
ABSTRACT
It is now becoming well established that vesicles are released from a broad range of cell types and are involved in cell-to-cell communication, both in physiological and pathological conditions. Once outside the cell, these vesicles are termed extracellular vesicles (EVs). The cellular origin (cell type), subcellular origin (through the endosomal pathway or pinched from the cell membrane) and content (what proteins, glycoproteins, lipids, nucleic acids, metabolites) are transported by the EVs, and their size, all seem to be contributing factors to their overall heterogeneity. Efforts are being invested into attempting to block the release of subpopulations of EVs or, indeed, all EVs. Some such studies are focussed on investigating EV inhibitors as research tools; others are interested in the longerterm potential of using such inhibitors in pathological conditions such as cancer. This review, intended to be of relevance to both researchers already well established in the EV field and newcomers to this field, provides an outline of the compounds that have been most extensively studied for this purpose, their proposed mechanisms of actions and the findings of these studies.
Introduction
Extracellular vesicles (EVs) are a heterogeneous group of vesicles released by cells under physiological and pathological conditions [Citation1–Citation3]. EVs are typically classified according to a range of properties including density, dimension and the biogenesis processes from which they originate. Two subclasses of EVs with different biogenesis pathways have been extensively reported on over the past number of years. These are exosomes, deriving from multi-vesicular bodies (MVB), and microvesicles (MVs), which bud directly from the cell membrane. Typically, exosomes are considered to be vesicles that fall within the size range of 30–150 nm, while MVs are often described as 100–1000 nm. However, it is evident that this classification is flawed, as these size ranges overlap and evidence suggests that vesicles in the size range of exosomes can also bud directly from the cell membrane rather than originate via the endosomal pathway.
Despite these limitations of the terminology, to adhere to the doctrine that those EVs derived through the endosomal pathway are exosomes and those pinching from the cell membrane are MVs, until such times as the majority of the EV research community agrees on other guidelines for nomenclature, here we refer to exosomes and MVs, respectively, according to their modes of biogenesis.
Exosomes biogenesis
Biogenesis of the EV subpopulation that are later termed exosomes occurs inside endosomes. Early endosomes, during their maturation towards late endosomes or MVBs, start to accumulate intraluminal vesicles (ILV) through endosomal membrane invaginations. These MVBs subsequently fuse with lysosomes and thus promote ILV destruction or, alternatively, they can either fuse with the cell membrane, releasing ILVs in the extracellular space. These vesicles are then termed exosomes [Citation4].
MVBs biogenesis is driven by at least two distinct mechanisms, i.e.. endosomal sorting complexes required for transport machinery (ESCRT)-dependent and ESCRT-independent pathways. Both pathways are schematised in which also includes drugs (manumycin A and GW4869, respectively) potentially able to block these mechanisms.
Figure 1. Exosomes are released from intracellular compartments known as multi-vesicular bodies (MVBs). MVBs biogenesis is associated with two different mechanisms: ESCRT-dependent and ESCRT-independent pathways.
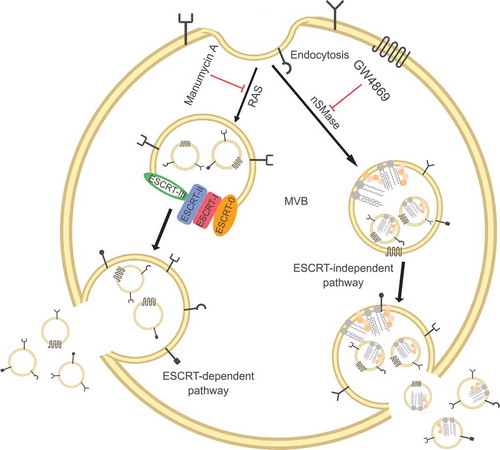
ESCRT consists of a multi-molecular machinery constituted by four main complexes (ESCRT-0, -I, -II and -III) and some additional associated proteins, such as those of the AAA ATPase Vps4 complex. These complexes orchestrate the ILVs formation in a stepwise fashion. ESCRT-0 and -I, interacting with ubiquitylated transmembrane cargos, form microdomain in the endosomal membrane and recruit ESCRT-III. This last complex subsequently interacts with the ESCRT-II complex, which is responsible for the vesicles abscission and release. In fact, the latter complex is able to form spirals that promote the inward budding and fission of vesicles in order to form MVBs [Citation5–Citation9]. The ESCRT pathway can also be recruited by other exosomes biogenesis mechanisms. Involvement of the axis syndecan-syntenin-ALIX in exosomes generation has been reported which, through the recruitment of the ESCRT accessory protein ALIX, is able to enrol the ESCRT-III complex and orchestrate ILVs formation. Based on this model, heparanases trim the heparin sulphate side chains of syndecans, facilitating the formation of syndecan microdomains able to interact with syntenin and, thus, draft the ESCRT machinery using ALIX as an intermediate [Citation10–Citation12].
MVBs production can also be achieved by ESCRT-independent processes, based on the presence of lipid rafts inside endosomal membranes. These lipid rafts are present at high levels of cholesterol and, most importantly, sphingolipids that can be subjected to the activity of the neutral sphingomyelinase family. These sphingomyelinase enzymes convert sphingolipids to ceramide, a cone-shaped rigid lipid that coalesces to form ever-larger microdomains inducing budding and formation of ILVs [Citation13–Citation17].
When MVBs are formed, as mentioned above, they can either fuse with lysosomes or result in exosomes release via fusion with the cell membrane. Their transport within cells and towards the cell membrane is dependent on interaction with actin and microtubules of the cytoskeleton and is regulated by many proteins. Particularly important here is the GTPase family of Rab proteins, although their involvement seems to be somewhat cell specific. For example, both Rab27a and Rab27b have been shown to be involved in exosomes release from HeLa cells, i.e. Rab27b regulates the motility of MVBs towards the cell membrane and Rab27a promotes their fusion. However, Rab27 isoforms are not ubiquitous. Thus, other cell types seem to regulate exosomes release using different Rab proteins, such as Rab11 in K562 cells (bone marrow chronic myelogenous leukaemia cells) and Rab35 in Oli-neu cells (oligodendroglial cell lines) [Citation18–Citation20]. SNARE (Soluble NSF Attachment Protein REceptor) protein family members are also involved in orchestrating the final exosomes release in response to membrane fusion. This is achieved through the formation of a SNARE complex composed of three-fourth coiled-coil helices proteins. SNARE activity is partially controlled by the phosphorylation state of these proteins, which influences their localisation and their interaction with SNARE partners, thus contributing to regulated exosomes release [Citation21–Citation23].
Microvesicles biogenesis
MVs biogenesis is modulated by lipid composition and organisation of the peripheral cytoskeleton, both of which are able to alter membrane fluidity and deformability [Citation24]. As for exosomes, mechanisms that generate or alter asymmetry of the cell membrane appear to be important during MVs generation. In fact, lipid rafts are fundamental to the budding of the cell membrane and cholesterol seems to play key roles, as cholesterol depletion reduces MVs release [Citation14–Citation17,Citation25]. Interestingly, MVs present unique lipid characteristics, such as externalisation of phosphatidylserine (a lipid normally contained in the internal leaflet of the membrane) and an enrichment of the sphingolipid sphingomyelin [Citation25,Citation26]. Enzyme involved in transferring lipids from one leaflet of the cell membrane to the other, such as calpain, scramblases, flippases and floppases, has been reported as important during MVs formation [Citation3,Citation27–Citation32]. Of note, the activity of acidic sphingomyelinases and the conversion of sphingomyelin to ceramide are also involved in MVs production (in a process similar to the one performed by neutral sphingomyelinases during exosomes biogenesis). Specifically, ceramide, being a cone-shaped lipid, induces membrane curvature and triggers MV release [Citation33–Citation38].
In addition to lipids, other cytoskeletal elements and their regulators are required for MVs biogenesis. Due to their involvement in regulating actin reorganisation and mediating the cytoskeleton contractility, Rho family members are fundamental players in this process. RhoA activity has been reported to promote MVs release from cancer cells, through its activity on ROCK and ERK [Citation39–Citation41]. Additionally, RhoA, ARF6 and ARF1, phosphorylating the myosin light chain at the MV necks in order to enhance myosin contractility, favour the fission and the pinching-off of the resulting MVs [Citation42–Citation45].
Interestingly, a partial overlapping mechanism for MVs with exosomes biogenesis has been proposed. Specifically, it seems that the ESCRT machinery may be involved in the production of nano-sized vesicles that are enriched in cell surface proteins, reflecting their cell membrane origin. Thus, consistent with the complexity of the EV world, MVs release may also be supported by this mechanism, although further research is needed to investigate this [Citation46–Citation48].
Pharmacological inhibitors of EVs release
EVs are involved in many pathophysiological conditions, and in some cases, their involvement relates to disease development and progression [Citation1,Citation3,Citation49]. Thus, many pharmacological agents are being explored to investigate how best to inhibit EV release, both as research tools and potentially as therapeutic approaches, if means can be identified that selectively affect EVs involved in pathology but not those performing necessary physiological roles. However, the complexity and heterogeneity of EV biogenesis is challenging the development of a single drug that would be able to completely block EVs production. Nevertheless, identifying and blocking the predominant EV subpopulation(s) associated with a particular disease, if such exists and if feasible, could be clinically useful. In this review, the main drugs evaluated thus far to block EV release (listed in ) are discussed, considering their proposed abilities to inhibit one EV population instead of another, such as GW4869 for exosomes or Y27632 for MVs. Thus, we have broadly categorised the compounds into two main groups according to their mechanisms-of-action, i.e. those that particularly affect EV trafficking (calpeptin, manumycin A and Y27632) and those that particularly affect lipid metabolism (pantethine, imipramine and GW4869).
