
ABSTRACT
Urea is authorised in the European Union (EU) as feed additive for ruminants. Because of its high molecular nitrogen content, it is a substance for potential protein adulteration in non-ruminant feed. The EU defines a spectro-colorimetric method as an official control method for the determination of urea in feed, whereas the Association of Official Analytical Chemists (AOAC) in the United States recommends an enzymatic method. Discrepancies between results obtained by these different approaches have been reported, especially at low concentrations. Therefore, we developed and validated two methods for urea determination in compound feed, including pet food, and yeast (Saccharomyces cerevisiae) over a wide concentration range using high-performance liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) and fluorescence detection (HPLC-FLD) and compared performance with a commercial enzyme kit. Limit of detection (LOD) and limit of quantification (LOQ) were found to be 3 and 8 mg kg−1 for LC-MS/MS and 2 and 7 mg kg−1 for HPLC-FLD, respectively. For both methods, the variation coefficients ranged between 1.4% and 7.2% in ruminant feed used as reference material as well as spiked samples of complete feed for chicken, pet food for dogs and cats, as well as yeast. Recovery rates for spiked samples ranged from 86% to 105%. For real samples of complete feed for poultry, wet and dry pet food for cats and dogs and yeast amounts of urea between < LOD and 200 mg kg−1 relative to a feedingstuff with a moisture content of 12% were found. In comparison with the enzyme kit, the newly developed methods proved to be less time-consuming in sample preparation and more stable regarding matrix effects.
Introduction
Urea (carbonyl diamide) is authorised in the EU as a feed additive for ruminants with a functional rumen. Regulation (EU) No 839/2012 sets a maximum content of 8,800 mg kg−1 in complete feed with a moisture content of 12% (EC Citation2012). Because of urea’s high molecular nitrogen content of about 46%, it is a potential substance for protein adulteration in non-ruminant feed. The addition of non-protein nitrogen mimics higher protein content, a quality characteristic of all kinds of feed, when using the Kjeldahl method for protein determination. Protein adulteration with substances high in molecular nitrogen is an issue in the field of food and feed integrity and safety. In 2007, cases of pet food adulterated with melamine and cyanuric acid were reported in the USA (Hilts and Pelletier Citation2009). In 2008, adulteration of milk and milk powder with melamine led to a food scandal in China (Xin and Stone Citation2008). Feed control authorities in the EU detected elevated urea content of up to 12% in 2016 and 2017 and again most recently in 2020 in yeast intended to be used in animal feed and poultry meal, a common ingredient for animal feed, leading to notifications in the Rapid Alert System for Food and Feed (RASFF) (EC Citation2020). The RASFF annual report of 2017 classified the incidents from 2016 to 2017 as cases of potential adulteration (EC Citation2018a). However, urea is also a natural substance of nitrogen metabolism (Depner Citation1991) as well as one of the world’s most widely used fertilisers (Heffer Citation2016). It is, therefore, necessary to be able to detect urea in a wide range of concentrations in feed, not only to monitor its legal application as feed additive but also to be able to distinguish between background presence and intentional adulteration.
Until now, Regulation (EC) No 152/2009 defines a spectro-colorimetric method as the official control method for the determination of urea in feed in the EU. No validation data are specified for this method and its scope of application is neither limited to a particular type of feed nor to a specific concentration range (EC Citation2009). In the USA, the Association of Official Analytical Chemists (AOAC) recommends an enzymatic method for the determination of urea in feed (AOAC Citation2000). In recent years, additional methods for determination of urea were developed, using high-performance liquid chromatographic (HPLC) separation before detection. Clark et al. (Citation2007) combined HPLC-separation with fluorescence-detection (HPLC-FLD) after derivatisation with xanthydrol to determine urea in urine and wine. Furthermore, the use of liquid chromatography tandem mass spectrometry (LC-MS/MS) was described for determination of urea in various matrices like milk and soy protein (Draher et al. Citation2014) and yeast (Flannelly et al. Citation2019).
Significant differences between the results of urea determination by different methods have been reported. Pibarot and Pilard (Citation2012) found results by the EU spectro-colorimetric method in pet foods much higher compared to those obtained by the AOAC enzymatic method and an LC-MS/MS approach, especially in samples rich in amino acids. They concluded that the spectro-colorimetric determination leads to an overestimation of the urea content. In a comparison of spectro-colorimetric, enzymatic and LC-MS/MS determinations of urea in yeast, Flannelly et al. (Citation2019) found results of the EU spectro-colorimetric determination lacking in repeatability. These findings for yeast are especially noteworthy taking into consideration that this matrix plays a role in all but one of the RASFF notifications regarding elevated urea content. In contrast to Pibarot’s and Pilard’s (Citation2012) findings, Flannelly et al. (Citation2019) reported results obtained by the AOAC enzymatic determination being considerably higher than those obtained by both LC-MS/MS and EU spectro-colorimetric methods. In a summary report of the Standing Committee on Plants, Animals, Food and Feed (EC Citation2018b), the European Union Reference Laboratory (EURL) for Feed Additives also showed its concern about the scope of application of the EU spectro-colorimetric method. It recommended LC-MS/MS and enzymatic methods instead for feeds, such as pet food, containing animal by-products. However, in the same meeting, another delegation stated concerns over the financial burden expensive equipment for LC-MS/MS-based methods represents (EC Citation2018b).
