIIIa binding were performed in triplicate (n = 3). FcRn binding kinetics to human and rat FcRn were obtained in quadruplicate and fit independently. For comparison, the steady state RU for the FcγRIIIa binding of the WT antibody is 142 ± 4 RU. Units: Biacore pH 6.0 hFcRn On Rate (x104 M−1 s−1), Off Rate (x10−1 s−1) and KD,app (x109 M); Biacore pH 6.0 rFcRn KD,app (x109 M); Biacore pH 7.4 hFcRn and rFcRn Steady State RU (RU), DSF Tm (°C); FcγRIIIa Binding (Steady State RU).
Figure 4. The Effect of Combination Mutations on the pH-Dependence of the FcRn Interaction. a. Representative FcRn affinity chromatography elution profiles of the single WT and LS (vertical dotted lines) compared to the single (dashed line), double (gray line), triple (dark gray line) and quadruple (black line) combination variants. b. Plot of the elution pH as a function of the off rate at pH 6.0; (R2 = 0.94). c. Sensorgrams of the FcRn binding at pH 7.4 using Biacore for the WT (black dotted line), LS (green line) and representative combination variants. The color scheme is the same as in a. c. Comparison of the residual FcRn binding at pH 7.4 with the FcRn binding affinity at pH 6.0. Lead combinations with improved FcRn binding properties occupy the lower left quadrant. The color scheme is the same as in b.
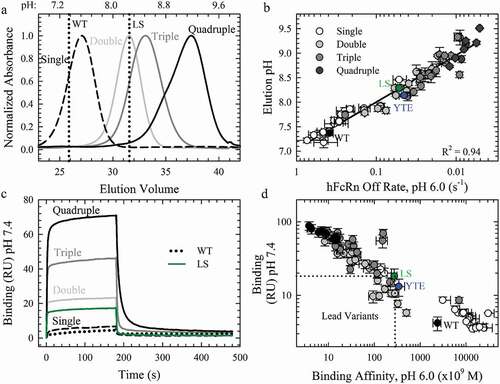
In order to directly compare the FcRn binding activity at pH 6.0 and under physiological conditions, binding experiments were performed at pH 7.4. In this experiment, the steady state response was used as a measure of binding, as WT and most of the single variants showed little to no FcRn binding at this pH (). Several of the lead single variants had slightly elevated FcRn binding compared to WT (4.3 ± 1.0 RU), but less than LS (18.5 ± 2.6 RU) and YTE (13.1 ± 1.6 RU), except for the N434F/Y mutations (), as expected from the FcRn affinity chromatography elution pH. The combination variants also possessed significant binding at pH 7.4 to an even greater extent than N434F/Y and LS (, ). Indeed, most of the variants identified in this study show significant FcRn binding enhancement regardless of the pH or FcRn species (, Sup. Figure 3). The key criteria for the selection of variants with an optimal pH profile for potential antibody half-life extension would be possession of both an increased FcRn binding affinity at pH 6.0 and FcRn binding at pH 7.4 that was comparable or reduced to the LS benchmark. A plot of the FcRn binding affinity at pH 6.0 and the residual binding at pH 7.4 to hFcRn highlighted the variants that satisfy these criteria in the lower left quadrant (). Multiple (7) combination variants () displayed these desired FcRn binding properties and were further characterized in vitro and in vivo. The inserted mutations in the WT sequence (M252/T256/T307/N434: MTTN) are highlighted in bold and underlined in .
Characteristics of the lead combination variants
All of the lead mutations are located in the CH2 domain, and may affect several major functions and characteristics of the Fc region,Citation24 including thermal stability,Citation25 effector function,Citation11 charge distribution and RF binding.Citation13 Differential scanning fluorimetry (DSF), FcγRIIIa binding, isoelectric focusing (IEF) and a RF-binding enzyme-linked immunosorbent assay (ELISA) was used to investigate the effect of the mutations on these key antibody attributes.
In comparison to WT and LS with melting temperatures (Tm) of 69.0 ± 0.2°C, the AAA and YTE benchmarks variants had lower thermal stabilities by ~8°C by DSF ( and , Sup. Figure 5). The lead combination variants showed a wide range of thermal stabilities (52.5–69.3°C) resulting from the compounded effects of each single mutation (Sup. Figure 5, Table 1). Six of the seven lead combinations had a comparable Tm to YTE, with MDQN similar to the WT. In an FcγRIIIa binding assay using Biacore, five combination variants showed a similar reduction as YTECitation11 (). Further investigation with the seven single mutations used for combinatorial variants revealed that M252Y significantly affected FcγRIIIa binding and translates this effect towards all combinations containing this mutation. The remaining six single mutations were WT-like or even slightly improved the binding to this receptor (, Sup. Figure 6).
Figure 5. Key Antibody Attributes of the Lead Variants. a-c. FcRn affinity chromatography elution profiles, DSF fluorescence profiles and FcγRIIIa binding sensorgrams of the WT, YTE (horizontal dotted lines), YD (dashed black line), DQ (solid black line) and DW (dotted black lines) variants. d. Homogeneous bridging RF ELISA of these variants compared to WT, LS and YTE. **p < .001, *p < .01.