Table 1. Most commonly used compounds reported to prevent EVs (MVs or exosomes) release and their biological properties.
Inhibition of EV trafficking
Compounds that are associated particularly with affecting EV trafficking include calpeptin, manumycin A and Y27632.
Calpeptin
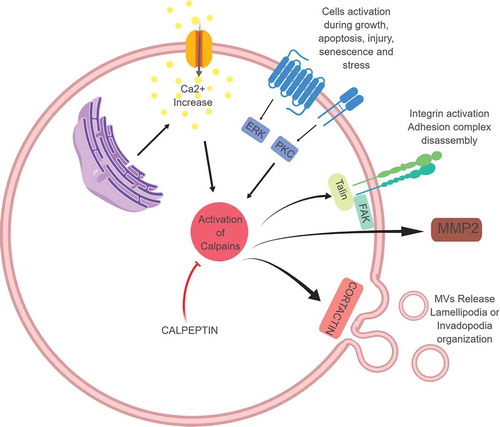
Calpain comprises a family of calcium-dependent neutral, cytosolic cysteine proteases. The most studied isoforms appear to be the µ- and m-calpains, which are ubiquitously expressed in cells and activated with calcium concentrations of µM to mM. µ- and m-calpains are hetero-dimers consisting of a large catalytic subunit (80 kDa) and a regulatory subunit (28 kDa) containing the calcium-binding site. When calcium levels rise, calcium binds and promotes a conformational change, allowing self-cleavage and activation of the pro-enzyme. Once activated, calpains can engage with numerous protein substrates, including G-proteins and cytoskeletal proteins, that contribute to the many processes in which calpains are involved. These activities include apoptosis, cell proliferation, migration, tumour invasiveness and cancer progression, as calpains are de-regulated in cancer cells [Citation50,Citation51]. Due to their activity on cytoskeleton remodelling, these proteins can apparently promote MVs shedding. Conversely, their inhibition, using calpain inhibitors, is reported to reduce the amount of MVs released by cells.
The most studied calpain inhibitor is calpeptin, a reversible semi-synthetic peptidomimetic aldehyde inhibitor developed by the chemical modification of a Streptomyces peptide aldehyde in which the N-terminal has been substituted with a hydrophobic cap group. Due to its inhibitory activity on calpain (see ), calpeptin has been quite extensively used as micro-vesiculation inhibitor.
Figure 2. Calpains, once activated through calcium binding, can activate different cellular processes including cell migration (through their interaction with Talin and FAK), cell invasion (thorough the promotion of MMP release and activation), and MVs formation and release (through their activity on cytoskeletal proteins including cortactin). Calpain inhibitors, of which calpeptin is one of the most studied, are under investigation for clinical application.
In resting platelets, compared to some other cell types, only a small percentage of MVs are produced, but this percentage increases upon platelet stimulation by agonists such as collagen, thrombin or the synthetic calcium ionophore A23187. Additionally, it has been demonstrated that prion protein fragment (106–126) (PrP) can activate platelets, favouring an increase of cytosolic calcium levels and leading to a concentration-dependent increased release of MVs. Specifically, PrP was shown to induce an 8–10 fold increase in calpain activity in the presence of 1 mM extracellular calcium, while pre-incubation of platelets with 80 µM calpeptin prevented calpain activation and MVs release [Citation52,Citation53].
Prior to these studies, Fox et al. [Citation54] demonstrated the involvement of calpain in the shedding of pro-coagulant-containing MVs released from activated platelets. Specifically, using a low concentration of calpeptin (10–20 µg/mL), calpain-mediated hydrolysis of the actin-binding protein was prevented, leading to reduced shedding of MVs. Pre-treatment of platelets with calpeptin substantially inhibited MVs formation (70% reduction), even after platelets activation with collagen, thrombin or a combination of these two agonists. However, increasing calpeptin concentration up to 200 µg/mL did not have any additional effect. In a subsequent experiment, Yano et al. [Citation55] showed the calpeptin activity to be concentration dependent (0–300 µM) and that the reduction of MVs shedding is mostly due to the reduced activity of calpain on actin-binding proteins, as the inhibition of MVs formation was observed 10 min after platelets incubation with thrombin and collagen.
Calpeptin effects have also been evaluated on other cell types. To investigate the involvement of MVs release during cell membrane repair, human embryonic kidney (HEK293) cells were treated with streptolysin-O (an agent known to damage cell membrane) and then treated with 60 µM calpeptin. Calpeptin substantially reduced MVs release and, consequently, streptolysin-O accumulated inside the cells and induced an increased lysis rate on calpeptin-treated cells compared to its effect on untreated cells [Citation56].
MVs have been implicated in anti-cancer drug-resistance [Citation57]. Using a prostate cancer cell line model, PC3, Jorfi et al. [Citation58] demonstrated that calpeptin’s inhibition of MVs release allowed docetaxel and methotrexate to accumulate inside the cells, resulting in significantly reduced cell proliferation and more cell death than that observed in the absence of calpeptin treatment. Subsequent pre-clinical in vivo studies showed reduced PC3 tumour growth in nude mice following combined therapy of either docetaxel or methotrexate and calpeptin (injected, 1 h before the chemotherapeutics, directly in the tumour mass at a dose of 10 mg/Kg) when compared to docetaxel or methotrexate treatment alone. This phenomenon was proposed to be the result of decreased intra-tumoural vascularisation, decreased proliferation and enhanced apoptosis of PC3 cells, which was enhanced by calpeptin’s inhibition of micro-vesiculation of the tumour cells.
Brought together, these studies support the hypothesis that calpeptin can inhibit shedding of particles/vesicles, described by the authors as microparticles in platelets studies and as microvesicles in the studies of HEK293, SH-SY5Y, and PC3 cell lines. However, as with many studies of EVs, there are challenges in meta-analysing the data and in drawing conclusions from the data as a whole, as different methods for EV separation were used for different studies. In fact, as summarised in , one study did not separate EVs from medium at all and instead evaluated treated platelets’ supernatant (after low-speed centrifugation) by FACS, counting the particles by comparing FITC-CD41a labelled events with bead events. It would have built confidence if the particles/vesicles had been characterised by more than one method, but this was not always the case. It should be noted that about half of these studies preceded the 2014 position statement from the International Society for Extracellular Vesicles (ISEV) that described minimal experimental requirements for the definition of EVs and their functions [Citation59]. Therefore, understandably there may not have been an awareness of what the ISEV community considers as necessary. Interestingly, unique amongst these studies, only Jorfi et al. [Citation58] included characterisation of the vesicles by TEM. Here the authors reported vesicles of their expected size (~250 nm). Numerous small structures are also visible in the image presented, the origin of which is unclear. Overall, however, based on the success of work done in the calpeptin studies reported thus far, future studies investigating its ability to inhibit EV release are warranted. To investigate if this is a somewhat selective effect on EV release, concentrations of the drug that are first proven to be non-toxic to cells should be evaluated.
Table 2. EV separation and characterisation methods used in the studies discussed in this review.
Manumycin A
Ras is a family of small GTPases able to regulate the function of other proteins and involved in many different cellular processes including cell differentiation, cytoskeletal integrity, cell adhesion and migration, cell proliferation, exosomes release and apoptosis. Due to their range of functions, it is not surprising that their de-regulation – which can occur in cancer cells – contributes to increased invasiveness, metastasise and reduced apoptosis.
Structurally, Ras is composed of two domains: the G domain that binds the GTP and the C-terminal membrane-targeting domain. To function properly, the C-terminal domain must be modified by the addition of a lipid chain, a modification mediated by the activity of a farnesyltransferase enzyme. Inhibition of this modification by farnesyltransferases inhibition, in turn, inhibits Ras activity and so prevents its down-stream effects. One of the most commonly used farnesyltransferases inhibitors is manumycin A, a cell-permeable antibiotic extracted from Streptomyces parvulus that is described as acting as a selective and potent inhibitor of Ras farnesyltransferases. Due to the involvement of Ras during exosomes release, manumycin A has been investigated as an inhibitor of exosomes secretion.
Using manumycin A, Hyun Jeong Oh et al. [Citation60] reported the involvement of exosomes in neuronal differentiation. Specifically, this study showed that exosomes carrying miRNA-193a and released from F11 cells (rat dorsal root ganglion cells), when in co-culture with undifferentiated F11 cells, stimulate neurogenesis of these previously undifferentiated recipient cells. This differentiation was inhibited by pre-treating the donor cells with 5 µM manumycin A and was linked to a reduction in the amount of released CD63-bearing exosomes.
Datta et al. [Citation61] reported the inhibitory activity of manumycin A specifically on the ESCRT-dependent exosomes biogenesis. Here, in prostate cancer cell lines (C4-2B, PC3 and 22Rv1) the activity of 250 nM manumycin A led to a reduction in exosomes production by 50–60% depending on the particular cell line; the effect was even more substantial when manumycin A was used in combination the nSMase inhibitor GW4869. This study also reported manumycin A’s mechanisms-of-action as not only inhibitory to Ras and its farnesyltransferases, but also to hnRNP H1, a protein belonging to the heterogeneous nuclear ribonucleoproteins and involved in the regulation of pre-mRNA biogenesis, metabolism and transport. Apparently, the additional inhibition of this protein enhances the inhibitory effect on exosomes biogenesis.