The aim of the presented study, therefore, was to in-house validate methods for urea determination in feed, including pet food, and yeast. An LC-MS/MS method was developed and the HPLC-FLD method by Clark (Citation2007) was modified for the matrix feed. Both in-house validated methods were applied on commercially available samples. Additionally, we compared the performance of an enzymatic approach using a commercial enzyme kit with the newly developed methods.
Materials and methods
Samples
In total, 34 different materials were analysed. Of these, 31 samples were purchased in local supermarkets and via local traders for feed in Berlin, Germany, between 2013 and 2020, consisting of four different complete feed for poultry (three complete feed for laying hens and one complete feed for fattening turkeys); five dry and six wet complete feed for cats; five dry and five wet complete feed for dogs; and six baker’s yeasts (Saccharomyces cerevisiae). The in-house validation was carried out with one sample each for complete feed for laying hens, wet complete feed for cats, dry complete feed for dogs and fresh yeast. Additionally, we analysed samples from Feedstuff Proficiency Tests (PT) organised by the Association of German Agricultural Analytic and Research Institutes (VDLUFA); one complementary feed for fattening cattle from PT No 433 and two complementary feed for dairy cows from PT Nos 449 and 458.
Chemicals and reagents
Water used for analysis was purified by a Millipore Milli-Q system from Merck Millipore (Darmstadt, Germany). Urea p.a., ≥ 99.5%, for preparation of standard solutions, 13C15N2-urea, xanthydrol and activated charcoal were obtained from Sigma-Aldrich (Taufkirchen, Germany). Hypergrade acetonitrile, gradient-grade ethanol and sodium acetate were obtained from Merck (Darmstadt, Germany). Ammonia acetate, 1-propanol and hydrochloric acid (3 M) were obtained from VWR (Langenfeld, Germany).
For the enzymatic method, a commercial assay for urea/ammonia determination (Art. No 10 542 946 035) was obtained by R-Biopharm (Darmstadt, Germany).
LC-ESI-MS/MS
For sample preparation, 2 g for dry feeds and dry yeast, 5 g for fresh yeast and 10 g for wet samples are weighed in a graduated 50 ml PE centrifugal tube (Sarstedt, Nürmbrecht, Germany); 0.5 g powdered activated charcoal and approximately 40 ml of water are added. After shaking for 30 min in an overhead shaker (Heidolph Instruments, Schwabach, Germany), the tubes are filled up to the mark and centrifuged for 15 min at 4500 g and 10°C using a centrifuge by Heraeus (Hanau, Germany). An aliquot of 1.8 ml supernatant is transferred into a 2 ml microcentrifuge tube (Eppendorf, Hamburg, Germany) and centrifuged again for 10 min at 20 000 g and 10°C, using a high-speed centrifuge (Eppendorf, Hamburg, Germany). To reduce matrix effects and competition of ionisation in the MS-system, as well as to match the calibration curve, if needed, extracts are diluted with water to contain a maximum of 10 mg l−1 of urea.
Aliquots of 50 µl of this extract are mixed with 925 µl of acetonitrile and 25 µl of a 1 mg l−1 solution of isotope-labelled 13C15N2-urea, shaken and are ready for injection. Chromatographic separation was achieved by a Kinetex HILIC (100 x 4.6 mm, 2.6 µm) column (Phenomenex, Aschaffenburg, Germany) on an Agilent 1200 HPLC system (Agilent Technologies, Waldbronn, Germany). Injection volume was 2 µl. Gradient elution was used with 2% acetonitrile in 5 mM ammonium acetate as eluent A and 100% acetonitrile as eluent B. The gradient starts with a flow rate of 0.3 ml min−1 with 95% of eluent B for 1 min and then decreased eluent B to 20% within 8 min. After 2 min with this composition, eluent B was increased back to 95% in 1 min and flow was increased to 0.5 ml min−1. This composition was held for 5 min. Then, flow was decreased back to 0.3 ml min−1, and the system was equilibrated for a further 3 min, resulting in a total run time of 20 min. Retention time of urea was 8.9 min.
