
ABSTRACT
Background
Vogt-Koyanagi-Harada (VKH) disease is a multisystemic autoimmune disorder characterized by granulomatous panuveitis. Gut microbiome has been considered to play a role in the pathogenesis of this disease but whether the alternation of gut microbiome was involved is unclear. This study was set up to identify abnormalities of gut microbiome composition in VKH disease.
Results
Depleted butyrate-producing bacteria, lactate-producing bacteria and methanogens as well as enriched Gram-negative bacteria were identified in the active VKH patients, as well as in VKH patients of Mix enterotype and Bacteroides enterotype. Changes of gut microbiome in the VKH patients were partially restored after an immunosuppressive treatment. The disease susceptibility genotype HLA-DRA was associated with Bacteroides sp.2.1.33B, Paraprevotella clara, Alistipes finegoldii and Eubacterium eligens. A microbial marker profile including 40 disease-associated species was established to differentiate patients from controls. Another microbial marker profile including 37 species was found to be associated with the response to treatment. An animal experiment showed that transfer of gut microbiome from VKH patients could significantly exacerbate disease activity clinically and pathologically in the recipient mice.
Conclusion
Our results revealed a distinct gut microbiome signature in VKH patients and showed an exacerbating effect of this gut microbiome on experimental autoimmune uveitis (EAU). We also developed two microbial marker profiles in differentiating VKH patients from healthy controls as well as predicting the effectiveness of treatment.
Introduction
Vogt-Koyanagi-Harada (VKH) disease is an autoimmune disorder affecting multiple organs such as eye, ear, skin and central nervous system (CNS). Bilateral granulomatous panuveitis is a hallmark of this disease. It is one of the major vision-threatening diseases in pigmented races, such as Asians and Native Americans .Citation1,Citation2 Autoimmunity, infection and genetic susceptibility have been proposed as possible etiologic factors of this disease.Citation2-Citation5 Our previous study revealed an increased expression of Toll like receptor (TLR)-3 and TLR-4 in active VKH patients.Citation6 Earlier experimental data have shown that TLRs are able to recognize ligands from pathogenic microorganisms which may subsequently trigger autoimmune responses.Citation7 These observations suggest that the host microbiome might play a role in the pathogenesis of VKH disease.
The gut microbiome has been shown to be involved in the pathogenesis of a number of immune or inflammatory diseases such as Behcet’s disease (BD),Citation8 rheumatoid arthritis (RA),Citation9,Citation10 psoriatic arthritis (PA),Citation10 inflammatory bowel disease (IBD),Citation11-Citation13 multiple sclerosis (MS),Citation14,Citation15 ankylosing spondylitis (AS)Citation16 and systemic lupus erythematosus (SLE).Citation17 Recent studies on experimental autoimmune uveitis (EAU) have shown that microorganisms in the gastrointestinal tract play a critical role in driving uveitis in this model.Citation18 Whether the composition and function of the gut microbiome is altered in VKH disease is not yet known and is the purpose of our study. In this study, we analyzed the phylotype profile and metabolic pathways of the gut microbiome in patients with VKH disease and constructed microbiome-associated gene marker sets that were useful in disease diagnosis and prognosis. Fecal microbiota transplant (FMT) using patients’ feces significantly increased the severity of disease in mice undergoing experimental autoimmune uveitis.
Materials & methods
Subjects
A total of 149 Chinese individuals (71 active VKH patients not yet receiving treatment, 11 paired inactive VKH patients after being treated with systemic corticosteroids combined with other immunosuppressive agents and 67 sex-, age- and BMI-matched healthy controls) were enrolled for this study (Table S1 and S2). After taking the stool specimens, the active VKH patients were treated with systemic corticosteroids and immunosuppressive drugs and their response to treatment was monitored over time. Individuals with other diseases such as diabetes, cardiovascular disease and infectious disease and/or taking antibiotics or probiotics within one month of the study were excluded. The diagnosis of VKH disease was made strictly according to the diagnostic criteria revised for VKH disease by an international committee on nomenclature.Citation19 Cells and flare in the anterior chamber, mutton-fat keratic precipitates, iris nodules and sunset glow fundus were considered as evidence of disease activity. None of the aforementioned signs was identified in inactive patients after treatment except sunset glow fundus. The demographic data (age, gender and BMI), intraocular and extraocular manifestations and treatment information of all participants were recorded.
Ethics, consent and permissions
All procedures followed the tenets of the Declaration of Helsinki and were approved by the Ethics Committee of Chongqing Medical University with written informed consent.
Sample collection and fecal DNA extraction
Fresh fecal samples were collected from all enrolled individuals and then stored at −80°C within two hours. Fecal DNA was extracted from frozen fecal samples using QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The concentration of DNA was measured by NanoDrop (Thermofisher, Waltham, MA, USA).
Metagenomic sequencing
The metagenomic DNA libraries were constructed with 2 μg genomic DNA according to the Illumina TruSeq DNA Sample Prep v2 Guide (Illumina, Inc; San Diego, CA, USA), with an average of 500 bp insert size. The quality of all libraries was evaluated using an Agilent bioanalyzer (Agilent Technologies, Wokingham, UK) with a DNA LabChip 1000 kit. Whole-genome shotgun sequencing of fecal samples collected was carried out on the Illumina Hiseq 4000 platform with 150 bp paired-end read length.
Raw paired-end reads of metagenomics sequencing were processed and included a quality control using the criteria below: (1) reads with adaptor contamination were removed; (2) reads were trimmed from the 3ʹ end using a quality threshold of 30; (3) reads containing more than 50% bases with low quality (Q30) were removed; (4) reads shorter than 70 bp were removed; (5) reads that mapped to human genome (alignment with SOAPaligner 2.21Citation20) were removed. On average, the proportion of high quality reads was 96.2% ± 1.4% in all samples.
De novo assembly and gene catalog construction
Employing the protocol which was used in the construction of the MetaHIT gene catalog,Citation21 we performed the assembly and gene prediction from the high quality reads by using SOAPdenovoCitation22 (version 2.04) and MetaGeneMarkCitation23 (version 3.26). To obtain a non-redundant gene of the human gut microbiome, the predicted ORFs (filtered by length of 100 bp) were clustered at 95% identity and 90% coverage using CD-HITCitation24 (version 4.5.7), and then the representative genes of each cluster (the longest one) were selected. The final non-redundant gene catalog contained 1,718,179 microbial genes, with an average length of 780 bp.