Furthermore, using manumycin A, Zhou et al. [Citation62] reported a role of exosomes in the wound-healing process. Their study initially correlated exosomes secreted by scratched BUMPT cells with a negative effect on the wound-healing process. To further support these findings, they progressed to reducing exosomes release. The results showed an increased wounded area after BUMPT cells scratch upon 10 µM GW4869 or 1 µM manumycin A treatment, confirming the deleterious effect of exosomes on this process.
Overall, as summarised in , these three studies reported in 2017 (so probably informed by the 2014 position statement from ISEV [Citation59]) used the commonly selected ultracentrifugation methods for EV separation and, to varying degrees, characterised the resulting vesicles. Hyun Jeong Oh et al. [Citation60] also investigated and confirmed the absence of the proposed negative marker, calnexin. However, as for the studies with calpeptin, not unexpectedly, not all the same characterising was done in all studies, which can make generalisation and overall conclusions challenging. Favourably, the limited number of studies of manumycin A reported thus far show relevance to progressing to further investigate the ability of this compound to inhibit EV release. Prior to such future studies, it would be necessary to do as Datta et al. [Citation61]did and establish concentrations of the drug that are non-toxic to the cells of interest to give confidence that any reduction in EV release is not simply as a consequence of the cells being destroyed by manumycin A.
Y27632
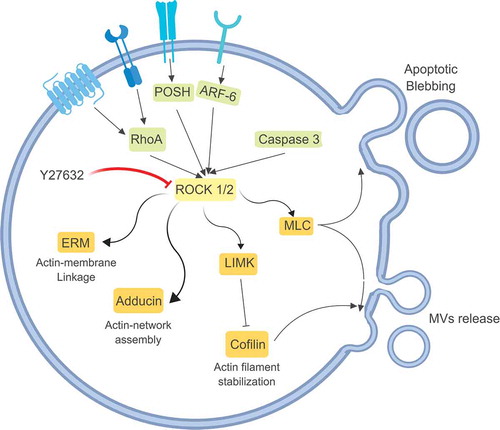
Rho-associated protein kinases (ROCK) are a family of serine-threonine kinases belonging to the PKA/PKG/PKC family and involved in regulating the shape and the movement of cells, by acting on the cytoskeleton (). This is relevant as re-organising the cytoskeleton and mediating cellular contractility through activity on actin filaments is important for MVs shedding. Y27632 is a commonly used cell-permeable, highly potent, competitive inhibitor of both ROCK1 and ROCK2 which is able to compete with ATP in binding the catalytic binding sites of these kinases.
Figure 3. Rho-associated protein kinases (ROCK) are serine-threonine kinases involved in cytoskeleton re-organisation. Once activated by multiple stimuli, ROCK regulate the shape and the movement of the cells, through the activation of Adducin or ERM (ezrin, radixin and moesin), but they can also interact with MLC (myosin light chain) and LIMK (LIM kinases, able to inactivate cofilin, fundamental for actin filament stabilisation) both involved in MVs release. Y27632 is a competitive inhibitor of both ROCK1 and ROCK2 and by blocking these proteins it can inhibit MVs release.
The involvement of ROCK kinases in MVs formation was investigating in triple-negative breast cancer (MDA-MB-231) and ovarian cancer (HeLa) cells with up-regulated levels of these kinases [Citation39]. Firstly, it was established that these cells, expressing the constitutively active RhoA mutant, had substantial MVs formation. This MVs release was suppressed using RhoA siRNA, which abolished not only the effect of the RhoA mutant but also the effect due to an external cell activation by epidermal growth factor (EGF). To confirm proteins causally involved, 5 µM Y27632 was used to block the activity of ROCK1 and ROCK2 and, subsequently, MVs were no longer visible at the cell membrane. Consequently, the medium conditioned by these cells contained a markedly reduced amount of MVs. Finally, investigating which enzymes were activated by ROCK1 and ROCK2, the main effect on MVs shedding was found to be due to the activation of LIMK and MYLK, which act on cofilin and myosin, thus favouring cytoskeleton re-organisation and filaments contraction.
Including Y27632 to elucidate the major player between ROCK1 and ROCK2 in promoting MVs release, Sapet et al [Citation63] worked with a microvascular endothelial cell line (HMEC-1) activated by thrombin. Here, thrombin was shown to stimulate an increase in MVs shedding from HMEC-1 cells, with substantially increased transcription of ROCK2 following 4-h treatment with thrombin. Pre-treatment of HMEC-1 cells with 1 µM Y27632 prevented the thrombin-induced MVs release. When specific siRNA targeting ROCK1 and ROCK2, respectively, were used, siRNA against ROCK1 had no effect on MVs shedding, while the siRNA against ROCK2 extensively suppressed the thrombin effect. This indicated the involvement of ROCK2, but not ROCK1, in MVs production.
Further using Y27632 to investigate and control the mechanisms by which MVs are released, Lathan et al. [Citation64] performed experiments with hCMEC/D3 cells (immortalised primary microvascular brain endothelial cells) after stimulation with tumour necrosis factor (TNF). Y27632 (5 µM) causes a reduction in the release of MVs in response to TNF occurred, as well as a change in cell surface morphology, with cells adapting a more cobblestone shape and presenting fewer fibrous protrusions. This effect correlated with the activity of the inhibitor which sustains activation of proteolytic enzymes, such as Stathmin and Calpain, that destabilised the cell membrane. This study also proposed a fundamental role for β- and γ-actin in facilitating membrane protrusion and MVs release.
The ability of Y27632 to efficiently inhibit MVs formation has been confirmed by many research groups. Tramontano et al. [Citation65] reported a reduction in CD105+ and CD31+ MVs release from human coronary artery endothelial cells (HCAEC) upon treatment with 10 µM Y27632 or 0.1 µM/mL statin (a well know anti-inflammatory molecule). Kim et al. [Citation66] highlighted the ability of isoflurane to activate endothelial cell Rho kinases, promoting an increase in CD73-bearing MVs production in vitro using immortalised human umbilical vein endothelial cells (EAhy9262) and immortalised mouse glomerular endothelial cells (GENC cells). Y27632 (10 µM) prevented this response to isoflurane in vitro. Additionally, Hussein et al. [Citation67] reported a correlation between inhibition of MVs release, using either 200 µM calpeptin, 30 µM Y27632 or the combination of the two drugs (Y27632 added after 1 h of pre-incubation with calpeptin), and increased endothelial cell (EC) apoptosis and detachment in response to external stress induced by staurosporine and IL-1α. This effect correlated with the inability of the cells to rid themselves of caspase 3, a pro-apoptotic protein that typically accumulates in MVs and so is removed from these cells. The, instead, accumulation of caspase 3 in the cytoplasm upon treatment with calpeptin and/or Y27632 led to cell detachment and apoptosis.
As concluded for the studies with other inhibitors, not all studies with Y27632 (and as summarised in ) actually include separating EVs from conditioned medium (CM) at all, i.e. Sapet et al. [Citation63] centrifuged their CM at 5000 g for 5 min and then proceeded with flow cytometry. This makes drawing collective conclusions very challenging, but it could be argued that it adds a different type of information. Even when EVs were separated in the studies involving Y27632, their characterisation was often limited when compared to the ISEV position paper [Citation59] and more recent MISEV2018 guidelines [Citation68] (but it is noteworthy that the Y27632 studies preceded the 2014 ISEV position paper). For example, the main – and sometimes the only – characterisation technique used was FACS, sometimes for annexin V alone. Techniques such as immunoblotting for EV markers and/or advanced microscopy were rarely used. So, here again, our conclusion is that although such studies are important to our fundamental understanding of how Y27632 might affect EVs, their interpretation can only be made in the context of pre- the ISEV position paper and MISEV2018 guidelines and not in the context of what the ISEV community now regard as ideal for EV characterisation. Thus, future studies should determine concentrations of Y27632 that are non-toxic to the cells being investigated to ensure that the effects claimed on EVs are not due to cellular cytotoxicity and then due consideration should be given to characterising the EVs in line with MISEV2018.
Inhibition of lipid metabolism
Compounds that are associated particularly with affecting lipid metabolism include pantethine, imipramine and GW4869.
Pantethine
Pantethine is a pantothenic acid (vitamin B5) derivate used as an intermediate in the production of co-enzyme A and it plays a role in the metabolism of lipids and reduction of total cholesterol levels. In fact, pantethine has been shown to inhibit (by 80%) cholesterol synthesis in cultured skin fibroblasts (GM0043), as well as total fatty acids synthesis [Citation69]. As the fluidity of the cell membrane is fundamental during membrane lipid bilayer re-organisation and thus MVs formation, pantethine may be used to impair MVs shedding. Specifically, this drug has been shown to block translocation of phosphatidylserine on the outer membrane leaflet, a process that is fundamental for MVs production.
Pantethine effect has been investigated using breast cancer MCF-7 cell variants that are resistant or sensitive to doxorubicin [Citation70]. After stimulation of these cells with the agonist A23187, resistant cells, but not doxorubicin-sensitive MCF-7, released higher amount of MVs. This effect was reduced upon pre-treatment with pantethine, obtaining an overall MV reduction of 24%.
Using pantethine, Kavian et al. [Citation71] investigated the role of MVs in systemic sclerosis (SSc). The main dysfunctions in SSc involves damage to ECs and fibroblasts, and so the effect of pantethine on ECs was investigated. Following addition of either 50 µM or 100 µM pantethine, the release of MVs from ECs decreased in a concentration-dependent manner. The same effect was observed in a pre-clinical in vivo model where mice develop SSc following daily intradermal injections of 200 µL of hypochlorous acid (HOCl). In these mice, pantethine substantially reduced the quantities of circulating ECs MVs. Mice treated orally with 150 mg/Kg of D-pantethine showed an attenuation of fibrosis and vascular alteration and this protection was associated with reduced quantities of circulating CD144+ MVs of endothelial origin, reflecting the level of endothelial damage.