For detection, the HPLC system was coupled with a Sciex QTRAP® 6500+ (Sciex, Darmstadt, Germany). The system is hence referred to as LC-MS/MS-1. The MS-system was operated in electrospray ionisation multiple reaction monitoring (ESI MRM) mode with positive polarity. Ion spray voltage was set at 4500 V with nitrogen as spray gas. Source temperature was 350°C, curtain gas was set at 40 psi, ion source flows 1 and 2 were 50 psi and 40 psi, respectively. Mass transitions used for the detection were 61 to 44 m/z for urea and 64 to 46 m/z for 13C15N2 urea. Entrance potential, declustering potential and collision energy were 10, 47 and 23 V, respectively, for both transitions. To quantify urea, a linear calibration function was prepared from standard solutions in solvent. The quotients of peak areas for urea and 13C15N2-urea were used for a least squares regression.
Additional measurements have been conducted on a second LC-MS/MS device, an UltiMate 3000 HPLC system coupled with a TSQ Quantiva (Thermo Fisher Scientific, Dreieich, Germany), referred to as LC-MS/MS-2. This system was also operated in ESI MRM mode with positive polarity. Ion spray voltage was set at 4000 V, source temperature at 450°C and ion transfer tube temperature at 333°C. Sheath gas, auxiliary gas and sweep gas were 45, 22 and 1 arbitrary units, respectively. Collision energy was set at 18 V for both transitions. The remaining parameters were the same as for LC-MS/MS-1.
HPLC-FLD
The same water extract of the sample as for the LC-MS/MS method is used; 125 µl of the water extract are mixed with 375 µl ethanol. For derivatisation, a solution of 0.02 M xanthydrol in 1-propanol is prepared by dissolving 0.4 g of xanthydrol in 100 ml of 1-propanol. This solution is acidified by adding 1.5 M hydrochloric acid at the ratio of one part acid and nine parts xanthydrol solution, prepared freshly every day; 300 µl of the acidified derivatisation solution is added to 500 µl of the extract-ethanol mixture and thoroughly mixed. Reaction takes place for 1 h at room temperature in the dark. After that, the solution is ready for injection onto the column. Injection volume was 5 µl. Separation was achieved with a ZORBAX Eclipse XDB-C18 column (150 × 4.6 mm, 5 µm) by Agilent Technologies (Waldbronn, Germany) with a system consisting of a P580A HPLC pump, an ASI-100 autosampler, a TCC-100 column oven and an RF-2000 fluorescence detector with a continuous lamp, all by Thermo Fisher Scientific (Dreieich, Germany). The instrument is hence referred to as HPLC-FLD-1. Gradient elution with 20 mM sodium acetate as eluent A and 100% acetonitrile as eluent B was used. The gradient starts with 50% of eluent B held for 3.3 min. The percentage of eluent B is increased to 100% within 0.7 min and held for 3 min. The percentage of eluent B was decreased back to 50% within 0.5 min, and the system was equilibrated for an additional 6 min, resulting in 13.5 min of total run time. The flow rate was set at 1 ml min−1 throughout the complete run. The retention time of urea was 3.0 min. Fluorescent excitation wavelength was set at 213 nm and emission wavelength at 308 nm. To quantify urea content, a linear calibration function was prepared from standard solutions of urea in solvent. The peak areas of the standard solutions were used for a least squares regression.
Additional measurements were performed on a second HPLC-FLD instrument, an UltiMate 3000 HPLC system with a FLD-3400RS fluorescence detector using a flash lamp, both from Thermo Fisher Scientific (Dreieich, Germany). The instrument is referred to as HPLC-FLD-2. The same parameters as for HPLC-FLD-1 were applied.
Enzymatic method
A commercial enzyme kit by R-Biopharm (Darmstadt, Germany) for the determination of urea/ammonia in foodstuffs and other materials was used. The method’s principle is based on the use of urease to degrade urea to ammonia and carbon dioxide. 2-oxoglutarate reacts with the generated ammonia in the presence of glutamate dehydrogenase, forming glutamate. The amount of NADH consumed during this second step is measured with a photometer and is proportional to the amount of urea.
As there is no fixed procedure given in the instructions for sample preparation for feed and yeast, the same water extract as described previously for LC-MS/MS and HPLC-FLD was used. To evaluate if the easy sample preparation of LC-MS/MS and HPLC-FLD could be transferred to the enzyme kit, we did not apply additional clean-up steps. We followed the kit’s instructions for the execution of analysis closely. Extinction was measured using a photometer from Analytik Jena (Jena, Germany) at 340 nm wavelength.
Moisture content
As the maximum limit of urea as a feed additive is set for feed with a moisture content of 12% in Regulation (EU) No 839/2012 (EC Citation2012) and to enhance comparability for commercial samples, values for urea are stated relative to a sample with 12% moisture content (corresponding to 88% dry matter). Moisture content was analysed according to Annexe III A of Regulation (EC) No 152/2009 for the determination of moisture in feed (EC Citation2009).