Taxonomic and gene profiling
Clean reads were mapped by SOAPalign2.21 to the microbial reference genomes downloaded from the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov), which consist of 65,770 bacterial, 898 archaeal, 1,508 fungal and 6,025 viral genomes. We selected the complete over draft genomes for strains with multiple genomes in NCBI. Taxonomic relative abundance profiles were generated with the procedure introduced in Qin.Citation25 Reads mapped to multiple taxa were allocated among them proportionally to reads counts uniquely mapped to these taxa (normalized by genome length). Gene abundance was calculated by the same strategy as used for the abundance profiling of the organisms.
MGS analysis
To cluster genes into Metagenomic Species (MGS), we followed the method described by Le ChatelierCitation26 and Nielsen.Citation27 Firstly, the differentially abundant gene markers were identified by Wilcoxon test with a P < .05. Secondly, we clustered the marker genes with a Spearman correlation coefficient (rho) >0.8 using single-linkage clustering according to their abundance variation across all samples, and then clusters with more than 25 genes were selected as MGS. The taxonomical annotation and abundance profile of MGS were performed according to the taxonomy and the relative abundance of its genes, as previously described.Citation25 Briefly, MGS assignment to a taxonomical level from strain to super kingdom level required >90% of the genes in this MGS with the best hit to the same phylogenetic group using blast with >95% identity and >90% overlap of query.
To construct the co-occurrence network of MGS, we computed the Spearman correlation coefficient between MGS using their abundances in all samples, and then visualized the co-occurrence network by Cytoscape3.0.2.
KEGG and eggNOG analysis
The metagenomics gene catalog was annotated by aligning gene sequences against the proteins in eggNOG 3.0 databaseCitation28 and KEGG database (version 20141209)Citation29 using BLASTP (e-value ≤1e −5). A gene was assigned to a OG or KO by the highest scoring annotated hit with at least one HSP (high-scoring segment pair) scoring >60. For each functional feature (OG in eggNOG or KO in KEGG database), we estimated its abundance by accumulating the relative abundance of all genes belonging to the same family.
Genomic DNA extraction and SNP genotyping
Peripheral blood samples were collected from 61 VKH patients. Genomic DNA was extracted from these blood samples using QIAamp DNA Mini Blood Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Five SNPs including rs78377598 for IL-23R-C1orf141, rs442309 and rs224058 for ADO-ZNF365-EGR2, rs3021304 for HLA-DRB1 and rs114800139 for HLA-DRA were performed using Sequenom MassARRAY system (Sequenom Inc.). Rs117633859 typing for IL-23R-C1orf141 was performed using TaqMan SNP genotyping assays (ID: AHVJKBP; Applied Biosystems, CA, USA) on the ABI 7500 Real-Time PCR system (Applied Biosystems).
Species-based classifier
To build the SVM (Support Vector Machine) classifier, extremely low abundant taxa (mean relative abundance less than 0.001) were removed first. To filter the redundant features, the mRMR algorithm (the side Channel Attack R package) and the leave-one-out cross-validation LDA (Linear Discriminant Analysis) (the paleoMAS R package) were applied. The feature set which has the highest MCC (Matthews Correlation Coefficient) was chosen to build the SVM classifier (the e1071 R package). The ROC curves of both discovery set and validation set were generated by pROC R package.
Fecal microbiota transplant and EAU induction
An antibiotic cocktail containing 1mg/ml ampicillin, 1mg/ml neomycin, 1mg/ml metronidazole and 0.5mg/ml vancomycin (Sigma-Aldrich, St. Louis, Mo, USA) was used to eliminate the indigenous microbiota community of B10RIII mice (Jackson Laboratory, Bar Harbor, ME, USA) as described previously.Citation8 Fecal samples from 5 randomly selected active VKH patients without any treatment (3 male, 2 female with an average age of 36.1 years) and 5 sex-, age- and BMI-matched healthy controls (3 male, 2 female with an average age of 35.3 years) were used to colonize the antibiotic-treated mice (n = 3 for active VKH patients, n = 3 for healthy controls and n = 3 for PBS). Each fecal sample (0.2 g) was resuspended in 1ml PBS, and pools were made from equal volumes of donor suspensions. Up to 200 µl of the pooled suspension was administered by gavage to each antibiotic-treated mouse once a day for 7 days, whereafter EAU was induced (see below).
FMT mice were immunized subcutaneously with 25μg human interphotoreceptor retinoid binding protein peptide spanning amino acid residues 161–180 (IRBP161–180, SGIPYIISYLHPGNTILHVD; Shanghai Sangon Biological Engineering Technology & Services Ltd. Co., Shanghai, China) peptide in 100 ml PBS, emulsified 1:1 v/v in complete Freund’s adjuvant (CFA; Sigma-Aldrich), supplemented with 1.0 mg/ml Mycobacterium tuberculosis (MTB; Sigma-Aldrich). We used a 25μg IRBP peptide dose since this dose causes a mild uveitis in the experimental animals (a full blown uveitis is observed when using a 50 μg dose). The clinical and histological scoring was performed as described previously.Citation30
All animals were treated according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the protocol was approved by the Ethics Committee of the First Affiliated Hospital of Chongqing Medical University.
Statistical analyses
To detect significant differences in relative abundance of metagenomics features, the nonparametric Wilcoxon test (wilcox.test in R) was performed with false discovery rate (FDR) <0.1 (Benjamini-Hochberg), and the enrichment group was then determined according to the higher rank-sum. Linear discriminant analysis (LDA) Effect Size (LEfSe) analysis was used to determine the features (organisms, KOs, or OGs) most likely to explain differences between the VKH and healthy controls (|LDA score| >2). Differentially enriched KO modules were identified according to their reporter scores from the Z-scores of individual KOs.Citation31,Citation32 A module with a reporter score of Z > 1.6 was defined as differentially enriched module.