With pantethine, Penet et al. [Citation72] explored the involvement of MVs in cerebral malaria. In vitro, using mouse brain endothelial cells (MBECs) and human umbilical vein endothelial cells (HUVECs) as model systems, a 51% reduction in quantities of MVs released was observed after pre-treatment with 1 mM of pantethine and subsequent stimulation with TNF when compared to treatment with TNF alone. Advancing to in vivo studies, mice received intraperitoneal injections of red blood cells infected with Plasmodium berghei ANKA and the cerebral syndrome occurred on day 7–8 after infection. However, administration of pantethine (30 mg for 5 days) prevented the occurrence of cerebral malaria, associated with the ability of this drug to prevent ECs’ MVs release. It should also be noted that MVs' release was inhibited only after administration of pantethine specifically, i.e. pantethine constituents, such as cystamine, cysteamine and pantothenic acid, did not affect MVs. This highlighted the importance of the disulphide bridge present in the pantethine molecular structure to exert this functional activity on MV release.
Again, as for the other compounds reported to reduce EV release, collectively there is substantial evidence from these studies that pantethine has shown beneficial results in the model systems evaluated. However, minimal characterisation (typically by FACS) was done in the context of both the ISEV position paper and MISEV2018. Furthermore, prior checks to ensure that the concentrations of pantethine used were non-toxic to the cells were not always included. Thus, building on the strengths of the earlier work, more extensive studies of pantethine and how EVs released are characterised in line with MISEV2018 are now warranted to fully understand its potential as a selective inhibitor of EVs.
Imipramine
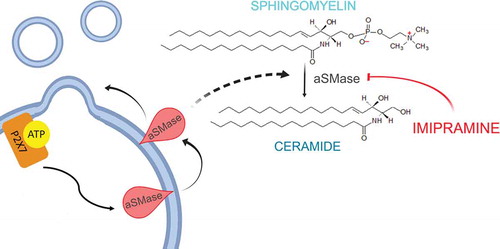
Imipramine is a tricyclic anti-depressant that has drawn attention due to its inhibitory activity on acid sphingomyelinase (aSMase). aSMase enzymes catalyse the hydrolysis of sphingomyelin to ceramide [Citation73], a process involved in both exosomes and MVs formation as it increases membrane fluidity, exosomes release and MVs generation (). Upon activation of the ATP receptor P2X7, MV shedding is associated with a rapid activation and translocation of aSMase to the outer leaflet of the cell membrane. After its internalisation inside the cells (especially inside endosomes and lysosomes), the basic imipramine molecule becomes protonated. In this form, it stimulates the proteolytic degradation of aSMase that, once it loses its negative charge, detaches from the membrane. Thus, imipramine is reported to prevent the translocation of aSMase, inhibiting MV and exosomes secretion.
Figure 4. MVs present unique lipid characteristics, including an enrichment of sphingomyelin and ceramide, thus every enzyme able to interfere with membrane composition, i.e. calpains, scramblases and acid sphingomyelinases, plays a key role in MVs biogenesis. Acid sphingomyelinases convert sphingomyelin into ceramide, a cone-shaped rigid lipid that forms micro-domains inside the cell membrane, inducing the budding of MVs. Imipramine, a well-known anti-depressant, can promote membrane fluidity by acting on aSMases, thus preventing MVs generation.
An early report on imipramine [Citation35] highlighted the ability of astrocytes and microglia cells to produce MVs in response to agonist activation of the receptor P2X7. This ability was blocked using different MVs inhibitors, such as BAPTA (equivalent of EGTA), cytochalasin D (an actin polymerisation inhibitor), and Y27632. However, the most substantial inhibitory effect on aSMase was achieved with 10 µM imipramine treatment or gen knock-out knocking of aSMase. With the latter, the MVs shedding was completely abrogated.
Using imipramine to investigate a potential correlation between osteoblast-derived MVs and bone loss, Deng et al. [Citation74] demonstrated that osteoblast (UAMS-32P) cells treated with this drug release a lower amount of MVs than do untreated cells. These cells were co-cultured with a macrophage cell line able to differentiate into osteoclast upon RANKL stimulation, but after 10 µM imipramine treatment, the number of osteoclasts recovered was lower than the imipramine-untreated co-culture. Advancing on this, imipramine was administered to ovariectomised mice for 45 days, and femur densitometry results showed an improved mineral density compared to mice not treated with imipramine, supporting the involvement of MVs in osteoclast differentiation and thus bone resorption.
Studies involving the prostate cancer cell line PC3 reported that imipramine blocked MVs and exosomes, but, of note, these vesicle types were not analysed separately [Citation75]. In this study, 25 µM imipramine treatment resulted in a 77% reduction in total EV release, included EVs of both <150 nm and >150 nm-sized vesicles. Incidentally, pantethine showed similar results to imipramine, although the effect on EVs release was less pronounced.
The latter two studies involving imipramine were reported in 2017, so post the ISEV position paper but prior to the publication of MISEV2018 guidelines. However, as for some studies with other compounds reported to inhibit EVs, characterisation of EVs typically falls somewhat short of that outlined in the ISEV position paper. To advance on these findings, more extensive studies of imipramine at proven subtoxic concentrations and with extensive characterisation of the EVs could be very useful.
GW4869
GW4869 is a cell-permeable, symmetrical dihydroimidazolo-amide compound that acts as a potent, specific, non-competitive inhibitor of membrane neutral sphingomyelinase (nSMase). nSMase is an ubiquitous enzyme that generates the bioactive lipid ceramide through the hydrolysis of the membrane lipid sphingomyelin [Citation33]. Quantitative analysis of lipid molecular species in the exosomes bilayer highlights an extensive enrichment in cholesterol, sphingolipids and ceramide with lower amounts of phosphatidylcholine than in the cellular membrane. This lipid composition is remarkably like that of lipid raft and treatment with exogenous SMase or application of C6-ceramide can induce vesicles formation, underlying the importance of ceramide in ESCRT-independent exosomes generation. Conversely, GW4869 has been reported to markedly reduce exosomes release. The cone-shaped structure of ceramide, released upon the SMase activity is, thus, important during the formation of spontaneous curvature, creating large lipid-raft domains involved in exosomes shedding [Citation13]. SMase is found in a range of compartments in all cells, including the Golgi apparatus, endosomes and cell membrane, so its activity is not only linked to exosomes generation but also MVs shedding. Specifically, using both the pharmacological inhibitor GW4869 and siRNAs against sphingomyelin phosphodiesterase 2/3 (SMPD2/3), a correlation between apparent exosomes inhibition and MVs release has been demonstrated. Treatment of SKBR-3 cells with 5 µM GW4869 (or the siRNA) resulted in increased quantities of vesicles with a size range of 100–200 nm, while the quantities of smaller vesicles released were much reduced. In keeping with this, overexpression of SMPD2 or SMPD3 decreased quantities of larger (100–200 nm) vesicles that were released and, instead, increased smaller vesicles release. This effect was also associated with a distinct lipid composition of MV bilayer, showing a higher content of sphingomyelin with the respect of the overall cell membrane lipid composition, that promotes the budding of cellular membrane. Thus deregulation of SMPD2/3 activity, interfering with lipid composition, may shift the secretion of cell cargo from exosomes to MVs or vice versa [Citation34].
GW4869 activity on vesicles has been extensively studied using fibroblasts. Lyu et al. [Citation76] reported that exosomes released from cardiac fibroblasts carry pro-hypertrophic molecules and that hypertrophy induced by cardiac fibroblasts CM was dramatically attenuated when its exosomes were depleted. Exosomes release was increased following cardiac fibroblasts’ activation using angiotensin II (AngII) and exosomes uptake led to up-regulation of renin, Angiotensin I, Angiotensin receptor 1 and 2 (AT1R and AT2R) in receiving cardiomyocytes. These effects by cardiac fibroblasts’ exosomes were substantially inhibited when neonatal rat cardiomyocytes co-cultured with neonatal rat cardiac fibroblasts were treated with 40 µM GW4869. These findings were also supported by in vivo experiments with C57BL/6N mice where AngII-induced myocardial hypertrophy and cardiac fibrosis were markedly reduced upon treatment with GW4869 (via intraperitoneal injection of 2.5 mg/kg body weight), supporting a causal role for exosomes during cardiac pathologic hypertrophy.