In-house validation of LC-MS/MS and HPLC-FLD methods
Our approach for in-house validation was mainly based on guidelines for single-laboratory validation by IUPAC (Thompson et al. Citation2002).
First, we compared standard calibration functions to matrix-matched calibration functions to determine if the sample matrix has an influence on instrumental response. We used complete feed for laying hens, dry complete pet food for dogs, wet complete pet food for cats and fresh yeast as matrices. The calibration approach described in DIN 32645 (Citation2008) was used to determine limit of detection (LOD) and limit of quantification (LOQ). No blank matrix was available as every analysed sample contained at least traces of urea close to the LOD. Therefore, pure water instead of a sample solution was used for fortification with levels equivalent to a sample solution from a sample with 0, 2, 4, 6, 8, 10, 12, 14, 16 and 18 mg kg−1 urea, (based on a sample amount of 2 g).
As no certified reference material was available for evaluation of trueness, recovery rate and precision, fortified matrix samples were used instead. The coefficient of variation (CV) was determined to evaluate repeatability. For this purpose, we conducted a sixfold analysis of four different matrices under repeatability conditions, each fortified with two different levels of urea ranging from 20 mg kg−1 to 50 000 mg kg−1. The wide concentration range was chosen to account for urea’s occurrence as natural background presence as well as urea as feed additive and possible adulterant. The same series of analysis was used for the calculation of recovery rate to estimate trueness. Additionally, we conducted a sixfold analysis of three different materials obtained from proficiency tests. The methods’ ruggedness against changes in instrumentation for LC-MS/MS and HPLC-FLD was tested by analysing selected samples on a second device.
Results
LC-MS/MS
An LC-MS/MS method using a HILIC-column for separation was developed for feed and yeast matrices. By using an HILIC-column, a sufficient retention of urea was achieved. Urea only has one mass transition for detection, 61 to 44 m/z. Baseline-noise for this transition was considerably higher than for the transition of the internal standard (64 to 46 m/z), but could be reduced by using acetonitrile without buffer salt as eluent B.
Chromatograms for a water blank without internal standard, a water blank containing internal standard and a standard solution of urea are shown in . The standard solution used for the lowest point of the calibration function (0.4 mg l−1 urea) resulted in a defined peak at 8.9 min retention time, clearly distinguished from the baseline noise. When comparing the water blank with and without internal standard, it is apparent that some unlabelled urea is also present in the internal standard solution of 13C15N2-urea.
Figure 1. Overlay of LC-MS/MS chromatograms of urea (m/z 61→44) as solid line and13C15N2-urea (m/z 64→46) as dotted line of (a) a water blank without internal standard, (b) a water blank with internal standard and (c) a standard solution with 0.4 mg l–1 urea in water
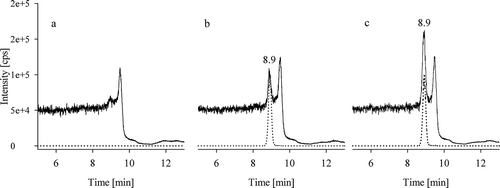
A comparison of standard calibration functions to matrix-matched calibration functions showed that there were no interfering signals in the analysed feed and yeast matrices. The slope of the calibration functions did not differ. The numeric values of the slope varied from 0.0016 to 0.0017 for standard calibration functions and from 0.0015 to 0.0017 for matrix-matched calibration functions. The LOD and LOQ in water were found to be 3 mg kg−1 and 8 mg kg−1, respectively.
The results of the sixfold analysis for determination of CV and recovery rate are shown in . As the matrices used for fortification contained different amounts of naturally occurring urea, we used a matrix-matched calibration function prepared with the respective matrix to correct results for recovery rate. For additional testing of the ruggedness of the method, selected samples were analysed on a second LC-MS/MS system.
Table 1. Coefficient of variation (CV) and recovery rate of a 6-fold analysis of fortified samples with different fortification levels of urea using LC-MS/MS on two different LC-MS/MS systems and HPLC-FLD on two different HPLC-FLD systems
With LC-MS/MS-1, CV was low, amounting to less than 10% for low and less than 5% for high concentration range. Recovery rates for analysed pet foods, complete feed for laying hens and yeast were all close to 100%, ranging from 91% to 105%. Results for LC-MS/MS-2 were similar, CV varies from 3.6% to 5.0% and recovery rates from 91% to 101%. The method, therefore, proved to be rugged against changes in measurement instrumentation.
The decision for adding the isotope-labelled internal standard after extraction was made for two reasons. First, it reduces the amount of 13C15N2-urea needed because of the much smaller volume of sample solution. This reduces the cost of the method, an important aspect for routine analysis in official control laboratories (OCL). Additionally, with this approach, the same sample preparation can be used for both LC-MS/MS and HPLC-FLD analysis. Recovery rates close to 100% demonstrate the feasibility of this approach ().