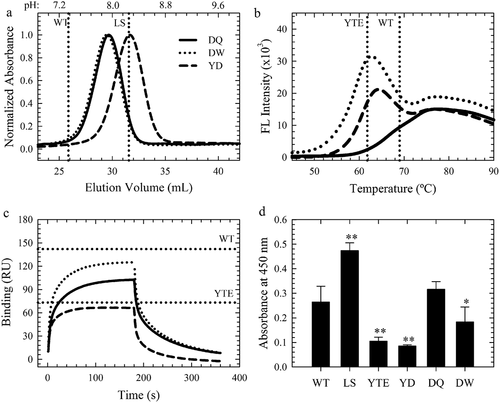
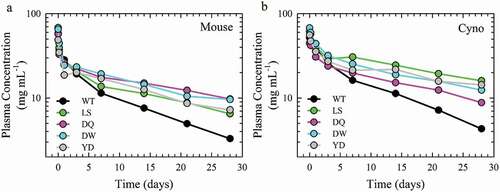
Figure 6. Antibody plasma half-life of the lead Fc variants. PK profiles of the plasma antibody concentration as a function of time of pooled samples from a single cohort of 6 hFcRn transgenic mouse (a) and n = 3 cynomolgus monkey (b) of the WT (black), LS (green), DQ (pink), DW (cyan) and YD (gray) antibodies.
Three of the seven lead combination variants were selected for further studies based on their FcRn binding properties, thermal stabilities and FcγRIIIa binding. MDQN (DQ: T256D/T307Q), MDWN (DW: T256D/T307W) and YDTN (YD: M252Y/T256D) each provided optimal FcRn binding properties as the LS variant (, ). However, each variant offers a diverse range of thermal stability and FcγRIIIa binding properties ( and , ) that provide a range of functionality. These variants were incorporated into an additional IgG1 antibody (mAb2) and a recombinant Fc fragment (Ab3). In each case, the pH-dependent human (Sup. Fig. 7), rat and cynomolgus monkey (data not shown) FcRn binding kinetics were highly similar, in addition to the elution pH, thermal stability and FcγRIIIa binding affinities (Sup. Table 1). These results indicate that the DQ, DW and YD variants confer their improved FcRn binding properties to other proteins consisting of an IgG1 Fc domain.
The isoelectric point and RF binding of the lead variants was investigated because these mutations may alter antibody surface charge and immunogenicity. More acidic antibodies have been thought to prolong antibody PK.Citation26 Compared to the WT and LS controls, all three leads resulted in a ~ 0.2 pH unit reduction in the isoelectric point (data not shown) as a result of the T256D substitution. FcRn enhancing mutations may simultaneous alter binding to host antibodies, such as RF,Citation27 due to overlapping interaction interfaces. A homogeneous bridging ELISA was adaptedCitation27 to measure the change in RF binding for the lead variants. Interestingly, LS and YTE show completely opposite shifts in RF binding compared to WT (). LS significantly increased the RF binding, while YTE (p < .001), as well as YD (p < .001) and DW (p < .01), showed a significant decrease. The DQ variant produced a similar RF binding response as the WT. These results indicated that the DQ, DW and YD variants may provide an immunogenic advantage compared to the LS variants. The YD, DW and DQ variants represent a range of key antibody characteristics that can be leveraged in conjunction with the improved FcRn binding properties over the benchmark YTE and LS variants.
The lead variants extend the in vivo plasma antibody elimination half-life
The PK of the DQ, DW and YD variants in mAb2 were examined for their effect on antibody circulation half-life with hFcRn transgenic mice (strain Tg32) and NHP (cynomolgus monkey), with WT and LS as controls. Those mutants displayed similar binding properties for cynomolgus, mouse and human FcRn (Sup. Fig. 8; Sup. Table 1). Each animal was intravenously injected with the WT, LS, DQ, DW or YD variants and the antibody concentration in the plasma was quantified over a period of two weeks (). The clearance rate and plasma half-life were determined from a non-compartmental model of the PK data. The plasma half-life of the WT antibody was 11.7 and 9.9 ± 0.5 days for the mouse and NHP models, respectively. All three lead variants and LS showed a significantly reduced clearance rate compared to WT in monkey (p < .001) (). Furthermore, the LS benchmark and variants identified here exhibited a significant increase of elimination half-life compared to WT in both species (1.7- and 2.5-fold increase in mouse and NHP, respectively). These three variants show a comparable extension of half-life to the LS benchmark (). Therefore, the DQ, DW and YD mutations identified here through saturation mutagenesis demonstrated significantly prolonged plasma half-life than their WT counterparts in both mouse and NHP animal models.
Table 3. Clearance Rates and Serum Half-Lives of the Lead Antibodies. The clearance rate and plasma-half-lives were determined for lead variants (DQ, DW, YD) and controls (WT, LS). *n = 2 due to anti-drug antibody formation, **n = 2 due to partial subcutaneous route of administration.
Discussion
FcRn binding is a critical index for antibody circulation half-life, and the modulation thereof is of key importance for the therapeutic use of antibodies and other Fc-based molecules. While a number of Fc variants have been engineered to enhance the FcRn interaction, only mAbs with the YTE and LS mutations have been validated in humans, showing a substantial 2–4-fold increase in serum retention compared to WT.Citation20,Citation21 Here, we describe three novel antibody Fc variants, DQ (T256D/T307Q), DW (T256D/T307W) and YD (M252Y/T256D), with improved binding properties to FcRn from multiple species, significantly prolonged in vivo circulation half-life in relevant animal models compared to WT, and overall acceptable in vitro properties.