Hu et al. [Citation77] reported that cancer-associated fibroblasts (CAFs) prime cancer stem cells (CSCs) in colorectal cancer, promoting their chemo-resistance. In these studies, exosomes separated from 18Co fibroblasts stimulated SW620 CSCs to form larger spheroids than those CSCs treated with the vehicle control (DMSO). Subsequently mice implanted with CSCs and treated subcutaneously with 18Co cells’ CM before intra-peritoneally injection with chemotherapeutic agents (1 µM 5-fluorouracil or oxaliplatin) had higher tumour incidence, faster tumour growth and larger tumours; demonstrating that 18Co cells’ CM protects xenograft tumours from chemotherapy, priming CSCs to become more resistant. However, when 18Co and CAF cells were treated with 10 µM GW4869 in vitro, the CM effect on CSCs was markedly reduced, supporting the relevance of exosomes in the CSCs priming process. Similarly, Richards et al. [Citation78] reported that CAFs exposed to 100 nM gemcitabine dramatically increased exosomes release that, in turn, up-regulated cell proliferation and survival of chemo-sensitive recipient pancreatic epithelial cancer cells (PDACs). However, removing exosomes from the CAF CM reduced its ability to offer gemcitabine-resistance. Additionally, when CAFs were co-cultured with chemo-sensitive pancreatic cancer cells (l3.6 cells), gemcitabine had reduced efficacy; conversely, blocking exosome secretion using 20 µM GW4869 significantly reduced these survival benefits. In vivo, in NOD/SCID mice bearing CAFs and ASPC1 cells, co-treatment with gemcitabine and GW4869 (100 mg/Kg body weight) resulted in remarkably decreased growth rate of the tumour 10 days after treatment, when compared to those treated with gemcitabine alone. While these studies of raw CM do not confirm that GW4869 inhibits exosomes release, it certainly seems to be producing benefit that could be attributed to blocking EVs.
Similarly using GW4869, the role of exosomes released by hepatic stellate cells (HSCs) has been investigated in relation to chronic liver injury [Citation79]. Here, pro-fibrogenic connective tissue growth factor (CCN2)-GFP transfected donor LX-2 hepatic stellate cells were incubated with control non-transfected LX-2 cells. Fluorescence was detected in the control cells after 48 h; this fluorescence was reduced by 55% upon pre-treatment of the donor cells with 10 g/mL GW4869. This was proposed to show the ability of exosomes to transport CCN2 to HSCs that may result in the amplification of fibrinogenic signalling in response to chronic liver injury. In another model system, Zhou et al. [Citation62] reported negative influences of exosomes released during wound healing in renal tubular cells. Specifically, when exosomes production from mouse proximal tubular cells (BUMPT) was inhibited using 10 µM GW4869 or 1 µM manumycin A, the wound healing process was promoted, suggesting that the released exosomes reduced the rate of healing. EGF was reported to be causally involved in exosomes release, i.e. EGF-activated BUMPT cells had dramatically decreased exosomes released, while suppression of EGFR activity using gefitinib promoted exosomes release. Similar results have been obtained with lung adenocarcinoma (PC9) cells [Citation80]. Here an increase in exosomes production occurred in response to gefitinib; conversely, pre-incubating cells with 0.5–20 µM GW4869 prior to gefitinib addition prevented gefitinib’s stimulatory effect on exosomes production.
Cao et al. [Citation81] reported the involvement of exosomal DNA methyltransferases (DNMTs) in transmitting cisplatin-resistance to SKOV3 ovarian cancer cells. Specifically, exosomes released from SKOV3 cells were found to carry a significantly higher amount of DNMTs than exosomes derived from normal endometrial stromal cell lines (ESCs). When SKOV3 cells were treated with these exosomes, the cells showed significant resistance to 3 µM cisplatin when compared to SKOV3 cells that were not treated with exosomes. The resistance to cisplatin, which was proposed to be due to exosomes-carried DNMTs, was remarkably reduced by GW4869.
Exosomes (and other EVs) have been implicated in tumour progression. In melanoma, Matsumoto et al. [Citation82] reported GW4869 (5 µg/mL) significantly decreased the growth of B16BL6 cells compared with that of untreated or vehicle-treated (DMSO) cells, which they proposed to show that GW4869 inhibition of exosomes secretion suppresses autocrine-regulated proliferation of murine B16BL6 cells. The same effect was observed in vivo in C57BL/6J mice subcutaneously inoculated with B16BL6 cells. Here, B16BL6-derived exosomes injected into mice promoted tumour growth compared to those mice treated with PBS, while GW4869 treatment (1 µg of drug injected inside the tumour mass every day) was associated with significantly reduced tumour growth and thus murine survival. Including GW4869 as a hypoxia study, Panigrahi et al. [Citation83] reported that prostate cancer cells (LNCaP, 22Rv1 and PC3 E006AA-hT) secrete more exosomes when cultured in this condition, compared to normoxic conditions, as a survival mechanism to remove metabolic waste including lactic acid encapsulated in the exosomes. Conversely, preventing exosomes production using the GW4869 inhibitor (10–20 µM) reduced cell viability compared to the untreated cells, supporting a fundamental role of exosomes in survival of these cancer cells.
GW4869 has been used to show that exosomes may play an essential role in immune regulation. For instance, exosomes have been associated with house dust mite allergen-induced airway inflammation [Citation84]. In a C57BL/6J mouse model, Th2-immune-targeted miRNAs were found to be selectively released into the airway lumen via exosomes during allergic inflammation. Pre-treatment of mice with GW4869 (1.25 mg/kg) or DMSO, injected intraperitoneally, decreased exosomes release in vivo and, in turn, decreased the numbers of eosinophils recruited and accumulated in the mucosa. Additionally, this treatment suppressed the production of IL-4 and IL-13, thus reducing the pro-inflammation signals. Exosomal miRNA content was further connected with different immune system-modulating pathways including T-helper cell differentiation, granulocytes adhesion, diapedesis and leukocyte extravasation.
Similarly, with the help of GW4869, Essandoh et al. [Citation85] reported on negative effects of exosomes during sepsis-induced inflammation and cardiac dysfunction. Here, in vitro study showed that RAW264.7 macrophages challenged with LPS increased exosomes release and that these exosomes carried pro-inflammatory cytokines, including TNF-α, IL-1β and IL-6. However, when these cells were treated with 10–20 µM GW4869, the quantity of exosomes released – and thus of pro-inflammatory cytokines present in their CM – was substantially decreased. A similar effect was also observed in vivo when C57BL/6 mice were intraperitoneally injected with 2.5 µg/g GW4869 for 1 h, followed by an intraperitoneal injection of 25 µg LPS. Those pre-treated with GW4869 presented a significant decrease in endotoxin-triggered production of exosomes and pro-inflammatory cytokines into their serum. As a consequence, sepsis-induced cardiomyopathy was prevented and animal survival was improved in the GW4869 pre-treated cohort of mice.
The neutral sphingomyelinases pathway is reported to be involved in prion protein packaging into exosomes, based on studies including GW4869. In a rat immortalised hypothalamic cell line, GT1-7, whether infected or not with a mouse-adapted strain of human prion (M1000), the quantity of exosomes released was inhibited upon treatment with 4 µM GW4869. Interestingly, a decrease of both cellular PrPTOT and PrPSC was observed. The lowered PrP levels in the cells, in turn, were also observed in their exosomes, indicating that packaging of PrP into exosomes is regulated by nSMase pathway [Citation38]. In OvRK13 cells infected with the ovine 127S prion strain, 10 µM GW4869 treatment did not affect infectivity levels in ovRK13 recipient cells. However, it did dramatically inhibit infectivity released into the CM, although GW4869 marginally inhibits the release of other exosomal proteins such as Alix, Flotilin-1 and Tsg101. This suggests the involvement of exosomes in PrP release. The same effects were observed using shRNA against Tsg101 of the ESCRT-dependent pathway. Together these data support both ESCRT-dependent and ESCRT-independent pathways participating in extracellular release of actively multiplying prions, and suggest that inhibiting these pathways could be beneficial to prevent prion spreading [Citation86].
While GW4869 has been relatively extensively studied and reported to inhibit exosomes, here again collating the data is complicated by the fact that some authors have not reported how exosomes were separated (if they were), some have used the well-accepted ultracentrifugation methods, while other have used less favoured kit-based approaches. Similarly, some studies have included quite extensive characterisation to support the authors claim of working with EVs, while in other studies, the characterisation used is substantially lacking. As for all other agents/drugs detailed in this review and summarised in , we strongly advise that efforts should initially be invested in determining concentrations of GW4869 that are non-toxic to the cells being investigated and due consideration should be given to methodologies for EV separation and characterisation, which are in line with MISEV2018 guidelines.
Other drugs used
Due to the EVs release mechanism complexity, it is not surprising that the same production pathway can be impaired using different drugs acting on diverse targets involved in the same signal cascade. For this reason, although the drugs mentioned above are the most commonly used, other drugs have been investigated for their ability to inhibit EV release. These include bisyndoylmaleimide I, U0126, clopidogrel, imatinib, NSC23766, dimethyl amiloride, glibenclamide, indomethacin, chloramidine, cytochalasin D and sulphisoxazole.
Bisyndoylmaleimide I is a highly selective, cell-permeable, reversible inhibitor of protein kinase C (PKC). It acts as a competitive inhibitor of the PKC ATP-binding site and it can inhibit different PKC isoforms, including PKCα-, βI-, βII-, γ-, δ-, and ε- isozymes. MVs release is dependent upon calcium release and externalisation of the phosphatidylserine, both of which are involved in PKC activation with diacylglycerol. Additionally, as outlined above, MVs release is dependent upon the ROCK family activation and, as PKC belongs to this family, its inhibition can block MVs release. Stratton and co-workers [Citation87] reported MV release to be inhibited by bisyndoylmaleimide I in prostate cancer cells (PC3). Specifically, they treat the cells with the sublytic membrane attack complex (MAC) used to increase intracellular calcium level, known to favour MVs release. However, bisyndoylmaleimide I prevented MVs shedding, independently from the increased calcium intracellular level.