Results
Dysbiosis of the gut microbiome in VKH patients
To characterize the gut microbiome in active VKH patients, we performed metagenomic sequencing on 107 fecal samples (55 from active untreated VKH patients and 52 from healthy controls) (Table S1) and generated 6.19 ± 1.92 gigabases (Gb) of cleaned reads per sample (Table S3). Clean reads were aligned to the reference genomes from the National Center for Biological Information (NCBI). The results showed that most of the clean reads could be mapped to bacteria (98.84%), followed by viruses (1.13%) and fungi (0.0029%). No difference was found in the α-diversity (Shannon index, observed species, P > .05) or β-diversity (Bray-Curtis distance metric of species abundance; ANOSIM, P > .05) of the gut microbiome between the active VKH patients and control group. However, there were apparent differences in the microbial compositions between the two groups. At genus level, we found that Ramularia, Alternaria and Rhizophagus were enriched, whereas Methanoculleus, Candidatus Methanomethylophilus and Azospirillum were depleted in the active VKH patients compared to the control group (), Table S4). In line with the results at genus level, one Ramularia species R. collo-cygni and one Alternaria species A. alternata were identified as VKH-enriched species and two Clostridium spp. (Clostridium sp. CAG:813 and Clostridium sp. CAG:349), one Methanoculleus species (Methanoculleus sp. CAG:1088), one Candidatus Methanomethylophilus species (Candidatus Methanomethylophilus alvus) and one Azospirillum species (Azospirillum sp. CAG:260) as VKH-depleted species (), Table S4).
Figure 1. Differentially abundant taxa between the VKH and healthy controls.
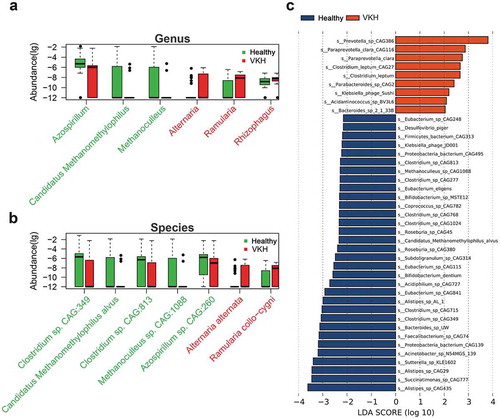
Linear discriminant (LDA) effect size (LEfSe) analysis was performed to further examine the different microbial features between active VKH patients and healthy controls and showed similar findings. Specifically, Paraprevotella spp. (Paraprevotella clara CAG116 and Paraprevotella_clara) were enriched in active VKH patients, whereas Clostridium spp. (Clostridium sp. CAG:349 and Clostridium sp.CAG:813), Bifidobacterium spp. (Bifidobacterium sp. MSTE12 and Bifidobacterium dentium), Candidatus Methanomethylophilus alvus and Methanoculleus sp. CAG:1088 were depleted ()).
Subsequent analyses on microbial genes revealed that a total of 11,720 microbial genes were enriched or depleted in the active VKH patients. These differential genes were clustered into 10 metagenomic species (MGSs) according to the abundance profiles (Fig. S1a, Table S5). We also analyzed the relationship between these VKH-associated MGSs based on their co-abundance. The cluster including Bacteroides, Paraprevotella and Eubacterium, which were positively correlated with each other, was enriched in the active VKH patients. In addition, another cluster with some MGSs including Faecalibacterium prausnitzii, Ruminococcus bromii and Alistipes was enriched in the control group (Fig. S1c).
Dysbiosis of the gut microbiome in different enterotypes in VKH patients
To investigate whether the dysbiosis of the gut microbiome in VKH patients was also present in subpopulations of enterotypes, we first identified the top 20 most abundant genera in active VKH patients and healthy controls (Fig. S2a-b). According to the relative abundance of the top genera, three enterotypes were identified both in VKH patients and healthy controls by hierarchical clustering analysis. Cluster 1 was characterized by a mix of genera including Bacteroides, Prevotella, Alisipes and Parabacteroides. Cluster 2 was characterized by a high relative abundance of Bacteroides but a low abundance of Prevotella. The third cluster was characterized by a high relative abundance of Prevotella but a low abundance of Bacteroides. Partitioning around medoids (PAM) clustering method using Jensen-Shannon distances confirmed the presence of these three enterotypes (Fig. S2c-d). The results showed that there was no difference between VKH patients and controls concerning the sample distributions across the three subgroups (P = .34, Fisher’s exact text).
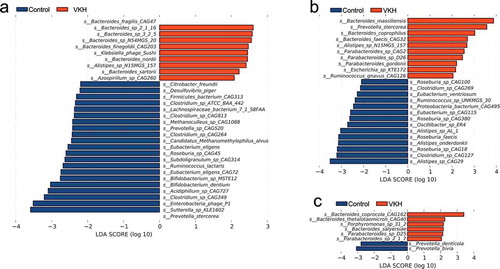
LEfSe analysis was performed to identify the differences of microbial composition in three Clusters between VKH patients and controls. The VKH-depleted species, which were identified in all VKH patients, including Clostridium spp. (Clostridium sp. CAG:349 and Clostridium sp.CAG:813), Bifidobacterium spp. (Bifidobacterium sp. MSTE12 and Bifidobacterium dentium), Candidatus Methanomethylophilus alvus and Methanoculleus sp. CAG:1088 were also depleted in patients from Cluster 1, the Mix enterotype. In Cluster 2, the Bacteroides enterotype, we identified an enriched Paracteroides sp. CAG2 and a depleted Alistipes spp. (Alistipes sp. AL.1 and Alistipes sp. CAG:29), Proteobacteria bacterium CAG:495, Eubacterium sp. CAG:115 and Roseburia sp. CAG:380 in active VKH patients, which were also in line with our LEfSe results in all patients. We also found some Parabacteroides spp. (Parabacteroides sp.2.1.7 and Parabacteroides sp.D25) and Bacteroides spp. (Bacteroides coprocola CAG:162 and Bacteroides thetaiotaomicron CAG:40) were enriched in VKH patients in Cluster 3, Prevotella enterotype ().
Figure 2. LEfSe analysis results comparing the VKH patients and healthy controls for 3 enterotype clusters.