A number of lead mutations with improved FcRn off rates identified in this study have overlap with previously reported variants for half-life extension, including M252Y and T256E in YTE,Citation4 T307A in AAACitation3 and N434F in the NHance platform.Citation16 Some of other mutations have been previously reported, including T307Q and N434Y, but generally in combination with a host (3+) of other mutations.Citation11,Citation16 The creation of unique combinations from the lead saturation mutations with optimal properties is the major strength of the saturation mutagenesis approach. Almost 90% of the combinations identified in our study surpassed the FcRn binding affinity of the YTE and LS benchmarks at pH 6.0 by orders of magnitude. However, a large portion of these variants would have limited application towards antibody half-life extension due to the simultaneous affinity improvement at pH 7.4.Citation17,Citation28 The N434F or N434Y mutations are responsible for the dramatic enhancement in the affinity at pH 7.4 and, as a result, are not present in any of the final lead variants.
Structural analysis of different variants indicated a few possible explanations for the affinity improvement observed in our work. In the complex of YTE with hFcRn and human serum albumin (Protein Data Bank code: 4n0u), T256E forms two additional hydrogen bonds with the β2-m subunit of hFcRn and is the main contributor to the increased affinity. The M252Y participates in the binding enhancement either through a hydrogen bond or increase in the contact surface area with the α-subunit of FcRn.Citation29 In our study, aspartic acid (T256D) was favored over T256E, which may be due to reduced steric clashes with the amino acids nearby as well as the presence of bulky T307Q and T307W substitutions in the DQ and DW combinations. Structural modeling with T307Q/W reveals possible hydrogen bond formation with β2-m along with T256D. In each case, hydrophobic interactions (M252Y/T307W)Citation30–Citation32 or hydrogen bonds (T256D/T307Q/W) may be the driving force behind our lead variants. While no structure is available for the LS mutation, modeling has indicated that M428L does not directly contact FcRn and that N434S was the driving force for the increased affinity. Interestingly, N434S alone in our screen weakened FcRn binding affinity, suggesting that mutations may enhance FcRn binding through multiple mechanisms depending on the nature and location of the substitutions. Structures of other Fc variants with prolonged half-lives in vivo and in clinical trials would provide key information for the rational design of FcRn binding variants that minimize the simultaneous affinity increase at both low and high pH. Additionally, the incorporation of histidine residuesCitation2 at the FcRn-binding interface may further optimize the pH-dependent binding profile and affect antibody half-life.
Engineering antibodies to optimize their FcRn binding can simultaneously alter other aspects of the Fc structure and function.Citation24,Citation33 In the case of YTE, the flexibility introduced by the mutations in M252-T256 loop directly contributed to the increased FcRn affinity, but also indirectly affected thermal stability, aggregation propensity, C1q and FcγRIIIa binding.Citation4 Indeed, M252Y was responsible for the reduction in FcγRIIIa binding (, Sup. Figure 6), despite lying far from the FcRn-binding interface.Citation34 Even though the Fc engineering had some impact on FcγRIIIa binding, the DQ and DW lead variants showed a less pronounced effect as compared with YTE. Binding to C1q and the other FcγRs may similarly have been affected, but were not investigated in this study. If Fc effector function is required, the DW variant would be preferable. Several mutations, such as T256D and T307W, lead to decreased thermal stability, a common caveat of affinity maturation. The DQ variant was comparable to WT, supporting its choice if other antibody engineering efforts affect this property, such as when affinity maturation or engineering multi-specific formats, is envisioned. Importantly, all three lead variants in our study maintain a WT-like or dramatically reduced binding to the RF autoantibody, a key marker for immunogenicity propensity,Citation12,Citation27 unlike LS and some other engineered variants. Approaches that address increased immunogenicityCitation12,Citation35 and aggregation propensity have been reported in the literature, but these require more extensive engineering and the incorporation of additional mutations.Citation12 Indeed, the full benefit of the Fc mutations with low immunogenicity propensity can only be recognized once the molecules reach the clinical stage.
Beyond antibodies, engineered Fc variants have applications in Fc fusions with other biomolecules of interest, such as drug conjugates,Citation36 peptidesCitation37 or protein domains, in order to prolong their half-lives. Recently, the design of small FcRn-binding sequences has shown successful competition with IgG,Citation38 pH-dependent FcRn binding, engagement with FcRn-mediated recycling in cell models,Citation39 and significantly prolonged the in vivo half-lives of these rapidly cleared moieties. The saturation mutagenesis strategies used in this study may also be adaptable to serum albumin fusions, due to its similar pH-dependent interaction with FcRn,Citation40–Citation41 as an alternative for the applications where longer half-life but no other Fc functions are desired.