U0126 is a potent, specific, non-competitive inhibitor of MEK 1 and MEK 2, two protein kinases belonging to the mitogen-activated protein kinase kinase (MAPKK) family. As one of the mechanisms involved in MVs generation requires the activation of ERK, a protein upstream MEK, its inhibition has been associated with MVs reduction. Here, Mingzhen and co-workers [Citation88] demonstrated that using tobacco smoke extract on THP-1 monocytic cell line and on primary human monocyte-derived macrophages (hMDMs) increased the release of pro-coagulant MVs, an activity subsequently associated with an overactivation of the MAPK cascade. Subsequently, using 10 µM U0126 to inhibit MEK activity, the MVs release and the pro-coagulant activity derived from the stimulated monocytes and macrophages were dramatically decreased.
Clopidogrel is used clinically, alone or in combination with aspirin, as an anti-platelet anti-coagulant to reduce the risk of heart disease and stroke. It is administered as pro-drug, activated through its metabolism by CYP450 enzymes and, once processed, it selectively inhibits the binding of ADP to its receptor P2Y12 present on platelet surface. Clopidogrel apparently provides a protective effect on ECs, although the mechanism involved is poorly understood. Jung-Hwa Ryu et al. [Citation89] investigated clopidogrel’s effect on HUVEC cells that were stimulated with indoxyl sulphate. Here, indoxyl sulphate overactivated the MAPK signalling cascade, leading to a massive production of MVs that was efficiently blocked by 10–50 µM clopidogrel, possibly via its inhibitory activity on p38 MAP kinase, downstream in the MAP kinase pathway.
Imatinib is clinically used to treat chronic myeloid leukaemia, due to its ability to interact and inhibit the bcr-abl tyrosine kinase enzyme deriving from the Philadelphia Chromosome. This drug, interacting with the ATP-binding site present in the catalytic site of the enzyme, blocks the protein in the close conformation of the catalytic site, resulting in the inactivation of the catalytic activity. Unfortunately, myeloid leukaemia cells develop resistance to imatinib, a reason that prompted the development of a new drug, dasatinib, which has a similar effect on bcr-abl kinase and is apparently effective on all the bcr-abl mutants. Mineo et al. [Citation90] reported that these drugs prevent exosomes production by K562 chronic myeloid leukaemia cells, with reduction of 58% and 56%, respectively, for imatinib and dasatinib.
NSC23766 is an inhibitor of Rac1, an enzyme belonging to the Rho small GTPase family and involved in many physiological processes, including cell growth, motility, cell-to-cell adhesion and cytoskeleton re-organisation. NSC23766 can block Rac1 activation by the guanine nucleotide exchange factors (GEF) Trio and Tiam1, without affecting its interactions with Cdc42 or RhoA. Wang et al. Citation[91 correlated the activity of Rac1 with MVs generation in sepsis-activated platelet. Specifically, their pre-clinical in vivo study demonstrated that platelets produce a higher amount of MVs during sepsis than under healthy conditions, and the use of the Rac1 inhibitor NSC23766 (5 mg/Kg body weight) reduces MV release by 87%, confirming the involvement of this small GTPase during MVs production.
Dimethyl amiloride (DMA) is a derivate of amiloride, a drug used to treat high blood pressure based on to its ability to inhibit H+/Na+ (NHE 1, 2 and 3) and the Na+/Ca2+ channels [Citation92]. As exosomes release is connected to intracellular calcium levels, and DMA impairs the function of channels connected with the calcium homoeostasis, this drug has been proposed as a possible exosomes blocker. Both in in vitro (using mouse colon carcinoma CT26, lymphoma EL4 and human lung adenocarcinoma H23 cell lines) and in vivo (using a panel of different mice including female C57BL/6, BALB/c and nude mice) studies, Chalmin et al. [Citation93] reported that DMA-reduced exosomes release into culture medium and blood serum, respectively. The in vivo studies indicated that DMA (1 µmol/kg body weight) enhances the anti-tumour efficacy of cyclophosphamide, suggesting it should be considered as part of a combination therapy for cancer.
Glibenclamide (glyburide) belongs to the sulphonylureas family and inhibits an ATP-binding cassette transporter involved in MVs release. Glibenclamide has been used as an anti-diabetic drug because of its activity on the SUR receptor (member of the ABC family) which is involved in insulin release via its ability to regulate potassium channels. However, this drug is not specific to SUR/Kir6.2 and interacts with other proteins including the ABCA1, which is implicated in the transfer and recycle of cholesterol and phospholipid from cells to the circulating apolipoproteins A1 (apoA-I) during their maturation in high-density lipoprotein (HDL) [Citation94]. As cholesterol seems to have a fundamental role in MVs and exosomes production, glyburide has been proposed as a possible EVs inhibitor. Henriksson et al. [Citation95] suggested glyburide to have an anti-coagulant effect on monocytes in vitro, reducing tissue factor expression and reducing the number of MVs released. This has been proposed to be due to inhibition of ABCA1, supporting the relevance of this drug to controlling MVs release. Conversely, however, Kosgodage et al. [Citation75] found glibenclamide to have no effect on MV release on prostate cancer (PC3) and breast cancer (MCF7) cells.
Indomethacin belongs to the non-steroidal anti-inflammatory drugs (NSAID) family and is used to decrease prostaglandins production during inflammation, due to its ability to non-selectively inhibit cyclooxygenase I and II. Indomethacin has also been shown to downregulate transcription of the ABCA3 transporter, an intracellular protein involved in lipid transport [Citation96]. As lipids are major players in exosomes biogenesis, inhibition of ABCA3 may have an impairing effect on exosomes release. Koch et al. [Citation97] illustrated an effect of indomethacin on exosomes release using lymphoma cell lines (DlBCL cell lines SU-DHL-4, OCl-Ly1 and OCl-Ly3). Specifically, these cells were reported to encapsulate doxorubicin and pixantrone in their released exosomes, resulting in cancer cell survival. Preventing exosomes export using 10 µM indomethacin maintained the cytotoxic effect of these chemotherapeutics, as they were then able to accumulate inside the cells’ nuclei.
Chloramidine is a cell-permeable compound that irreversibly binds the calcium-bound form of peptidyl-arginine deiminases (PAD) enzymes, leading to their inactivation. Six PAD isoforms are known to exist in human, expressed in different cells and with different cell localisation. These enzymes are involved in many cellular processes, including chromatin rearrangement and protein deimination [Citation98]. PAD enzymes are overexpressed in many different pathological processes including cancer and, due to their ability to interact with cytoskeletal proteins, they have been proposed as possible players in MVs release. Kholia and co-workers [Citation99] demonstrated that micro-vesiculation promotion in prostate cells leads to increased levels of deiminated (i.e. removal of an amine group) of cytoskeletal proteins, particularly β-actin and actin-α1. These deiminations were found to be performed by PAD enzymes and cell pre-incubation with chloramidine substantially prevented actin deimination and, thus, MVs release.
Cytochalasin D is an alkaloid produced as a toxin by many fungi. Cytochalasin D can bind the edges of actin filaments preventing subunits association or dissociation, thus inhibiting and avoiding actin polymerisation. Given the importance of actin and cytoskeleton re-organisation during MVs budding and release or MVBs trafficking towards the cell membrane, cytochalasin D may possibly be used to prevent EVs release. Using a range of cancer cell lines including cervical carcinoma (HeLaS), pancreatic carcinoma (Panc1) and prostate carcinoma (PC3), Salma Khan et al. [Citation100] reported that cancer cells can release exosomes carrying the anti-apoptotic protein survivin. Cytochalasin D treatment reduced exosome release, in turn reducing the amount of survivin present in the tumour environment.
Finally, sulphisoxazole, a short-acting sulphomanide exerting an antibiotic activity against a wide range of gram-positive and gram-negative bacteria, has shown an anti-cancer activity in breast cancer (in a study including MCF10A, MCF7, MDA-MB-231 cells). Using both in vitro and pre-clinical in vivo studies, Im et al. [Citation101] reported the ability of this drug to efficiently reduce the release of small vesicles through its activity on the ESCRT-dependent pathway. This drug proved to inhibit the expression of several RABs and ESCRT-related components such as VPS4B and Alix, without affecting other vesicles-producing pathways such as the ESCRT-dependent pathways (neutral sphingomyelinases proved to be not affected) or modifying intracellular calcium levels. Sulphisoxazole efficiently inhibited the endothelin receptor type A, suggested this to be causally involved in small vesicles release.
Considering side-effects
As outlined earlier, even if some of these drugs – that are already formulated and used as therapeutic agents – are found to reliably, robustly and reproducibly, inhibit release of EVs, substantial efforts would still be needed to investigate their influence on EV release from healthy cells. Approaches to selectively deliver them to cancer cells may be required. Of course, the drugs that are already approved for use in humans, for some indication(s), would likely have a more straightforward pathway to utility than those are molecules that have never been developed as therapeutics. Notwithstanding that, even for currently used as therapeutics, their side-effects (whether or not related to their influence on EVs) must also be considered. For example, known side-effects of imipramine include blood disorders/suppression of immune cells and associated infections, disorientation, dizziness, tiredness, nausea and vomiting, low blood pressure, among others. Side-effects of pantetheine include – but are not limited to – nausea, diarrhoea and possible impaired blood clotting. Similarly, side-effects of others of these drugs such as imatinib, glibenclamide, and indomethacin are well established. However, it must also be remembered that no drugs in clinical use are without some side-effects and so decisions must ultimately be made on the benefit/risk ratio to decide upon the appropriateness of use.
Critical considerations in the context of MISEV guidelines
As indicated in , in the studies of inhibitors reported to date, a relatively broad range of methodologies have been used to separate and characterise vesicles. It is evident that some studies reported do not comply with the MISEV guidelines released in 2014 or 2018 [Citation59,Citation68]. However, as the scope here was to review the proposed abilities of compounds thus far reported to decrease the amount of EVs released, we elected not to bias the review by eliminating studies that did not fully comply with MISEV guidelines, particularly as some such studies were performed prior to the MISEV2018 guidelines being published. However, we believe that some observation in this regard should be considered.