HPLC-FLD
The HPLC-FLD method from Clark et al. (Citation2007) was modified with regard to the conditions for derivatisation, as with the original setup our results were poor. We varied ethanol concentration, xanthydrol-sample ratio and derivatisation time. Finally, derivatisation time was found to be critical to obtain stable results. We, therefore, kept ethanol concentration and xanthydrol-sample ratio similar to the original method, but extended derivatisation time to one hour in the dark. Chromatograms of a water blank, a non-derivatised wet pet food for cats and a standard solution are shown in . With the analysis of the non-derivatised pet food sample, we additionally checked if there are naturally occurring substances present in the matrix before derivatisation which might interfere with fluorescence detection.
Figure 2. HPLC-FLD chromatograms of (a) a water blank with xanthydrol-derivatisation, (b) a cat feed sample without xanthydrol-derivatisation and (c) a standard solution with 0.4 mg l–1 urea with xanthydrol-derivatisation
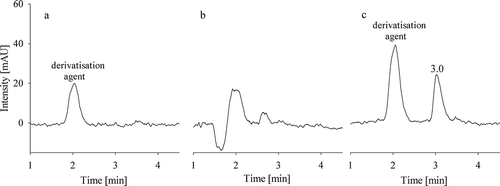
No interfering signals were detected in the blank and the non-derivatised cat feed sample. The standard solution used for the lowest point of the calibration function (0.4 mg l−1 urea) resulted in a clearly defined peak at 3.0 min retention time.
Again, a comparison of a standard calibration function to matrix-matched calibration functions showed no interfering signals and the slope of the calibration functions matched closely. The numeric values of the slope varied from 0.0116 to 0.0122 for standard calibration functions and from 0.0112 to 0.0127 for matrix-matched calibration functions. The LOD and LOQ in water were determined to be 2 mg kg−1 and 7 mg kg−1, respectively.
The method’s performance parameters for detecting urea in small amounts proved to be comparable to LC-MS/MS. Results for CV and recovery rates of fortified samples with different fortification levels are shown in . To test the ruggedness, selected samples were analysed using two different HPLC-FLD systems.
With HPLC-FLD-1, CV was low for all analysed matrices, never exceeding 5%. Recovery rates were close to 100% for all analysed matrices, ranging from 86% to 99%. Results for HPLC-FLD-2 were similar, CV varying from 1.4% to 2.8% and recovery rates from 86% to 99%. The method, therefore, proved to be rugged against changes in measurement instrumentation. Hence, CV and recovery rates for the fluorescence-based method were found to be close to the ones determined for LC-MS/MS.
Application of LC-MS/MS and HPLC-FLD to ruminant feed
To estimate bias of the new methods, three ruminant feeds from proficiency tests with known concentration for urea were analysed. Results are shown in .
Table 2. Results of a 6-fold analysis by LC-MS/MS and HPLC-FLD of ruminant feed from proficiency tests (PT) by the Association of German Agricultural Analytic and Research Institutes
LC-MS/MS results are closer to the assigned concentration but all results from both methods lay within the assigned concentration ranges. The results for the two different methods deviate less than 10% from each other for all analysed samples. Determined CV values ranged between 1.1% and 5.3%.
Application of the enzyme kit to feed, including pet food, and yeast
An enzyme kit for urea determination in food was tested for its suitability for feed, including pet food, and yeast matrices. shows results for CV and recovery rates for sixfold analysis of different fortified samples.
Table 3. Coefficient of variation (CV) and recovery rate of a 6-fold analysis of fortified samples with different fortification levels of urea using an enzyme kit
CVs were in the same range as the ones determined for LC-MS/MS and HPLC-FLD, ranging from 1.6% to 7.3%. Recovery rates for the enzyme kit were generally close to 100% except for fresh yeast fortified with 20 mg kg−1, showing a recovery rate of only 62%.
Furthermore, additional matrices were analysed with different fortification levels to prove the enzyme kit’s applicability to feed matrices in low concentration ranges. Results are shown in .
Table 4. Results of a one-fold analysis of different matrices fortified with five different urea concentrations using an enzyme kit
For complete feed for laying hens, the results for recovery rates were close to 100% for every fortification level. With fresh yeast, recovery rates for fortification levels 100, 200 and 300 mg kg−1 were close to 100%. Results for the fresh yeast fortified with 400 and 500 mg kg−1 tended to be lower, but still amounting to 79% and 73%, respectively. For pet food matrices, however, the method without additional clean-up resulted in questionable recovery rates. For dry pet food for dogs, recovery rates far exceeded 100%, ranging from 326% to 472%. On the other hand, for wet pet food for cats, recovery rates were very low. For wet pet food for cats with fortification levels of 400 and 500 mg kg−1, no urea content could be detected. During sample preparation, both pet food for cats and dogs led to sample solutions with a slight opacity. Additionally, the solutions of the wet pet food for cats were strongly coloured brown.