Evidence has suggested that antibody half-life can highly depend on FcRn binding affinity at both low and high pH.Citation11,Citation18 Antibodies with too low of an FcRn binding affinity at pH 6.0 or aberrantly post-translationally modified antibodies, which have an impaired interaction with FcRn,Citation6 are rapidly cleared through normal lysosomal degration.Citation42 Conversely, Borrok et al. have proposed a lower threshold for the FcRn binding affinity at physiological pH, whereby any further optimization is negated by increased antibody turnover. Indeed, the residual FcRn binding at pH 7.4 was a key determinant in this study for the selection of the DQ, DW and YD as the final combinations with improved FcRn binding profiles for extended half-life. A considerable FcRn binding affinity at high pH, such as the ABDEG variant,Citation16 limits the amount of FcRn molecules available for subsequent recycling,Citation43 and leads to the rapid degradation of the circulating antibodies, which is a concept used for a promising treatment of autoimmune diseases.Citation44,Citation45 Furthermore, SELDEGs can selectively degrade antigen-specific antibodies through lysosomal degradation without affecting other host antibodies.Citation46 Although not within the scope of this study, variants identified here could have desired or improved properties for application in other types of disease treatment beyond antibody half-life extension. FcRn affinity chromatography has emerged as a complementary technique to probe the pH-dependence of the FcRn interaction and has been used to compare the elution pH with antibody half-life in a set of chimeric antibodies. However, a detailed analysis between the elution pH and the in vivo PK profiles of FcRn enhanced antibodies has yet to be conducted.
In summary, using rational design and saturation mutagenesis approaches, we have identified novel Fc variants that can be widely applied to therapeutic antibodies to extend their circulation half-life, which allow increased efficacy with reduced dosage and administration frequency.
Materials and methods
Protein reagents
The following proteins were expressed and isolated in-house: mAb1 antigen with a C-terminal octahistidine tag; rat FcRn (rFcRn, UniProt: P1359, p51 subunit: residues 23–298; UniProt: P07151, β2-m: residues 21–119); biotinylated cynomolgus FcRn (UniProt: Q8SPV9, p51 subunit: residues 24–297 with a C-terminal Avi-tag; UniProt: Q8SPW0, β2-m: residues 21–119); biotinylated human FcRn (hFcRn, UniProt: P55899, p51 subunit: residues 24–297 with a C-terminal Avi-tag; UniProt: P61769, β2-m: residues 21–119); Human CD16a (UniProt: P08637, FcγRIIIa: residues 17–208 with C-terminal HPC4 tag and valine at position 158 (V158)). The H435A and H310A/H435Q heavy chain variants were obtained in-house from HEK293 conditioned media. mAb2 variants were cloned by Evitria and purified from suspension CHO K1 conditioned media using mAbSelect SuRe affinity columns (GE Healthcare) and buffer exchanged into phosphate-buffered saline (PBS) pH 7.4 for subsequent experiments.
Saturation library construction
The WT heavy and light chains with leader DNA sequences were incorporated into mammalian expression plasmids for the IgG1 model system, mAb1. The saturation library was created with the Lightning Site Directed Mutagenesis Kit (Agilent) and NNK (N = A/C/G/T, K = G/T) and WWC (W = A/T) primers (IDT Technologies) to introduce all possible amino acids at the following positions: M252, I253, S254, T256, K288, T307, K322, E380, L432, N434 and Y436 (Eu Numbering). The heavy chain DNA sequences of the three control variants in mAb1, AAA (T307A/E380A/N434A), LS (M428L/N434S) and YTE (M252Y/S254T/T256E), were constructed into the pBH6414 vector by LakePharma.
The combination saturation library was obtained through site-directed mutagenesis of the mAb1 heavy chain with the Q5 Mutagenesis Kit (NEBiolabs) and T256D, T256E, T307Q, T307W, N434F and N434Y primers with the WT and M252Y templates in the PCR reaction. All possible combinations of the seven mutations at the four positions were obtained, 18 double, 20 triple and 8 quadruple variants. Mutation incorporation into the Fc fragment, Ab3, was performed using the Q5 Mutagenesis kit (NEBiolabs) with M252Y, T256D, T307Q and T307W primers. The creation of all Fc variants was confirmed through Sanger Sequencing (Genewiz, Inc.).
Recombinant antibody expression and purification
For conditioned media screening, DNA containing the mutant heavy chain and the WT light chain were transfected into 1 mL of Expi293 mammalian cells (Invitrogen) for expression according to the manufacturer’s instructions. The cells were incubated at 37°C, 5% carbon dioxide and 80% humidity with shaking at 900 rotations per minute (RPM) in a 2 mL 96-well plate (Griener Bio-one) and sealed with an aerated membrane. The conditioned media was collected five days post-transfection and stored at −80°C until use. The lead variants were expressed on a 30 mL scale in 125 mL flasks with 0.2 μm vented caps (Corning). The 125 mL culture flasks were shaken at 125 RPM during the entire expression duration. The conditioned media was collected five days post-transfection and filtered through 0.22 μm, 50 mL conical filters (Corning) at stored at 4°C until purification.
Isolation of the antibodies or Fc fragments was performed using 1 mL mAbSelect SuRe HiTrap columns (GE Healthcare). Following a wash step of PBS pH 7.4 for 10 column volumes, the antibodies were eluted with five column volumes of 0.1 M citric acid pH 3.0 (Sigma) and neutralized with 0.5 column volumes of 1 M tris base pH 9.0 (Sigma). The eluted antibodies were buffer exchanged against PBS pH 7.4 and concentrated to >1 mg mL−1 using 30 kDa MWCO Amicon concentrators (Millipore) for subsequent studies. The concentration of all purified antibodies was determined from their UV absorbance at 280 nm with an appropriate extinction coefficient.