Firstly, on the methodologies used to recover and subsequently characterise EVs: many studies in which for example calpeptin and Y27632 activity were evaluated used Annexin V test as characterisation method. While analysis of Annexin V can help to identify the presence of some vesicles subpopulations would be unreliable for identifying all MVs and to confidentially identify MVs compared to apoptotic bodies, if the latter be present. This could then lead to potentially under- or overestimating MV presence. Furthermore, where Annexin V immunoaffinity is used as an EV separation method, procedural bias may be unavoidable. Similarly, for some studies using ultracentrifugation without density gradients for EV separation. Here small, medium and large EVs may be pooled, masking influences of potential inhibitors on a specific subpopulation evaluated. Furthermore, it would be prudent for studies where 0.2 µm filters were used and resulting vesicles are described as MVs to consider that this filtration would likely have eliminated MVs.
Secondly, considering the presence of all EVs subpopulations would be ideal. However, some studies claim a reduction in MVs or microparticles released, but without considering if there were any associated changes in other EV subpopulations, such as small EVs. It would be useful to consider if blocking MVs release in turn either positively or negatively affects the release of exosomes, and vice versa.
Thirdly, we would advise that consideration always be given to the concentrations of inhibitors used and whether quantities of EVs released are evaluated also in the context of the number of cells releasing those EVs. As these studies include inhibitors interacting with fundamental cellular events, it is reasonable to propose that these compounds, depending on the concentrations used, will affect cell viability and proliferations. Although some studies report the use of “safe” concentrations that do not affect cellular viability and proliferations (i.e. Datta et al. and Panigrahi et al. [Citation61,Citation83]), the majority have given no consideration to this. So, it is impossible to determine whether or not the associated changes in EV amounts reported may be at least partly associated with a cytotoxic effect of the compounds on cells.
Conclusion
Many in vitro studies including cell lines and a limited number of pre-clinical in vivo studies indicate that many compounds have the ability to block, or at least limit, the formation and release of exosomes and/or microvesicles. These include calpeptin, Y27632, pantethine, imipramine, GW4869, manumycin A, bisyndoylmaleimide I, U0126, clopidogrel, imatinib, NSC23766, dimethyl amiloride, glibenclamide, indomethacin, chloramidine, cytochalasin D and sulphisoxazole. More extensive studies are now warranted to investigate the activities of these drugs – singly and in combination – on a much broader range of cell line and pre-clinical in vivo models. Being mindful of how we now define EVs, in the context of the recently published minimal information for studies of EVs [Citation68], such studies should give due consideration to concentration-response effects of these compounds on vesicle release; their off-target effects, if any, and their potential to somehow selectively block vesicles from cells representing disease (such as cancer) rather than vesicles from normal healthy cells, in order to truly understand their potential uses as research tools and as future therapeutics for controlling EV release.
Disclosure of interest
The authors have no interest to declare.
Additional information
Funding
References
- Yuana Y, Sturk A, Nieuwland R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013:27:31–39. doi:10.1016/j.blre.2012.12.002
- van der Pol E, Böing AN, Harrison P, et al. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64:676–22.
- Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol. 2015;40:41–51.
- Huotari J, Helenius A. Endosome maturation. Embo J. 2011;30:3481–3500.
- Chiaruttini N, Redondo-Morata L, Colom A, et al. Relaxation of loaded ESCRT-III spiral springs drives membrane deformation. Cell. 2015;163:866–879.
- Christ L, Raiborg C, Wenzel EM, et al. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem Sci. 2017;42:42–56.
- Henne WM, Stenmark H, Emr SD. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol. 2013;5:a016766-a016766.
- Lee I-H, Kai H, Carlson L-A, et al. Negative membrane curvature catalyzes nucleation of endosomal sorting complex required for transport (ESCRT)-III assembly. Proc Natl Acad Sci U S A. 2015;112:15892–15897.
- McCullough J, Clippinger AK, Talledge N, et al. Structure and membrane remodeling activity of ESCRT-III helical polymers. Science. 2015;350:1548–1551.
- Baietti MF, Zhang Z, Mortier E, et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012;14:677–685.
- Roucourt B, Meeussen S, Bao J, et al. Heparanase activates the syndecan-syntenin-ALIX exosome pathway. Cell Res. 2015;25:412–428.
- Friand V, David G, Zimmermann P. Syntenin and syndecan in the biogenesis of exosomes. Biol Cell. 2015;107:331–341.
- Trajkovic K, Hsu C, Chiantia S, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247.
- Skotland T, Sandvig K, Llorente A. Lipids in exosomes: current knowledge and the way forward. Prog Lipid Res. 2017;66:30–41.
- Record M, Poirot M, Silvente-Poirot S. Emerging concepts on the role of exosomes in lipid metabolic diseases. Biochimie. 2014;96:67–74.
- Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667.
- Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50.
- Hsu C, Morohashi Y, Yoshimura S-I, et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J Cell Biol. 2010;189:223–232.
- Ostrowski M, Carmo NB, Krumeich S, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12:19–30. sup pp 1–13.
- Savina A, Vidal M, Colombo MI. The exosome pathway in K562 cells is regulated by Rab11. J Cell Sci. 2002;115:2505–2515.
- Pfeffer SR. Unsolved mysteries in membrane traffic. Annu Rev Biochem. 2007;76:629–645.
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166.
- Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci CMLS. 2018;75:193–208.
- Muralidharan-Chari V, Clancy JW, Sedgwick A, et al. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci. 2010;123:1603–1611.
- Del Conde I, Shrimpton CN, Thiagarajan P, et al. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106:1604–1611.
- Haraszti RA, Didiot M-C, Sapp E, et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J Extracell Vesicles. 2016;5:32570.
- Clark MR. Flippin’ lipids. Nat Immunol. 2011;12:373–375.
- Hugel B, Martínez MC, Kunzelmann C, et al. Membrane microparticles: two sides of the coin. Physiol Bethesda Md. 2005;20:22–27.
- Al-Nedawi K, Meehan B, Micallef J, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–624.
- Piccin A, Murphy WG, Smith OP. Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 2007;21:157–171.
- Sedgwick AE, D’Souza-Schorey C. The biology of extracellular microvesicles. Traffic Cph Den. 2018;19:319–327.
- Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. 2017;27:172–188.
- Shamseddine AA, Airola MV, Hannun YA. Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv Biol Regul. 2015;57:24–41.
- Menck K, Sönmezer C, Worst TS, et al. Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J Extracell Vesicles. 2017;6:1378056.
- Bianco F, Perrotta C, Novellino L, et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. Embo J. 2009;28:1043–1054.
- Awojoodu AO, Keegan PM, Lane AR, et al. Acid sphingomyelinase is activated in sickle cell erythrocytes and contributes to inflammatory microparticle generation in SCD. Blood. 2014;124:1941–1950.
- Hoehn RS, Jernigan PL, Japtok L, et al. Acid sphingomyelinase inhibition in stored erythrocytes reduces transfusion-associated lung inflammation. Ann Surg. 2017;265:218–226.
- Guo BB, Bellingham SA, Hill AF. The neutral sphingomyelinase pathway regulates packaging of the prion protein into exosomes. J Biol Chem. 2015;290:3455–3467.
- Li B, Antonyak MA, Zhang J, et al. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene. 2012;31:4740–4749.
- McConnell RE, Higginbotham JN, Shifrin DA, et al. The enterocyte microvillus is a vesicle-generating organelle. J Cell Biol. 2009;185:1285–1298.
- Sedgwick AE, Clancy JW, Olivia Balmert M, et al. Extracellular microvesicles and invadopodia mediate non-overlapping modes of tumor cell invasion. Sci Rep. 2015;5:14748.
- D’Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol. 2006;7:347–358.
- Muralidharan-Chari V, Clancy J, Plou C, et al. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol CB. 2009;19:1875–1885.
- Schlienger S, Campbell S, Claing A. ARF1 regulates the Rho/MLC pathway to control EGF-dependent breast cancer cell invasion. Mol Biol Cell. 2014;25:17–29.
- van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–228.
- Wang Q, Lu Q. Plasma membrane-derived extracellular microvesicles mediate non-canonical intercellular NOTCH signaling. Nat Commun. 2017;8:709.
- Nabhan JF, Hu R, Oh RS, et al. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc Natl Acad Sci U S A. 2012;109:4146–4151.
- Dreyer F, Baur A. Biogenesis and functions of exosomes and extracellular vesicles. Methods Mol Biol Clifton NJ. 2016;1448:201–216.
- Lowry MC, Gallagher WM, O’Driscoll L. The role of exosomes in breast cancer. Clin Chem. 2015;61:1457–1465.
- Siklos M, BenAissa M, Thatcher GRJ. Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm Sin B. 2015;5:506–519.
- Leloup L, Wells A. Calpains as potential anti-cancer targets. Expert Opin Ther Targets. 2011;15:309–323.
- Mallick RL, Kumari S, Singh N, et al. Prion protein fragment (106–126) induces prothrombotic state by raising platelet intracellular calcium and microparticle release. Cell Calcium. 2015;57:300–311.
- Crespin M, Vidal C, Picard F, et al. Activation of PAK1/2 during the shedding of platelet microvesicles. Blood Coagul Fibrinolysis. 2009;20:63–70.