Urea content in various commercial feeds, including pet food, and yeast
To compare LC-MS/MS and HPLC-FLD with each other and to determine urea content in commercially available feed, including pet food, and yeast matrices, different brands of five different feedstuffs and yeast were examined in a twofold analysis by the two methods. Selected chromatograms are shown in . Results for urea content are shown in .
Figure 3. Selected chromatograms for different matrices. Upper row: HPLC-chromatograms; Lower row: LC-MS/MS chromatogram overlays of urea (m/z 61→44) as solid line and13C15N2-urea (m/z 64→46) as dotted line. (A1/A2) 2 g dry complete feed for dogs (approx. 128 mg kg–1 urea) (B1/B2) 10 g wet complete feed for cats (approx. 173 mg kg–1 urea); (C1/C2) 2 g complete feed for turkeys (approx. 10 mg kg–1 urea); Approx. urea contents are given relative to a feed with a moisture content of 12 %
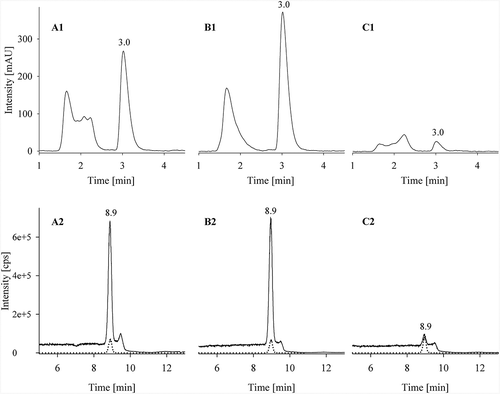
Figure 4. Comparison of LC-MS/MS and HPLC-FLD single measurement results for urea content of different feed and yeast matrices in mg kg–1 relative to a feed with a moisture content of 12 %; Horizontally striped bar: First LC-MS/MS analysis; black dotted bar: second LC-MS/MS analysis; white bar: first HPLC-FLD analysis; diagonally striped bar: second HPLC-FLD analysis; LODMS: 3 mg kg–1; LOQMS: 8 mg kg–1; LODFLD: 2 mg kg–1; LOQFLD: 7 mg kg–1
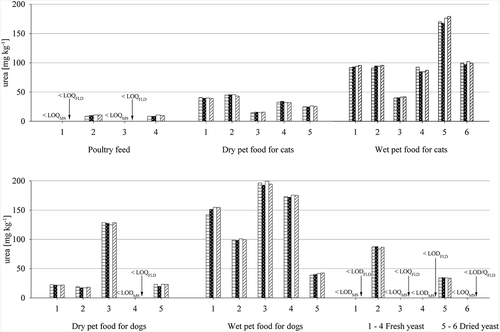
Results for LC-MS/MS and HPLC-FLD were in very good accordance. Determined urea content for poultry feed, dry pet food for cats and dogs and yeast was low, with only one pet food and one yeast exceeding 50 mg kg−1 relative to a feed with a moisture content of 12%. Results for wet pet food for cats and dogs were higher with nine out of eleven samples ranging between 80 and 200 mg kg−1 relative to a feed with a moisture content of 12%. Two of the poultry feed samples, one of the dry pet foods for dogs and four of the six analysed yeast samples resulted in values below the LOQ or LOD of both methods.
Discussion
Due to the lack of general performance criteria for the analysis of feed additives and undesirable substances in feed in the EU, to assess our validation data, we compared the performance of our methods with performance criteria from Regulation (EC) No 401/2006 for single-analyte methods for determination of mycotoxins in food (EC Citation2006). In particular, we chose the performance criteria for the highest concentration level of deoxynivalenol (DON) and fumonisins, as they come closest to the concentrations of urea in non-ruminant feed. The regulation sets a limit of 20% for relative standard deviation under repeatability conditions (RSDr) for both mycotoxins and recovery rates from 70% to 120% for DON and 70% to 110% for fumonisins (EC Citation2006) for concentrations above 0.5 mg kg−1. Both LC-MS/MS and HPLC-FLD conform with these criteria, CV values, equivalent to RSDr, never exceeding 8% for material from proficiency tests and fortified samples (ranging from 1.4% to 7.2%) and recovery rates ranging from 86% to 105%. Additionally, the results for the analysis of ruminant feed from proficiency tests were within the assigned concentration ranges for all analysed samples for both methods.
We, therefore, conclude that both LC-MS/MS and HPLC-FLD prove to be a suitable approach for the determination of urea in feed, including pet food and yeast. Both methods give reliable results for a wide concentration range (8 mg kg−1 and 7 mg kg−1 to 50,000 mg kg−1 for LC-MS/MS and HPLC-FLD, respectively). Hence, they are suitable for monitoring the background presence of urea as well as controlling the legal application of urea as a feed additive.