Dysfunction of the gut microbiome in VKH patients
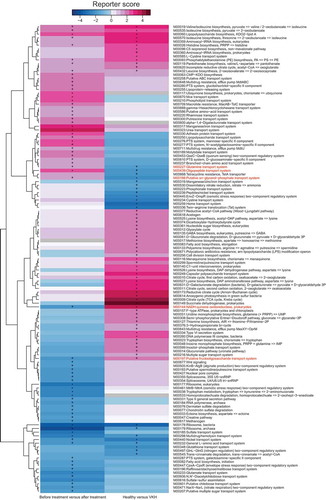
The aforementioned data showed differences in microbiome composition between the active VKH patients and controls. To further investigate the functional role of the gut microbiome in VKH disease, we constructed a gene set of 1,718,179 non-redundant genes from VKH patients and healthy controls and then functionally annotated them using KEGG (Kyoto Encyclopedia of Genes and Genomes database) and eggNOG (evolutionary genealogy of genes: Non-supervised Orthologous Groups database). LEfSe analysis showed that 6 KEGG orthologues (KO) and 6 eggNOG orthologues (OG) were significantly different between the active VKH patients and healthy controls (Table S6). At the pathway level, 10 metabolic pathways including that of oxidative phosphorylation (ko00190) and lipopolysaccharide biosynthesis (ko00540) were enriched in active VKH patients (Table S7). In addition, 54 modules including lipopolysaccharide biosynthesis (M00060), type VI secretion system (M00334), and reductive acetyl-CoA pathway (M00377) were found to be more abundant in active VKH patients (, Table S8).
Figure 3. KO modules overrepresented between the VKH and healthy controls, or patients before and after treatment.
Comparison of the gut microbiome between VKH and Behcet’s disease patients
Our recent study showed that BD,Citation8 another uveitis entity common in China, had a characteristic composition of the gut microbiome. To investigate whether there are differences and similarities in the gut microbiome composition between BD and VKH, we compared the disease-associated species between these two uveitis entities. As shown in Fig. S3a, 9 species were found to be associated with both BD and VKH disease. Only Paraprevotella clara CAG:116 was enriched in both diseases. Three Clostridium spp., Acidiphilium sp. CAG: 727, Subdoligranulum sp. CAG:314, Candidatus Methanomethylophilus alvus, Methanoculleus sp. CAG:1088 and Coprococcus sp. CAG:782 were all depleted in both VKH patients and BD patients (Fig. S3b).
Altered gut microbiome composition is associated with HLA-DRA in VKH patients
Using Genome-wide association analysis (GWAS), our previous study identified three susceptibility loci in VKH disease including IL-23R-C1orf141, ADO-ZNF365-EGR2 and HLA-DRA/HLA-DRB1.Citation33 To investigate whether there was any correlation between these susceptibility genotypes and gut microbiome components, we genotyped 54 active VKH patients according to the six major SNPs for these three susceptibility loci (rs78377598 and rs117633859 for IL-23R-C1orf141, rs3021304 for HLA-DRB1, rs114800139 for HLA-DRA, rs442309 and rs224058 for ADO-ZNF365-EGR2). The VKH patients were divided into groups with and without the risk allele for each susceptibility locus. The microbiome was then compared between patients with and without a given susceptibility locus by LEfSe analysis. The results showed that HLA-DRA risk alleles were positively correlated with 2 VKH-enriched species (Bacteroides sp.2.1.33B and Paraprevotella clara) but negatively correlated with 2 VKH-depleted species (Alistipes finegoldii and Eubacterium eligens) (Fig. S1d). We did not observe a detectable correlation between any gut microbiome component and IL-23R-C1orf141, HLA-DRB1 or ADO-ZNF365-EGR2.
Immunosuppressive treatment partly changes the gut microbiome in VKH patients
Immunosuppressive agents are used in the treatment of VKH disease and have been shown to effectively control the ocular inflammation.Citation2 LEfSe analysis was designed to investigate the influence of immunosuppressive treatment on the gut microbiome composition in a group of 11 VKH patients before (with active intraocular inflammation) and after treatment with systemic corticosteroids combined with cyclosporine A. This treatment was effective since patients were subsequently free of intraocular inflammation. The results showed that 9 genera and 27 species were enriched after the treatment when compared with before treatment, whereas 15 species were decreased (Fig. S4). Of these differential features, a VKH-enriched species Acidaminococcus sp.BV3L6 was significantly decreased after the treatment, whereas 3 VKH-depleted species Proteobacteria bacterium CAG495, Azospirillum sp.CAG260 and Alistipes sp.CAG435 were remarkably increased (Fig. S4). MGS analysis showed that 5 MGSs were enriched in VKH patients before the treatment and 3 MGSs including Faecalibacterium spp. were enriched in VKH patients after the treatment (Fig. S1b, Table S9). A further analysis was made to investigate the functional profile of the microbiome alterations following the treatment. Up to 36 modules were enriched and 35 modules were depleted in VKH patients after the treatment as compared to the profile found before treatment. Of these modules that exhibited significant differences between these patients before and after treatment levels, 3 VKH-depleted modules were increased and 2 VKH-enriched modules were reduced (, Table S10); in other words, their responses to the treatment mirrored their VKH association.
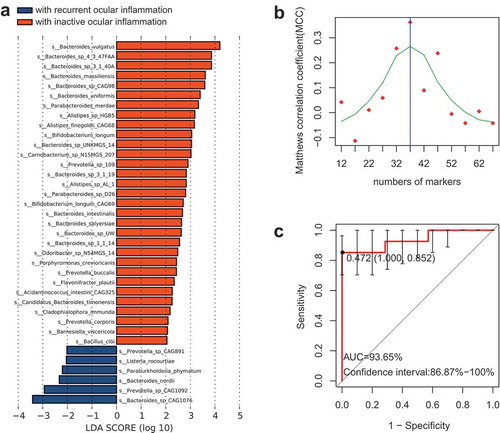
To examine whether the gut microbiome is associated with the response to treatment, we reviewed 50 active VKH patients’ clinical data after treatment and then compared the gut microbiome between the patients with inactive ocular inflammation (30 patients) and those with a relapse ocular inflammation after treatment (20 patients). LEfSe analysis identified 70 species to be different between these two groups (Table S11). Of these, 5 Alistipes spp. and Bacteroides sp. UW were associated with a good response to treatment (), Table S11).