- Fox JE, Austin CD, Reynolds CC, et al. Evidence that agonist-induced activation of calpain causes the shedding of procoagulant-containing microvesicles from the membrane of aggregating platelets. J Biol Chem. 1991;266:13289–13295.
- Yano Y, Shiba E, Kambayashi J-I, et al. The effects of calpeptin (a calpain specific inhibitor) on agonist induced microparticle formation from the platelet plasma membrane. Thromb Res. 1993;71:385–396.
- Atanassoff AP, Wolfmeier H, Schoenauer R, et al. Microvesicle shedding and lysosomal repair fulfill divergent cellular needs during the repair of streptolysin O-induced plasmalemmal damage. PloS One. 2014;9:e89743.
- Namee NM, O’Driscoll L. Extracellular vesicles and anti-cancer drug resistance. Biochim Biophys Acta. 2018. DOI:10.1016/j.bbcan.2018.07.003
- Jorfi S, Ansa-Addo EA, Kholia S, et al. Inhibition of microvesiculation sensitizes prostate cancer cells to chemotherapy and reduces docetaxel dose required to limit tumor growth in vivo. Sci Rep. 2015;5:13006.
- Lötvall J, Hill AF, Hochberg F, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the international society for extracellular vesicles. J Extracell Vesicles. 2014;3:26913.
- Oh HJ, Shin Y, Chung S, et al. Convective exosome-tracing microfluidics for analysis of cell-non-autonomous neurogenesis. Biomaterials. 2017;112:82–94.
- Datta A, Kim H, Lal M, et al. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017;408:73–81.
- Zhou X, Zhang W, Yao Q, et al. Exosome production and its regulation of EGFR during wound healing in renal tubular cells. Am J Physiol Renal Physiol. 2017;312:F963–F970.
- Sapet C, Simoncini S, Loriod B, et al. Thrombin-induced endothelial microparticle generation: identification of a novel pathway involving ROCK-II activation by caspase-2. Blood. 2006;108:1868–1876.
- Latham SL, Chaponnier C, Dugina V, et al. Cooperation between β- and γ-cytoplasmic actins in the mechanical regulation of endothelial microparticle formation. FASEB J. 2013;27:672–683. https://doi.org/10.1096/fj.12-216531.
- Tramontano AF, O’Leary J, Black AD, et al. Statin decreases endothelial microparticle release from human coronary artery endothelial cells: implication for the Rho-kinase pathway. Biochem Biophys Res Commun. 2004;320:34–38.
- Kim M, Ham A, Kim KY-M, et al. The volatile anesthetic isoflurane increases endothelial adenosine generation via microparticle ecto-5ʹ-nucleotidase (CD73) release. PloS One. 2014;9:e99950.
- Abid Hussein MN, Böing AN, Sturk A, et al. Inhibition of microparticle release triggers endothelial cell apoptosis and detachment. Thromb Haemost. 2007;98:1096–1107.
- Théry C, Kenneth WW, Elena A, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750.
- Ranganathan S, Jackson RL, Harmony JA. Effect of pantethine on the biosynthesis of cholesterol in human skin fibroblasts. Atherosclerosis. 1982;44:261–273.
- Roseblade A, Luk F, Ung A, et al. Targeting microparticle biogenesis: a novel approach to the circumvention of cancer multidrug resistance. Curr Cancer Drug Targets. 2015;15:205–214.
- Kavian N, Marut W, Servettaz A, et al. Pantethine prevents murine systemic sclerosis through the inhibition of microparticle shedding. Arthritis Rheumatol Hoboken NJ. 2015;67:1881–1890.
- Penet M-F, Abou-Hamdan M, Coltel N, et al. Protection against cerebral malaria by the low-molecular-weight thiol pantethine. Proc Natl Acad Sci U S A. 2008;105:1321–1326.
- Arenz C. Small molecule inhibitors of acid sphingomyelinase. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2010;26:1–8.
- Deng L, Peng Y, Jiang Y, et al. Imipramine protects against bone loss by inhibition of osteoblast-derived microvesicles. Int J Mol Sci. 2017;18:1013.
- Kosgodage US, Trindade RP, Thompson PR, et al. Chloramidine/Bisindolylmaleimide-I-mediated inhibition of exosome and microvesicle release and enhanced efficacy of cancer chemotherapy. Int J Mol Sci. 2017;18:1007.
- Lyu L, Wang H, Li B, et al. A critical role of cardiac fibroblast-derived exosomes in activating renin angiotensin system in cardiomyocytes. J Mol Cell Cardiol. 2015;89:268–279.
- Hu Y, Yan C, Mu L, et al. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. Plos One. 2015;10:e0125625.
- Richards KE, Zeleniak AE, Fishel ML, et al. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36:1770–1778.
- Charrier A, Chen R, Chen L, et al. Exosomes mediate intercellular transfer of pro-fibrogenic connective tissue growth factor (CCN2) between hepatic stellate cells, the principal fibrotic cells in the liver. Surgery. 2014;156:548–555.
- Li X-Q, Liu J-T, Fan -L-L, et al. Exosomes derived from gefitinib-treated EGFR-mutant lung cancer cells alter cisplatin sensitivity via up-regulating autophagy. Oncotarget. 2016;7:24585–24595.
- Cao Y-L, Zhuang T, Xing B-H, et al. Exosomal DNMT1 mediates cisplatin resistance in ovarian cancer. Cell Biochem Funct. 2017;35:296–303.
- Matsumoto A, Takahashi Y, Nishikawa M, et al. Accelerated growth of B16BL6 tumor in mice through efficient uptake of their own exosomes by B16BL6 cells. Cancer Sci. 2017;108:1803–1810.
- Panigrahi GK, Praharaj PP, Peak TC, et al. Hypoxia-induced exosome secretion promotes survival of African-American and Caucasian prostate cancer cells. Sci Rep. 2018;8:3853.
- Gon Y, Maruoka S, Inoue T, et al. Selective release of miRNAs via extracellular vesicles is associated with house-dust mite allergen-induced airway inflammation. Clin Exp Allergy J Br Soc Allergy Clin Immunol. 2017;47:1586–1598.
- Essandoh K, Yang L, Wang X, et al. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim Biophys Acta BBA - Mol Basis Dis. 2015;1852:2362–2371.
- Vilette D, Laulagnier K, Huor A, et al. Efficient inhibition of infectious prions multiplication and release by targeting the exosomal pathway. Cell Mol Life Sci CMLS. 2015;72:4409–4427.
- Stratton D, Moore C, Zheng L, et al. Prostate cancer cells stimulated by calcium-mediated activation of protein kinase C undergo a refractory period before re-releasing calcium-bearing microvesicles. Biochem Biophys Res Commun. 2015;460:511–517.
- Li M, Yu D, Williams KJ, et al. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2010;30:1818–1824.
- Ryu J-H, Kim S-J. Clopidogrel effectively suppresses endothelial microparticle generation induced by indoxyl sulfate via inhibition of the p38 mitogen-activated protein kinase pathway. Blood Purif. 2011;32:186–194.
- Mineo M, Garfield SH, Taverna S, et al. Exosomes released by K562 chronic myeloid leukemia cells promote angiogenesis in a Src-dependent fashion. Angiogenesis. 2012;15:33–45.
- Wang Y, Luo L, Mörgelin M, et al. Rac1 regulates sepsis-induced formation of platelet-derived microparticles and thrombin generation. Biochem Biophys Res Commun. 2017;487:887–891.
- Savina A, Furlán M, Vidal M, et al. Exosome release is regulated by a calcium-dependent mechanism in K562 cells. J Biol Chem. 2003;278:20083–20090.
- Chalmin F, Ladoire S, Mignot G, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. 2010;120:457–471.
- Nieland TJF, Chroni A, Fitzgerald ML, et al. Cross-inhibition of SR-BI- and ABCA1-mediated cholesterol transport by the small molecules BLT-4 and glyburide. J Lipid Res. 2004;45:1256–1265.
- Henriksson CE, Hellum M, Haug KBF, et al. Anticoagulant effects of an antidiabetic drug on monocytes in vitro. Thromb Res. 2011;128:e100–106.
- Aung T, Chapuy B, Vogel D, et al. Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proc Natl Acad Sci U S A. 2011;108:15336–15341.
- Koch R, Aung T, Vogel D, et al. Nuclear trapping through inhibition of exosomal export by indomethacin increases cytostatic efficacy of doxorubicin and pixantrone. Clin Cancer Res Off J Am Assoc Cancer Res. 2016;22:395–404.
- Mohanan S, Cherrington BD, Horibata S, et al. Potential role of peptidylarginine deiminase enzymes and protein citrullination in cancer pathogenesis. Biochem Res Int. 2012;2012:1–11.
- Kholia S, Jorfi S, Thompson PR, et al. A novel role for peptidylarginine deiminases in microvesicle release reveals therapeutic potential of PAD inhibition in sensitizing prostate cancer cells to chemotherapy. J Extracell Vesicles. 2015;4:26192.
- Khan S, Jutzy JMS, Aspe JR, et al. Survivin is released from cancer cells via exosomes. Apoptosis Int J Program Cell Death. 2011;16:1–12.
- Im E-J, Lee C-H, Moon P-G, et al. Sulfisoxazole inhibits the secretion of small extracellular vesicles by targeting the endothelin receptor A. Nat Commun. 2019;10:1387.
- Colombo F, Bastoni M, Nigro A, et al. Cytokines stimulate the release of microvesicles from myeloid cells independently from the P2X7 receptor/acid sphingomyelinase pathway. Front Immunol. 2018;9:204.