LC-MS/MS proved to be without interference from matrix effects and rugged against changes in instrumentation. However, low-resolution LC-MS/MS usually requires at least two mass transitions for identification of an analyte, as for example laid down in Commission Decision 2002/657/EC (EC Citation2002). Urea’s molecular structure only provides one non-specific mass transition for MS detection in MRM mode, a potential problem for using LC-MS/MS as a single method for urea determination.
With performance characteristics similar to LC-MS/MS, HPLC-FLD seems to be a reliable and economically feasible method for urea determination as a first choice method, especially for laboratories limited in expensive LC-MS/MS equipment. Thus, it may be an answer to concerns stated in a summary report by the Standing Committee on Plants, Animals, Food and Feed (EC Citation2018b) over the financial burden LC-MS/MS might pose.
HPLC-FLD as a second, orthogonal method is also a possible answer to the lack of selectivity of LC-MS/MS. Both methods can be used side by side, for example HPLC-FLD for routine and LC-MS/MS for verification of questionable findings. Using the same sample preparation for both methods saves time. Recoveries close to 100% and very good accordance between LC-MS/MS and HPLC-FLD results for spiked as well as real samples prove the applicability of this approach.
The results of the enzyme kit proved to be highly dependent on the analysed matrix. Matrices like feed for laying hens, consisting mainly of grains, resulted in satisfactory results. For pet food samples with their mixture of raw materials from plant and animal origin, however, results were poor. As mentioned before, we did not apply the clean-up steps specified in the kit’s instruction for coloured samples and samples rich in protein and fat. This was to investigate whether the kit is applicable without any time-consuming clean-up and, hence, capable of competing with the easy sample preparation of LC-MS/MS and HPLC-FLD. We presume that questionable recoveries were the result of sample solutions being both coloured and turbid, a sign for the presence of matrix substances, potentially interfering with photometric measurements. This would also explain why there were no problems in highly fortified samples, independent from the matrix. The high dilution used in these samples for enzymatic determination mitigates the effect of interfering substances and, therefore, leads to plausible results. To use the enzyme kit for low concentration ranges in different matrices, more efforts need to be put into optimisation of sample preparation. However, considering the quick and easy sample preparation for LC-MS/MS and HPLC-FLD, the use of these two methods is a preferable option, especially for the reliable determination of small amounts of urea in feed. As demonstrated, both LC-MS/MS and HPLC-FLD have the potential to supplement the spectro-colorimetric method currently required by Regulation (EC) No 152/2009 (EC Citation2009).
The analysis of commercially available feed and yeast samples showed urea contents ranging from < LOD up to 200 mg kg−1 relative to a feed with a moisture content of 12%. Nine out of ten dry pet food samples resulted in urea content below 50 mg kg−1. This is in accordance with Pibarot and Pilard (Citation2012), who reported concentrations in five different dry pet foods between 10 and 100 mg kg−1, determined by LC-MS/MS. The reason for the elevated urea content (>125 mg kg−1) in one of the dry pet food for dogs remains unclear, as its labelled ingredients were similar to other samples of this category. Our findings of low urea content in baker’s yeast are in accordance with Flanelly et al. (Citation2019). None of the four different baker’s yeasts Flannelly et al. (Citation2019) analysed exceeded the LOD of their method of 50 mg kg−1 and neither did any of our six analysed yeast samples exceed 50 mg kg−1 urea in fresh weight (Note that values are given relative to a moisture content of 12% in ). As urea is a natural metabolite in animal and plant metabolism, small amounts of urea are not conclusive evidence for adulteration. To access whether urea adulteration is of major concern in the feed sector, further data on occurrence are needed. It is recommended to focus first on yeast intended for feed production, as most RASFF notifications concerning elevated urea content are dealing with this matrix (EC Citation2020). For a potential implementation of a regulatory limit for urea in feed, the background presence of urea in different feeds has to be established. With their low LOD of 8 and 7 mg kg−1 for LC-MS/MS and HPLC-FLD, respectively, both validated methods are suitable for this purpose.
Additionally, during method development, we found first indications that urea content is not stable over time for spiked yeast samples and for water extracts of selected feed samples. For yeast, a possible explanation might be active metabolism in living yeast cells. In literature, the use of urea as a nitrogen source in the process of growing fodder yeast is described (Boze et al. Citation1992). The metabolism might also be the reason for the low recovery rate of 62% of the yeast sample fortified with 20 mg kg−1 during enzymatic determination. In feed, plant urease might be the reason for the degradation of urea over time. Especially Leguminosae like soybean, widely used in animal nutrition for their high-protein content, are known for containing urease (Follmer Citation2008). There might be enzyme activity still present after processing as specified for soy feedstuff in Regulation (EU) No 68/2013 (EC Citation2013). Therefore, when analysing urea in biological matrices, one should consider these issues.