Development of a gut microbial marker profile for VKH disease
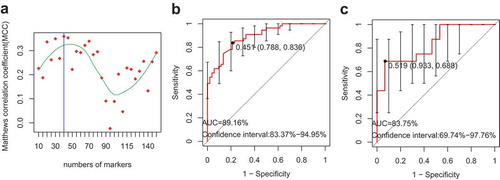
To develop a marker profile of the gut microbiome in VKH disease, a training set that included 55 active VKH patient and 52 healthy controls was used. Firstly, we removed the extremely low abundant species (mean relative abundance < 0.001) and then filtered the redundant features with the mRMR algorithm and leave-one-out cross-validation LDA (Linear Discriminant Analysis). The final feature set with the highest MCC (Matthews Correlation Coefficient) was chosen to build the SVM (Support Vector Machine) classifier, which consists of 40 species. The area under the receiver operating characteristic curve (AUC) of the classifier was 89.16% and the 95% CI was 83.37%-94.95% ()). The classifier was also validated in 16 VKH patients and 15 controls. The result showed an AUC value of 83.75% (95% CI: 69.74%-97.76%), demonstrating the usefulness of these markers in differentiating VKH patients from healthy controls ()).
Figure 4. The classifier used to distinguish VKH patients from healthy controls.
Another microbial marker profile based on 37 inflammation prognosis-associated species ()) was selected using the SVM classifier. Results showed that the classifier was able to distinguish the patients with a good prognosis from their counterparts showing relapse (AUC = 93.65%, 95% CI = 86.87%-100%) ()).
Figure 5. Species used to predict the prognosis.
Fecal transfer of VKH patient microbiota exacerbates experimental autoimmune uveitis (EAU)
To investigate whether gut microbiome composition in VKH patients contributes to the development of this disease, we examined the effect of fecal microbiota transplantation (FMT) on the course of EAU, an animal model which has many features resembling human uveitis including VKH disease.Citation34 Three groups of antibiotic-treated B10RIII mice, each containing three animals, were colonized with a pooled fecal sample from five randomly selected active VKH patients or a pooled fecal sample from five randomly selected healthy controls or were treated with PBS, respectively. To investigate whether the indigenous microbiota community was adequately knocked-down by antibiotic treatment, we performed 16S rRNA gene sequence analysis on the fecal samples from antibiotic-treated mice and human fecal-colonized mice first. Shannon diversity index ()) and PCoA plot analysis ()) showed that the diversity of antibiotic-treated mice was extremely low and the gut microbial phenotypes of human fecal-colonized mice were totally different from antibiotic-treated mice. Furthermore, the VKH-depleted genus Alistipes was also found to be depleted in the VKH-recipient group when compared with the control-recipient group (Table S12).
Figure 6. Effect of FMT using active VKH patient microbiota on EAU.
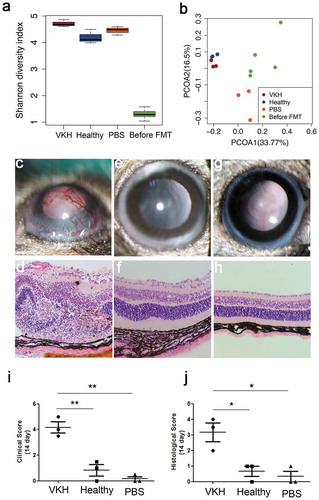
EAU was successfully induced in the VKH-recipient group following immunization with the retinal protein peptide IRBP161-180 combined with complete Freund’s adjuvant (CFA), evidenced by conjunctival hyperemia, ciliary injection, corneal edema, posterior synechiae, aqueous flare and cells (clinical scores: 3.5 to 5) ()). A minor case of uveitis manifested by conjunctival hyperemia was observed in the control-recipient group (clinical scores: 0 to 1.5) ()) and PBS-treated group (clinical scores: 0 to 0.5) ()). Histological analysis also showed severe uveitis symptoms in the VKH-recipient group, evidenced by destruction of the retinal architecture with severe folding and detachment, inflammatory cells throughout the retinal and choroid and intensive retinal vasculitis (histological scores: 2 to 4) ()). However, in the control-recipient group ()) and PBS-treated group ()), none or minimal derangement of the retinal architecture was observed (histological scores: 0 to 1). Both our clinical and histological results showed that the scores in the VKH-recipient group was significantly higher than that in the control-recipient group as well as PBS treated-group ).
Discussion
In the present study, we showed that the gut microbiome composition was different (both microbial composition and function) either between VKH patients with active intraocular inflammation and healthy controls or among the three enterotypes, namely Mix enterotype, Bacteroides enterotype and Prevotella enterotype. Up to 4 VKH-associated species were found to be associated with HLA-DRA, a disease susceptibility genotype. We also found that immunosuppressive treatment in the VKH patients led to partial restoration of the gut microbiome alterations. Gut microbiome-based markers were constructed and showed a high specificity for VKH, as well as for disease prognosis. Fecal transplants into mice subjected to EAU significantly worsened the severity of intraocular inflammation in these animals.
The gut microbiome, as an environmental factor, has been proposed to be involved in host innate and adaptive immune responses and the pathogenesis of a number of immune mediated diseases.Citation35 In this study, we investigated whether the gut microbiome was implicated in the development of VKH disease. Since the microbiome may be influenced by various factors such as medication, dietary habits, environment and diseases, we made the following efforts to eliminate possible confounding factors. Firstly, VKH disease was strictly diagnosed according to the diagnostic criteria revised for VKH disease that was issued by an international committee on nomenclature.Citation19 The enrolled VKH patients and controls did not have any underlying diseases. Secondly, only patients with active ocular inflammation who did not receive any medication including antibiotics and/or probiotics for at least one month before the start of the project were enrolled for our study. Finally, healthy controls were strictly matched with the enrolled patients based on similarity in dietary habits, living conditions, age, sex and body mass index (BMI).
Gut microbial gene repertoire of VKH disease was established by metagenomic analysis. VKH patients were enriched in Gram-negative bacteria in their gut such as Bacteroides spp., Paraprevotella spp., Prevotella spp. and Parabacteroides spp. but depleted with butyrate-producing bacteria (BPB, such as Clostridium spp.and Roseburia spp.), lactate-producing bacteria (LPB, such as Bifidobacterium spp.) and methanogens (such as Candidatus Methanomethylophilus spp. and Methanoculleus spp.). These results were partially consistent with those reported recently in BD,Citation8 showing that BPB and methanogens were depleted in patients with the two inflammatory eye diseases, although most VKH-associated species were specific for this uveitis entity. These results indicate that the similar clinical manifestations, each disease has a unique microbiome composition.