Octet conditioned media screening and analysis
Screening of conditioned media containing the mAb1 variants was performed on an Octet QK 384 with Ni-NTA biosensors (PALL Life Sciences). His-tagged antigen for the IgG1 model system was captured at 15 μg mL−1 for 300 s in PBS, 0.1% bovine serum albumin (BSA, Sigma) and 0.01% Tween-20 (Sigma) pH 7.4 (PBST-BSA 7.4) followed by a 20 s wash with PBST-BSA pH 7.4. The antibodies were then captured for 200 s in conditioned media diluted 1:1 with PBST-BSA pH 7.4. Following buffer wash steps in pH 6.0 buffer, FcRn binding kinetics were obtained using 200 nM rFcRn for association and dissociation times of 150 and 200 s, respectively, at pH 6.0. rFcRn was used for the Octet screening to maximize the observable changes in the FcRn binding off rates for several benchmark variants, AAA, LS and YTE, compared to hFcRn. At all steps during the Octet screening, the temperature was 30°C with a shake speed of 1000 RPM. The rFcRn binding kinetic profiles were corrected to the initiation of the FcRn association phase and fit with a 1:1 binding model using the Octet 7.1 Analysis Software. Variants with a significant improvement in the FcRn off rate compared to WT was defined as three standard deviations slower than the mean off rate of the WT antibody (n = 3).
FcRn binding kinetics
FcRn binding kinetics at pH 6.0 and pH 7.4 were measured using a Biacore T200 instrument (GE Healthcare) using modified protocols with either the direct immobilization of FcRnCitation22 or the biotin CAPture kit (GE Healthcare). For the direct immobilization, biotinylated FcRn, at concentrations of 20 μg mL−1, was immobilized for 180 s at 10 μL min−1 in 10 mM sodium acetate pH 4.5 (GE Healthcare) to ~20 RU on the surface of a C1 sensor chip through amine coupling chemistry (GE Healthcare). With the biotin CAPture kit, the CAPture reagent was captured on the CAP chip surface to a surface density of >2,000 RU, followed by 0.1 μg mL−1 FcRn in the appropriate channels for 24 s at 30 μL min−1 to a final surface density of ~2 RU. The running buffer for the FcRn binding kinetics experiments was PBS with 0.05% Surfactant P-20 (PBS-P+, GE Healthcare) at pH 6.0 or 7.4. A concentration series of a 4-fold serial dilution from 1000 nM antibody was performed in quadruplicate for each variant, including a 0 nM control. Kinetic measurements were obtained for association and dissociation times of 180 and 300 s, respectively, at a flow rate of 10 μL min−1. The C1 and CAP sensor chips were regenerated with 10 mM sodium tetraborate, 1 M NaCl pH 8.5 (GE Healthcare) for 30 sec at 50 μL min−1 or 6 M guanidine hydrochloride, 250 mM sodium hydroxide (GE Healthcare) for 120 s at 50 μL min−1, respectively, followed by an additional 60–90 s stabilization step in PBS-P+ pH 6.0. Steady state RU measurements at pH 7.4 were obtained for all variants at 1000 nM in triplicate using the same C1 or CAP sensor chip and kinetic parameters as described above, except for the capture level of FcRn was increased 10–20-fold for both methods.
Kinetic parameters for the concentration series at pH 6.0 were fit to a bivalent model and averaged using the Biacore T200 Evaluation Software due to avidity effects. Each concentration series was fit independently to obtain the average rates and binding affinities. The apparent binding affinity was calculated from the first on and off rates from the bivalent model. The residual binding at pH 7.4 was measured using 1000 nM of each antibody in triplicate at the same time for response comparison. The steady state response of each replicate was averaged to obtain the mean and standard deviation.
FcRn affinity chromatography
The FcRn affinity column was adapted from Schlothauer et al. with biotinylated hFcRn on a 1 mL Streptavidin HP HiTrap column (GE Healthcare). The column was injected with 300 μg of each antibody in low pH buffer (20 mM 2-(N-morpholino)ethanesulfonic acid (MES; Sigma) pH 5.5; 150 mM NaCl) on an AKTA Pure System (AKTA). The antibodies were eluted by a pH gradient created with low and high pH buffer (20 mM 1,3-bis(tris(hydroxymethyl)methylamino)propane (bis tris propane; Sigma) pH 9.5; 150 mM NaCl) over 30 column volumes (CV) at 0.5 mL min−1 and monitoring the absorbance and pH. The creation of a linear pH gradient (linear regression, R2 > 0.99) was achieved through the following stepwise format: 0–30% high pH buffer over 9 CV, 30–70% over 16.5 CV and 70–100% over 4.5 CV. The column was re-equilibrated with low pH buffer for subsequent runs. All variants were performed in triplicate. The FcRn affinity column elution profiles were fit to a single Gaussian distribution in Sigmaplot 11 (Systat Software, Inc.) to determine the elution volume, full width at half maximum (FWHM) and pH from at the UV280 maximum.