The presented study introduces two methods for urea determination over a wide concentration range in different feed matrices with a quick and easy sample clean-up. The practicability of both methods has been proven by an in-house validation as well as the method’s application to real samples.
Declaration of interest
The authors declare no conflict of interest.
Acknowledgments
The authors would like to thank Kerstin Mietle and Mandy Schwieters for their help with the work in the laboratory.
Additional information
Funding
References
- [AOAC] Association of Official Analytical Chemists. 2000. Official method 941.04. Washington (DC): AOAC.
- Boze H, Moulin G, Galzy P. 1992. Production of food and fodder yeasts. Crit Rev Biotechnol. 12(1–2):65–86. doi:https://doi.org/10.3109/07388559209069188.
- Clark S, Francis PS, Conlan XA, Barnett NW. 2007. Determination of urea using high-performance liquid chromatography with fluorescence detection after automated derivatisation with xanthydrol. J Chromatogr A. 1161(1–2):207–213. doi:https://doi.org/10.1016/j.chroma.2007.05.085.
- Depner TA. 1991. Prescribing hemodialysis: a guide to urea modeling. Boston (MA): Springer.
- [DIN] Deutsches Institut für Normung. 2008. DIN. Vol. 32645. p. 2008–2011. Berlin (Germany): Beuth.
- Draher J, Pound V, Reddy TM. 2014. Validation of a rapid method of analysis using ultrahigh-performance liquid chromatography - tandem mass spectrometry for nitrogen-rich adulterants in nutritional food ingredients. J Chromatogr A. 1373:106–113. doi:https://doi.org/10.1016/j.chroma.2014.11.019.
- [EC] European Commission. 2002. Commission decision of 12 August 2002 implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (2002/657/EC). Off J Eur Union L. 221:8–36.
- [EC] European Commission. 2006. Commission Regulation (EC) No 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off J Eur Union L. 70:12–54.
- [EC] European Commission. 2009. Commision Regulation (EC) No 152/2009 of 27 January 2009 laying down the methods of sampling and analysis for the official control of feed. Off J Eur Union L. 54(52):27–29.
- [EC] European Commission. 2012. Commission Implementing Regulation (EU) No 839/2012 of 18 September 2012 concerning the authorisation of urea as a feed additive for ruminants. Off J Eur Union L. 252(55):11–13.
- [EC] European Commission. 2013. Commission Regulation (EU) No 68/2013 of 16 January 2013 on the Catalogue of feed materials. Off J Eur Union L. 29(56):24–26.
- [EC] European Commission. 2018a. RASFF - The rapid alert system for food and feed – 2017 annual report. Luxembourg City (Luxembourg): Publications Office of the European Union.
- [EC] European Commission. 2018b. Summary report of the standing committee on plants, Animals, Food and Feed held in Brussels on 24 April 2018-25 April 2018.
- [EC] European Commission. 2020. RASFF Portal. [accessed 2020 May 15]. https://webgate.ec.europa.eu/rasff-window/portal/?event=searchResultList .
- Flannelly L, Dillon GP, Moran CA. 2019. A comparison of analytical methods for the determination of urea in yeast-based food and feed ingredients. Food Addit Contam Part A. 36(10):1467–1474. doi:https://doi.org/10.1080/19440049.2019.1644457.
- Follmer C. 2008. Insights into the role of structure of plant ureases. Phytochemistry. 69(1):18–28. doi:https://doi.org/10.1016/j.phytochem.2007.06.034.
- Heffer PM 2016. Global nitrogen fertiliser demand and supply: trend, current level and outlook. 2016 International Nitrogen Initiative Conference; Melbourne, Australia.
- Hilts C, Pelletier L 2009. Background paper on occurrence of melamine in foods and feed. https://www.who.int/foodsafety/fs_management/Melamine_3.pdf .
- Pibarot P, Pilard S. 2012. Analysis of urea in petfood matrices: comparison of spectro-colorimetric, enzymatic and liquid chromatography electrospray ionization high resolution mass spectrometry methods. Am J Anal Chem. 3(9):613–621. doi:https://doi.org/10.4236/ajac.2012.39080.
- Thompson M, Ellison SLR, Wood R. 2002. International union of pure and applied chemistry: harmonized guidelines for single-laboratory validation of methods of analysis. Pure Appl Chem. 74(5):835–855. doi:https://doi.org/10.1351/pac200274050835.
- Xin H, Stone R. 2008. Tainted milk scandal. Chinese probe unmasks high-tech adulteration with melamine. Science. 322(5906):1310–1311. doi:https://doi.org/10.1126/science.322.5906.1310.