Previous studies revealed that the gut microbiome can be classified into different clusters termed “enterotypes” based on the dominant taxa.Citation36 Different enterotypes were shown to be associated with different dietary habitsCitation37,Citation38 and life style.Citation39,Citation40 Recent studies on Type II diabetes,Citation41 neuro-Behcet’s and multiple sclerosisCitation42 both confirmed that enterotype analysis provided crucial biological insight. In the present study, we identified three enterotypes, namely Mix enterotype, Bacteroides enterotype and Prevotella enterotype, in VKH patients and healthy controls. Subsequent analysis showed enrichment of Gram-negative bacteria and depletion of BPB, LPB and methanogens in both the Mix enterotype and Bacteroid enterotype, which was in line with our whole-cohort analysis. The subcluster stratified analysis results suggest that the disease state may be the only reason in explaining the differences observed in the VKH gut microbiome.
In line with the observation that Gram-negative bacteria were more abundant in VKH, increased KEGG pathways and modules in lipopolysaccharide (LPS) biosynthesis, capsular polysaccharide transport system and bacterial secretion system (type VI) were enriched in patients. Interaction of LPS from gram-negative bacteria and the Toll-like receptor 4 (TLR4) can trigger strong inflammatory responses in host immune cells.Citation6 In a previous study we reported an over activation of the LPS/TLR4 pathway in monocyte-derived macrophages from active VKH patients.Citation6
Bifidobacterium spp. are considered beneficial bacteria and many strains in the genus are commonly used as probiotics. Previous studies have shown that Bifidobacterium spp. plays a role in the treatment or prevention of IBD,Citation43 inhibits bacterial translocation,Citation44 exerts antimicrobial activity,Citation45 and enhances the intestinal epithelial barrier (IEC) function.Citation46 Taken together, these results suggest that Bifidobacterium spp., which were significantly depleted in active VKH patients, are beneficial in maintaining the health of the gastrointestinal tract. Butyrate also promotes gut health and has a role in controlling the permeability of the IEC.Citation47 In the present study, the abundance of BPB Clostridium spp. was significantly decreased in active VKH patients, which is in line with earlier observations in other autoimmune or autoinflammatory diseases such as BDCitation8 and IBD.Citation48,Citation49 The depleted Clostridium spp. in VKH patients is in agreement with the findings concerning purine metabolism modules, showing the role of these species in purine ribonucleoside degradation.Citation50 Bifidobacterium spp., Clostridium spp. and methanogens, which are characterized by their ability to produce methane, were also significantly depleted in active VKH patients. Earlier studies in animal models have confirmed the regulatory effect of methane on oxidative stress damage as well as on the control of inflammation.Citation51 In the present study, we observed an overabundance of the oxidation phosphorylation pathway in active VKH patients, which has also been observed in patients with systemic lupus erythematosus (SLE), and is thought to be related to abnormal oxidative stress in the digestive tract.Citation52 These observations collectively suggest that the patients with VKH disease might have a more “harmful” gut environment compared to healthy individuals. Whether this is the cause or effect of the disease is subject of further investigation.
Numerous studies have shown that host genetic factors, especially HLA polymorphism, are associated with VKH disease.Citation5,Citation53,Citation54 Our study found that gut microbiome and the disease susceptibility genotype HLA-DRA, which are two possible factors associated with VKH disease, also showed a correlation. These results support the hypothesis that gut microbiome composition could be dependent on host genotype, which is supported by earlier studies on IBD.Citation55,Citation56 How interactions between host genes and gut microbiome contribute to the development of VKH disease is still unknown. One hypothesis is that a susceptible genotype may influence the gut micro-ecology, leading to the alteration of the gut microbiome, which ultimately leads to disease development. Alternatively, individuals with a susceptible genotype may develop disease, which is followed by alteration of their gut microbiome to generate a disease specific pro-inflammatory profile.
Immunosuppressive agents are the first choice in the treatment of VKH disease. Our results showed that the altered gut microbiome in VKH could be partially restored after immunosuppressive treatment, which coincides with resolution of intraocular inflammation. An increased abundance of bacteria negatively associated with VKH such as Alistipes sp.CAG435, Azospirillum sp.CAG260 and Proteobacteria bacterium CAG495 was observed after the treatment. A beneficial alteration seems to occur in the gut environment after the immunosuppressive treatment, although the exact mechanism is still unknown.
Previous studies have shown the potential of gut microbiome composition in disease classification.Citation9,Citation25,Citation41 Our results confirmed that a disease-associated microbiome marker profile could distinguish the VKH patients from the healthy control group and that the use of another specific microbiome profile might facilitate the prediction of treatment effects. Further study is needed to investigate whether gut microbiome composition can be useful in disease prevention, diagnosis or prognosis in a large cohort.
FMT was performed in the present study to determine whether the gut microbiome composition might play a causal role in the pathogenesis of VKH disease. Transplantation of feces from active VKH patients to mice undergoing EAU resulted in a more severe manifestation of the intraocular inflammation in these animals. These results are similar to our recent study on BD, where we showed that the gut microbiome from BD patients could also exacerbate disease activity in mice receiving a fecal transplant.Citation8 While the exact mechanism involved is still unknown, these results together indicated that the gut microbiome from uveitis patients might play a role in the pathogenesis of the intraocular inflammatory disease.
The present study still has several limitations. Recruitment of all VKH patients and healthy controls was limited to a Chinese Han population. Our findings should therefore be confirmed in other ethnic populations. Furthermore, only a limited alteration of the gut microbiome was identified in Prevotella-dominant VKH patients. This might be due to the relatively small cohort size (number of Prevotella enterotype: VKH = 11, N = 7) and further study on more patients after enterotype stratification should be performed.
In conclusion, we found changes in the gut microbial composition and function in active VKH patients, as well as in VKH patients of different enterotypes. These changes could partly be restored after immunosuppressive treatment. Differences in the gut microbiome were correlated with disease susceptibility genotypes. Microbial-associated gene profiles could be used to differentiate VKH patients from controls and could also be used to predict response to treatment. Moreover, an animal experiment suggests that gut microbiome composition might be a causal factor in the development of this disease.
Author contributions
Z. Y., C. W., N. Z., N.Q. and P. Y. conceived and directed the study; N.Q., C.W. and X.X. analyzed the data; P. Y. made clinical diagnoses and performed treatment; N. Z., X. H., J. T., Q. C., L. D., F. L. and C. Z. collected the samples; Z. Y., X. H., J. T., and Q. W. extracted the fecal DNA; Z. Y., X. H. and J. T. extracted the genomic DNA and contributed the SNP genotyping; Q. X. and X. X. performed the 16S rRNA and Metagenomic sequencing; Z. Y., Q. W. and X. H. performed the animal experiment; Z. Y., C. W., and N. Z. drafted the manuscript; P. Y., N. Q., and A. K. reviewed data analysis and edited the manuscript. All authors reviewed the manuscript.