Differential scanning fluorimetry
DSF experiments were performed on a BioRad CFX96 real time system thermal cycler (BioRad) with 20 μL reactions. The antibody samples and 5000x stock of Sypro Orange dye (Invitrogen) were diluted to 0.4 mg mL−1 and 10x, respectively, in PBS pH 7.4. The antibodies and Sypro Orange were mixed in a 1:1 ratio in 96-well PCR plates and sealed with adhesive microseal (BioRad) to final concentrations of 0.2 mg mL−1 of each antibody and 5x Sypro Orange dye. All antibody variants were performed in triplicate. The thermal cycler program consisted of a 2 min equilibration step at 20°C followed by constant temperature ramping rate of 0.5°C/5 s to a final temperature of 100°C. Fluorescence measurements of each well were acquired using the FAM excitation wavelength (485 nm) and ROX emission (625 nm) detectors suitable for Sypro orange fluorescence. The DSF fluorescence intensity profiles were exported from the BioRad CFX Manager and analyzed in Sigmaplot 11. The Tm was defined as the midpoint of the first transition of the fluorescence intensity profile.
FcγRIIIa binding kinetics
FcγRIIIa binding kinetics and affinity were measured using a Biacore T200 instrument (GE Healthcare). Anti-HPC4 antibody (Roche), at 50 μg mL−1 in sodium acetate pH 4.5, was coupled to the surface of CM5 sensor chip for 600 s at 10 μL min−1 with amine chemistry to a final density of >20,000 RU. The running buffer for the FcγRIIIa binding kinetics experiments was HEPES Buffered Saline with 0.05% Surfactant P-20 (HBS-P+, GE Healthcare) and 2 mM calcium chloride (CaCl2, Fluka) at pH 7.4. Each kinetic trace was initialized with the capture of 1.25 μg mL−1 HPC4-tagged FcγRIIIa-V158 for 30 s at 5 μL min−1. Association and dissociation kinetics were measured for 180 s at 5 μL min−1. The surface was regenerated with HBS-P+ buffer supplemented with 10 mM EDTA (Ambion). Prior to the next kinetic measurements, the CM5 chip was washed for 120 s with HBS-P+ with CaCl2.
The FcγRIIIa kinetic experiments were analyzed in a similar manner as described for the FcRn binding at pH 7.4 using the average steady state binding response. For all variants, the steady state RU of 300 nM antibody was determined in triplicate and averaged. The fold change in response change relative to WT (Response Fold Change) was determined for comparison between the variants in each backbone.
Isoelectric focusing
The isoelectric point (pI) of the lead variants was determined using capillary electrophoresis on a Maurice C (Protein Simple). Each 200 μL sample contained 0.35% methyl cellulose (Protein Simple), 4% pharmalyte 3–10 (GE Healthcare), 10 mM arginine (Protein Simple), 0.2 mg mL−1 antibody and the 4.05 and 9.99 pI markers (Protein Simple). The sample was loaded into the capillary for 1 min at 1500 V, followed by a separation phase for 6 min at 3000 V and monitored using tryptophan fluorescence. The pI for each variant was determined using the Maurice C software and defined as the pH at the fluorescence maximum for the major species.
Homogeneous bridging rheumatoid factor ELISA
Antibodies were biotinylated and digoxigenin-labeled using the EZ-Link Sulfo-NHS-LC-Biotin and Mix-n-Stain™ Digoxigenin Antibody Labeling Kits (Biotium) according to the manufacturer’s instructions. A stock solution containing 4 μg mL−1 of the biotinylated and digoxigenin-labeled antibodies was prepared for each variant and mixed in a 1:1 ratio with 300 U mL−1 RF from an anti-RF IgM ELISA kit (Abcam; ab178653). Following incubation at room temperature for 20 hours, 100 μL of each antibody-RF mixture was added to Streptawell plates (Sigma-Aldrich) and incubated at room temperature for 2 hours. The plate was washed three times with PBS pH 7.4 with 0.05% Tween-20 and 100 μL of a 1:2000 dilution of horseradish peroxidase-conjugated anti-digoxigenin secondary antibody (Abcam; ab420) was added to each well. After a 2-hour incubation at room temperature, the wells were washed and treated with 100 μL of the TMB substrate (Abcam) for 15 min at room temperature. The reaction was stopped with 100 μL of the stop solution (Abcam) and the absorbance was measured at 450 nm on a SpectraMax plate reader. A well containing no antibody-RF mixture provided the blank subtraction and the experiment was repeated three times. P-values were determined using the student’s t-test.
In vivo pharmacokinetics
PK studies were conducted in homozygous hFcRn transgenic mouse (Tg32 strain, Jackson Laboratory, Bar Harbor, ME) and NHPs (cynomolgus monkey). In hFcRn mice (n = 6), the WT, LS, DQ, DW and YD variants in mAb2 were administered as single intravenous dose of 2.5 mg kg−1 into the tail vein with a dose volume of 5 mL kg−1. At each time point 20 µL of blood was collected from saphenous vein using prefilled heparin capillaries. Collected blood samples were transferred into microtubes and centrifuged at 1500 x g for 10 min and at 4°C. Plasma samples were collected, pooled for each time point (6 mice/sample), and stored at −80°C prior to analysis. In the NHP studies, the variants were administered as a single intravenous dose of 2.5 mg/kg into the brachiocephalic vein with a dose volume of 1.5 mL/kg, to three treatment-naive male cynomolgus. Blood samples (0.5 mL) were collected by venipuncture of saphenous vein at 8 sampling times post dosing: 0.0035, 0.17, 1, 3, 7, 14, 21 and 28 days. Once collected, blood samples were centrifuged at 4°C for 10 minutes at 1500g and stored at −80°C.
All in vivo studies were conducted in compliance with the Sanofi institutional animal care policy. The monkey and mouse studies were approved by French “Ministère de l’Enseignement Superieur et de la Recherche” and German “Regierungspraesidium Darmstadt.”