Availability of data and materials
Metagenomic sequencing data for all samples have been deposited in NCBI with accession number of PRJNA356225.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Ethics approval and consent to participate
All procedures followed the tenets of the Declaration of Helsinki and were approved by the Ethics Committee of Chongqing Medical University with written informed consent.
Abbreviations
| AS | = | Ankylosing spondylitis |
| AUC | = | Area under the receiver operating characteristic curve |
| BMI | = | Body mass index |
| CNS | = | Central nervous system |
| EAU | = | Experimental autoimmune uveitis |
| eggnog | = | Evolutionary genealogy of genes: Non-supervised Orthologous Groups database |
| Gb | = | Gigabases |
| GWAS | = | Genome-wide association analysis |
| IBD | = | Inflammatory bowel disease |
| KEGG | = | Kyoto Encyclopedia of Genes and Genomes database |
| KO | = | KEGG orthologues |
| LPS | = | Lipopolysaccharide |
| MGS | = | Metagenomic Species |
| MS | = | Multiple sclerosis |
| NCBI | = | National Center for Biological Information |
| OG | = | EggNOG orthologues |
| ORFs | = | Non-redundant open reading frames |
| OUT | = | Operational taxonomic unit |
| PA | = | Psoriatic arthritis |
| PCoA | = | Principal component analysis |
| RA | = | rheumatoid arthritis |
| SLE | = | systemic lupus erythematosus |
| VKH | = | Vogt-Koyanagi-Harada |
Supplemental Material
Download Zip (6.4 MB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Yang P, Ren Y, Li B, Fang W, Meng Q, Kijlstra A. Clinical characteristics of Vogt-Koyanagi-Harada syndrome in Chinese patients. Ophthalmology. 2007;114:606–614. doi:10.1016/j.ophtha.2006.07.040.
- Du L, Kijlstra A, Yang P. Vogt-Koyanagi-Harada disease: novel insights into pathophysiology, diagnosis and treatment. Prog Retin Eye Res. 2016;52:84–111. doi:10.1016/j.preteyeres.2016.02.002.
- Chi W, Yang P, Li B, Wu C, Jin H, Zhu X, Chen L, Zhou H, Huang X, Kijlstra A. IL-23 promotes CD4+ T cells to produce IL-17 in Vogt-Koyanagi-Harada disease. J Allergy Clin Immunol. 2007;119:1218–1224. doi:10.1016/j.jaci.2007.01.010.
- Moorthy RS, Inomata H, Rao NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol. 1995;39:265–292. doi:10.1016/S0039-6257(05)80105-5.
- Weisz JM, Holland GN, Roer LN, Park MS, Yuge AJ, Moorthy RS, Forster DJ, Rao NA, Terasaki PI. Association between Vogt-Koyanagi-Harada syndrome and HLA-DR1 and -DR4 in Hispanic patients living in southern California. Ophthalmology. 1995;102:1012–1015. doi:10.1016/S0161-6420(95)30920-7.
- Liang L, Tan X, Zhou Q, Tian Y, Kijlstra A, Yang P. TLR3 and TLR4 But not TLR2 are Involved in Vogt-Koyanagi- Harada Disease by triggering proinflammatory cytokines production through promoting the production of mitochondrial reactive oxygen species. Curr Mol Med. 2015;15:529–542. doi:10.2174/1566524015666150731095611.
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi:10.1038/ni.1863.
- Ye Z, Zhang N, Wu C, Zhang X, Wang Q, Huang X, Du L, Cao Q, Tang J, Zhou C, et al. A metagenomic study of the gut microbiome in Behcet’s disease. Microbiome. 2018;6:135. doi:10.1186/s40168-018-0520-6.
- Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 2015;21:895–905. doi:10.1038/nm.3914.
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. doi:10.7554/eLife.01202.
- Nani E, Amato G, Lavitola A, Morgantetti F. [Evaluation of a semiautomatic system (ABAC-ABACTOR III) for the identification of Enterobacteriaceae]. Quad Sclavo Diagn. 1985;21:56–61.
- Forbes JD, Van Domselaar G, Bernstein CN. Microbiome survey of the inflamed and noninflamed gut at different compartments within the gastrointestinal tract of inflammatory bowel disease patients. Inflamm Bowel Dis. 2016;22:817–825. doi:10.1097/MIB.0000000000000684.
- Naftali T, Reshef L, Kovacs A, Porat R, Amir I, Konikoff FM, Gophna U. Distinct microbiotas are associated with ileum-restricted and colon-involving crohn’s disease. Inflamm Bowel Dis. 2016;22:293–302. doi:10.1097/MIB.0000000000000662.
- Tremlett H, Fadrosh DW, Faruqi AA, Zhu F, Hart J, Roalstad S, Graves J, Lynch S, Waubant E, Centers U. Gut microbiota in early pediatric multiple sclerosis: a case-control study. Eur J Neurol. 2016;23:1308–1321. doi:10.1111/ene.13026.
- Cantarel BL, Waubant E, Chehoud C, Kuczynski J, DeSantis TZ, Warrington J, Venkatesan A, Fraser CM, Mowry EM. Gut microbiota in multiple sclerosis: possible influence of immunomodulators. J Investig Med. 2015;63:729–734. doi:10.1097/JIM.0000000000000192.
- Costello ME, Ciccia F, Willner D, Warrington N, Robinson PC, Gardiner B, Marshall M, Kenna TJ, Triolo G, Brown MA. Intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol. 2014.
- Lopez P, Sanchez B, Margolles A, Suarez A. Intestinal dysbiosis in systemic lupus erythematosus: cause or consequence? Curr Opin Rheumatol. 2016;28:515–522. doi:10.1097/BOR.0000000000000309.
- Horai R, Zarate-Blades CR, Dillenburg-Pilla P, Chen J, Kielczewski JL, Silver PB, Jittayasothorn Y, Chan CC, Yamane H, Honda K, et al. Microbiota-dependent activation of an autoreactive T cell receptor provokes autoimmunity in an immunologically privileged site. Immunity. 2015;43:343–353. doi:10.1016/j.immuni.2015.07.014.
- Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001; 131:647–652. DOI: 10.1016/S0002-9394(01)00925-4
- Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi:10.1093/bioinformatics/btn025.
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi:10.1038/nature08821.
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:1–6. doi:10.1186/2047-217X-1-18.
- Noguchi H, Park J, Takagi T. MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006;34:5623–5630. doi:10.1093/nar/gkl723.
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi:10.1093/bioinformatics/btl158.
- Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59–64. doi:10.1038/nature13568.
- Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi:10.1038/nature12506.
- Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, Plichta DR, Gautier L, Pedersen AG, Le Chatelier E, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotech. 2014;32:822–828. doi:10.1038/nbt.2939.
- Jensen LJ, Julien P, Kuhn M, von Mering C, Muller J, Doerks T, Bork P. eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008;36:D250–D4. doi:10.1093/nar/gkm796.
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D80. doi:10.1093/nar/gkh063.
- Caspi RR. Experimental autoimmune uveoretinitis in the rat and mouse. Curr Protoc Immunol. 2003; Chapter 15:Unit 15 6. doi:10.1002/0471142735.im1506s53.
- Patil KR, Nielsen J Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:2685–2689. DOI 10.1073/pnas.0406811102
- Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, Zhang D, Xia H, Xu X, Jie Z, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. 2015;6:6528. doi:10.1038/ncomms7528.
- Hou S, Du L, Lei B, Pang CP, Zhang M, Zhuang W, Zhang M, Huang L, Gong B, Wang M, et al. Genome-wide association analysis of Vogt-Koyanagi-Harada syndrome identifies two new susceptibility loci at 1p31.2 and 10q21.3. Nat Genet. 2014;46:1007–1011. doi:10.1038/ng.3061.
- Caspi RR. A look at autoimmunity and inflammation in the eye. J Clin Invest. 2010;120:3073–3083. doi:10.1172/JCI42440.
- Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. doi:10.1016/j.cell.2014.03.011.
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi:10.1038/nature11053.
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi:10.1126/science.1208344.
- Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, Tremaroli V, Bakker GJ, Attaye I, Pinto-Sietsma SJ, et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat Med. 2018;24:1526–1531. doi:10.1038/s41591-018-0160-1.
- Gupta VK, Paul S, Dutta C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front Microbiol. 2017;8:1162. doi:10.3389/fmicb.2017.01162.
- Petersen LM, Bautista EJ, Nguyen H, Hanson BM, Chen L, Lek SH, Sodergren E, Weinstock GM. Community characteristics of the gut microbiomes of competitive cyclists. Microbiome. 2017;5:98. doi:10.1186/s40168-017-0320-4.
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi:10.1038/nature11450.
- Oezguen N, Yalcinkaya N, Kücükali CI, Dahdouli M, Hollister EB, Luna RA, Türkoglu R, Kürtüncü M, Eraksoy M, Savidge TC, et al. Microbiota stratification identifies disease-specific alterations in neuro-Behcet’sdisease and multiple sclerosis. Clin Exp Rheumatol. 2019 May 30. PMID: 31172918. Epub ahead of print.
- Tanabe S. The effect of probiotics and gut microbiota on Th17 cells. Int Rev Immunol. 2013;32:511–525. doi:10.3109/08830185.2013.839665.
- Romond MB, Colavizza M, Mullie C, Kalach N, Kremp O, Mielcarek C, Izard D. Does the intestinal bifidobacterial colonisation affect bacterial translocation? Anaerobe. 2008;14:43–48. doi:10.1016/j.anaerobe.2007.09.003.
- Lievin V, Peiffer I, Hudault S, Rochat F, Brassart D, Neeser JR, Servin AL. Bifidobacterium strains from resident infant human gastrointestinal microflora exert antimicrobial activity. Gut. 2000;47:646–652. doi:10.1136/gut.47.5.646.
- Kleessen B, Blaut M. Modulation of gut mucosal biofilms. Br J Nutr. 2005;93(Suppl 1):S35–40. doi:10.1079/BJN20041346.
- Peng L, Li ZR, Green RS, Holzman IR, Lin J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J Nutr. 2009;139:1619–1625. doi:10.3945/jn.109.104638.
- Geirnaert A, Calatayud M, Grootaert C, Laukens D, Devriese S, Smagghe G, De Vos M, Boon N. Van de Wiele T. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci Rep. 2017;7:11450. doi:10.1038/s41598-017-11734-8.
- Wang W, Chen L, Zhou R, Wang X, Song L, Huang S, Wang G, Xia B. Increased proportions of Bifidobacterium and the Lactobacillus group and loss of butyrate-producing bacteria in inflammatory bowel disease. J Clin Microbiol. 2014;52:398–406. doi:10.1128/JCM.01500-13.
- Vogels GD. Van der Drift C. Degradation of purines and pyrimidines by microorganisms. Bacteriol Rev. 1976;40:403–468.
- Ye Z, Chen O, Zhang R, Nakao A, Fan D, Zhang T, Gu Z, Tao H, Sun X. Methane attenuates hepatic ischemia/reperfusion injury in rats through antiapoptotic, anti-inflammatory, and antioxidative actions. Shock. 2015;44:181–187. doi:10.1097/SHK.0000000000000385.
- Hevia A, Milani C, Lopez P, Cuervo A, Arboleya S, Duranti S, Turroni F, Gonzalez S, Suarez A, Gueimonde M, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio. 2014;5:e01548–14. doi:10.1128/mBio.01548-14.
- Hou S, Yang P, Du L, Zhou H, Lin X, Liu X, Kijlstra A. Small ubiquitin-like modifier 4 (SUMO4) polymorphisms and Vogt-Koyanagi-Harada (VKH) syndrome in the Chinese Han population. Mol Vis. 2008;14:2597–2603.
- Kim MH, Seong MC, Kwak NH, Yoo JS, Huh W, Kim TG, Han H. Association of HLA with Vogt-Koyanagi-Harada syndrome in Koreans. Am J Ophthalmol. 2000;129:173–177. doi:10.1016/S0002-9394(99)00434-1.
- Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17:179–184. doi:10.1002/ibd.21339.
- Rausch P, Rehman A, Kunzel S, Hasler R, Ott SJ, Schreiber S, Rosenstiel P, Franke A, Baines JF. Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc Natl Acad Sci U S A. 2011;108:19030–19035. doi:10.1073/pnas.1106408108.