The concentration of each variant at each time point was determined by a bottom-up LC-MS/MS assay. After a precipitation of a plasma aliquot, the plasma pellet was subjected to a protein denaturation, reduction, alkylation, trypsin digestion and solid phase extraction prior to analysis of surrogate peptides. Calibration standards were prepared by spiking the antibody variant into the plasma at 1.00, 2.00, 5.00, 10.0, 20.0, 50.0, 100, 200 and 400 µg mL−1. Peptide separation was performed on a Waters Acquity UPLC system with a reverse phase XBridge BEH C18 column (2.1x150 mm, 3.5 µM, 300Å, Waters) at a flow rate of 300 µL min−1 in a step-wise gradient of 0.1% formic acid in water and 0.1% formic acid in acetonitrile. For detection, a Sciex API5500 mass spectrometer was used in positive ion mode, with the source temperature at 700°C, the ionspray voltage at 5500 V, curtain and nebulizer gases at 40 and the collision gas at mid. The dwell times were 20 ms and the entrance potential was at 10 V for each transition. The multiple reaction monitoring transitions for two unique surrogate peptides of the mAb2 backbone were used for concentration determination relative to the standards and controls using the peak area from the MQIII integration algorithm of the Analyst software. The clearance rate and serum half-life were obtained from a non-compartmental modelCitation14 of the antibody concentration as a function of time using the Phoenix Software (Certara). All time points showing a sharp reduction in concentration were excluded from the mean plasma concentration due to presumed target-mediated drug disposition and/or anti-drug antibody interference.
Abbreviations
Supplemental Material
Download Zip (1.7 MB)Acknowledgments
We thank Sagar V. Kathuria, Denise M. Honey, Ronnie Wei for assistance with technical details and many fruitful discussions, Paul Ferrari for FcRn preparations, Ingo Focken and Jochen Beninga for help with Fc variants. We are grateful to the other members of Sanofi Biologics Research and DMPK teams for the numerous insightful suggestions and sample characterizations.
Disclosure Statements
The authors declare that they have no conflicts of interest with the contents of this article.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol. 2003;170:3528–33. doi:10.4049/jimmunol.170.7.3528.
- Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature. 1994;372:379–83. doi:10.1038/372379a0.
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–604. doi:10.1074/jbc.M009483200.
- Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006;281:23514–24. doi:10.1074/jbc.M604292200.
- Rodewald R. pH-dependent binding of immunoglobulins to intestinal cells of the neonatal rat. J Cell Biol. 1976;71:666–69. doi:10.1083/jcb.71.2.666.
- Medesan C, Matesoi D, Radu C, Ghetie V, Ward ES. Delineation of the amino acid residues involved in transcytosis and catabolism of mouse IgG1. J Immunol. 1997;158:2211–17.
- Kuo TT, Aveson VG. Neonatal Fc receptor and IgG-based therapeutics. MAbs. 2011;3:422–30. doi:10.4161/mabs.3.5.16983.
- Latvala S, Jacobsen B, Otteneder MB, Herrmann A, Kronenberg S. Distribution of FcRn Across Species and Tissues. J Histochem Cytochem. 2017;65:321–33. doi:10.1369/0022155417705095.
- Ward ES, Zhou J, Ghetie V, Ober RJ. Evidence to support the cellular mechanism involved in serum IgG homeostasis in humans. Int Immunol. 2003;15:187–95. doi:10.1093/intimm/dxg008.
- Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179:4580–88. doi:10.4049/jimmunol.179.7.4580.
- Dall'Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol 2002;169:5171–80.
- Maeda A, Iwayanagi Y, Haraya K, Tachibana T, Nakamura G, Nambu T, Esaki K, Hattori K, Igawa T. Identification of human IgG1 variant with enhanced FcRn binding and without increased binding to rheumatoid factor autoantibody. MAbs 2017:9:844–53.
- Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol 2006;18:1759–69.
- Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol 2010;28:157–9.
- Ober SWED, TX, US. Immunoglobulin molecules with improved characteristics. Austin (TX, US): United States: The Board of Regents of the University of Texas System; 2012.
- Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005;23:1283–88. doi:10.1038/nbt1143.
- Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos 2011;39:14697–77.
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Jiang W, Wroblewski VJ. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: relationship to pharmacokinetics in mice and primates. Drug Metab Dispos. 2007;35:86–94. doi:10.1124/dmd.106.011734.
- Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N. An engineered human IgG1 antibody with longer serum half-life. J Immunol. 2006;176:346–56. doi:10.4049/jimmunol.176.6.3635.
- Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Wroblewski VJ. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem. 2007;282:1709–17. doi:10.1074/jbc.M607161200.
- Zhu Q, McLellan JS, Kallewaard NL, Ulbrandt ND, Palaszynski S, Zhang J, Moldt B, Khan A, Svabek C, McAuliffe JM, et al. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci Transl Med. 2017;9:eaaj1928. doi:10.1126/scitranslmed.aaj1928.
- Abdiche YN, Yeung YA, Chaparro-Riggers J, Barman I, Strop P, Chin SM, Pham A, Bolton G, McDonough D, Lindquist K, et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. MAbs. 2015;7:331–43. doi:10.1080/19420862.2015.1008353.
- Huang X, Zheng F, Zhan CG. Binding structures and energies of the human neonatal Fc receptor with human Fc and its mutants by molecular modeling and dynamics simulations. Mol Biosyst. 2013;9:3047–58. doi:10.1039/c3mb70231f.
- Rabia LA, Desai AA, Jhajj HS, Tessier PM. Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochemical Engineering Journal. 2018;137:365–74. doi:10.1016/j.bej.2018.06.003.
- Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 2011;3:568–76. doi:10.4161/mabs.3.6.17922.
- Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem. 2010;21:2153–63. doi:10.1021/bc100261d.
- Araujo J, Zocher M, Wallace K, Peng K, Fischer SK. Increased rheumatoid factor interference observed during immunogenicity assessment of an Fc-engineered therapeutic antibody. J Pharm Biomed Anal. 2011;55:1041–49. doi:10.1016/j.jpba.2011.03.008.
- Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A. 2006;103:18709–14. doi:10.1073/pnas.0606304103.
- Oganesyan V, Damschroder MM, Woods RM, Cook KE, Wu H, Dall’acqua WF. Structural characterization of a human Fc fragment engineered for extended serum half-life. Mol Immunol. 2009;46:1750–55. doi:10.1016/j.molimm.2008.12.022.
- Young L, Jernigan RL, Covell DG. A role for surface hydrophobicity in protein-protein recognition. Protein Sci. 1994;3:717–29. doi:10.1002/pro.5560030613.
- Moreira IS, Fernandes PA, Ramos MJ. Hot spots–a review of the protein-protein interface determinant amino-acid residues. Proteins. 2007;68:803–12. doi:10.1002/prot.21474.
- Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–58. doi:10.1006/jmbi.1996.0158.
- Avery LB, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, Kavosi MS, Penn S, Cirelli D, Kurz JC, et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs. 2018;10:244–55. doi:10.1080/19420862.2017.1417718.
- Caaveiro JM, Kiyoshi M, Tsumoto K. Structural analysis of Fc/FcgammaR complexes: a blueprint for antibody design. Immunol Rev. 2015;268:201–21. doi:10.1111/imr.12365.
- Levin D, Golding B, Strome SE, Sauna ZE. Fc fusion as a platform technology: potential for modulating immunogenicity. Trends Biotechnol. 2015;33:27–34. doi:10.1016/j.tibtech.2014.11.001.
- Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16:315–37. doi:10.1038/nrd.2016.268.
- Molineux G, Newland A. Development of romiplostim for the treatment of patients with chronic immune thrombocytopenia: from bench to bedside. Br J Haematol. 2010;150:9–20.
- Mezo AR, McDonnell KA, Hehir CA, Low SC, Palombella VJ, Stattel JM, Kamphaus GD, Fraley C, Zhang Y, Dumont JA, et al. Reduction of IgG in nonhuman primates by a peptide antagonist of the neonatal Fc receptor FcRn. Proc Natl Acad Sci U S A. 2008;105:2337–42. doi:10.1073/pnas.0708960105.
- Sockolosky JT, Tiffany MR, Szoka FC. Engineering neonatal Fc receptor-mediated recycling and transcytosis in recombinant proteins by short terminal peptide extensions. Proc Natl Acad Sci U S A. 2012;109:16095–100. doi:10.1073/pnas.1208857109.
- Cantante C, Lourenco S, Morais M, Leandro J, Gano L, Silva N, Leandro P, Serrano M, Henriques AO, Andre A, et al. Albumin-binding domain from streptococcus zooepidemicus protein zag as a novel strategy to improve the half-life of therapeutic proteins. J Biotechnol. 2017;253:23–33. doi:10.1016/j.jbiotec.2017.05.017.
- Chaudhury C, Brooks CL, Carter DC, Robinson JM, Anderson CL. Albumin binding to FcRn: distinct from the FcRn-IgG interaction. Biochemistry. 2006;45:4983–90. doi:10.1021/bi052628y.
- Stracke J, Emrich T, Rueger P, Schlothauer T, Kling L, Knaupp A, Hertenberger H, Wolfert A, Spick C, Lau W, et al. A novel approach to investigate the effect of methionine oxidation on pharmacokinetic properties of therapeutic antibodies. MAbs. 2014;6:1229–42. doi:10.4161/mabs.29601.
- Swiercz R, Chiguru S, Tahmasbi A, Ramezani SM, Hao G, Challa DK, Lewis MA, Kulkarni PV, Sun X, Ober RJ, et al. Use of Fc-engineered antibodies as clearing agents to increase contrast during PET. J Nucl Med. 2014;55:1204–07. doi:10.2967/jnumed.113.136481.
- Patel DA, Puig-Canto A, Challa DK, Perez Montoyo H, Ober RJ, Ward ES. Neonatal Fc receptor blockade by Fc engineering ameliorates arthritis in a murine model. J Immunol. 2011;187:1015–22. doi:10.4049/jimmunol.1100967.
- Li X, Kimberly RP. Targeting the Fc receptor in autoimmune disease. Expert Opin Ther Targets. 2014;18:335–50. doi:10.1517/14728222.2014.877891.
- Devanaboyina SC, Khare P, Challa DK, Ober RJ, Ward ES. Engineered clearing agents for the selective depletion of antigen-specific antibodies. Nat Commun. 2017;8:15314. doi:10.1038/ncomms15314.