
ABSTRACT
Macrophage is essential for host anti-bacterial defense by directly eliminating invading microbes and inducing a series of immune reactions. Here we identified a Streptococcus pneumoniae protein, PepO, as a TLR2/TLR4 bi-ligand. We found that PepO enhances macrophage unspecific phagocytosis and bactericidal activity, which is related to the induction of autophagy in macrophage, for the inhibition of autophagy significantly decreased the phagocytosis and bactericidal activity of PepO-treated macrophage. We confirmed that these effects of PepO are dependent on interacting with both TLR2 and TLR4. The tlr2 or tlr4 deficiency partially abolished the effect of PepO while tlr2/tlr4 deficiency abolished it completely. In vivo study demonstrated that PepO reduced the bacteria load in WT mice significantly, while the depletion of macrophage or tlr2/tlr4 deficiency abrogated the effect of PepO. Our findings suggested the therapeutic potential of PepO and provided experimental evidence for immunotherapy against infectious disease.
Introduction
Manipulating host immune system to fight against invading pathogen has a long history. There are several immunomodulators applied in clinical practice for preventing and treatment against bacterial infection [Citation1,Citation2]. However, these agents are usually lysate of multiple bacteria, which made it hard to explain their mechanism and thus restrained their application.
The best-characterized receptors participate in the elimination of invading pathogen by innate immunocytes are Toll-like receptors (TLRs) [Citation3]. TLRs are essential pattern recognition receptors expressed in innate immunocytes such as macrophage and dendritic cells, which initiate innate immune response by recognizing PAMPs, participate in multiple immune reactions including phagocytosis, bacteria killing, antigen presentation, and cytokine production [Citation4]. Pharmaceutical agents targeting TLRs demonstrated therapeutic potential for various diseases [Citation5,Citation6]. An artificial lipid derivate mimics LPS specifically targeting TLR2 to switch macrophage from pro-tumor M2 phenotype to anti-tumor M1 phenotype [Citation7]. TLR7 agonist Imiquimod [Citation8] and TLR7/8 agonist Resiquimod [Citation9] had been used for virus infection treatment. As for bacterial infection treatment, the artificial TLR4 ligand AGPs protect mice from Listeria monocytogenes and Yersinia pestis infection [Citation10]. TLR2/6 agonist MALP-2 promotes macrophage phagocytosis and significantly reduced the mortality of the coinfection of influenza virus and S. pneumoniae [Citation11], while protected against P. aeruginosa infection [Citation12]. These studies have proved the therapeutic potential of TLRs agonist for bacterial infection.
Streptococcus pneumoniae endopeptidase O, PepO, a 72kD virulence protein, was reported to be capable of inducing strong innate immune response in a TLR2/TLR4 dependent manner [Citation13]. Thus, we assume that PepO is a potential immunomodulator which is conducive to host anti-infection defense. We reported in our previous study that PepO enhances macrophage phagocytosis through TLR2/miR155 pathway [Citation14]. In this study, we found that the PepO-induced phagocytosis enhancement is also related to TLR4; however, the underlying mechanism is not clear yet.
Autophagy is a universal existing, evolutionary conserved physiology process in eukaryotes by which malfunctioning organelles, misfolding protein, and internalized microorganism are degraded [Citation15]. Many research had revealed that the activation of autophagy in macrophage is quite important in the elimination of pathogen [Citation16–Citation18], while the crosstalk between TLRs signaling and autophagy is also confirmed by accumulating evidences [Citation19–Citation21]. TLR2 induces autophagy via Lyn and NF-κB [Citation22] while TLR4 induces autophagy via Warburg effect mediated by AMPK [Citation23]. It is noticeable that different TLRs may activate autophagy by different mechanisms, and those mechanisms may be independent or synergistic. For example, MyD-88/NF-κB [Citation24] and PAI-2 [Citation25] signaling is related to both TLR2 and TLR4. Previously we reported that PepO manipulates PI3 K/Akt signal pathway [Citation13], a main regulatory axis in autophagy regulation, in a TLR2/4 dependent manner, which inspired us the possibility that PepO triggers macrophage autophagy.
In this study, we confirmed the interaction between PepO with TLR2 and TLR4, respectively, which led to the enhancement of macrophage phagocytosis and bactericidal activity. In tlr2 or tlr4 deficiency macrophage, the effect of PepO is partially abrogated while tlr2/tlr4 double-deficiency abrogated it completely. Further investigation revealed that the enhanced macrophage function is related to autophagy, which is induced by the suppression of mTOR phosphorylation. In vivo study showed that PepO unspecific reduced bacteria load in pulmonary infection, which is dependent on macrophage and TLR2/TLR4. Taken together, these results suggested that PepO may be a potential therapeutic agent for pulmonary infection therapy and provided experimental evidence for infectious disease immunotherapy by targeting TLRs.
Materials and methods
Animal model and treatment
tlr2−/- and tlr4−/- gene-deficient mice were purchased from Jackson Lab. tlr2/4−/- gene-deficient mice were obtained by tlr2−/- and tlr4−/- gene-deficient mice hybridization. All gene-deficient mice were confirmed according to the genotyping protocol provided by Jackson Lab. Female WTC57/BL6 micebe between 7 ~ 8 weeks old or gene-deficient C57/BL6 mice weighing 21 ~ 23 g were treated with PBS, PepO (0.33 mg/mL, 30 μL per mouse) or LytR (0.17 mg/mL, 30 μL per mouse) intranasally 6 h before infection, the concentration of LytR was adjusted to equate the molarity of PepO. For infection, mid-exponential phase cultures of Streptococcus pneumoniae D39 and Pseudomonas aeruginosa were pelleted, washed with PBS and then resuspended with PBS. Treated mice were infected with S. pneumoniae D39 (1 × 108) and P. aeruginosa (5 × 107) intranasally, respectively. Twenty-four hours after infection, the blood was obtained, lungs were harvested and homogenized in 1 mL PBS with tissue homogenizer. The blood and lung homogenate were then diluted and seeded on Columbia blood agar. The colony-forming unit (CFU) was counted on the second day.
Bacterial strains and culture
S. pneumoniae D39 (NCTC 7466), P. aeruginosa PAK and S. aureus 502A were preservation of our laboratory. P. aeruginosa MDR strain was isolated in Children’s Hospital of Chongqing Medical University. The strains were seeded on Columbia Blood Agar plates and cultured overnight at 37°C supplemented with 5% CO2, single colony of S. pneumoniae D39 was then inoculated into C + Y culture medium and other strains were cultured in Luria Bertani broth at 37°C supplemented with 5% CO2. Bacteria were harvested at mid-log phase for subsequent procedure.
Construction of D39ΔpepO mutants
The D39ΔpepO mutant is constructed according to the protocol described in our earlier research [Citation26]. Briefly, the cassette containing pepO upstream sequence, erythromycin-resistant encoding sequence, and pepO downstream sequence is generated by Long Flank Homology-PCR using the primers listed in Supplemental Table.1. The constructed fragment is transformed into S. pneumoniae D39 according to the established protocol [Citation27]. The erythromycin-resistant colonies were selected and identified with PCR and gene sequencing.
Primary macrophage harvest and cell culture
For primary peritoneal derived macrophage harvest, 1 mL sterilized liquid paraffin was injected to mice 6 ~ 8 weeks old C57/BL6 mice. The treated mice were sacrificed by cervical dislocation on the 4th day after injection. The peritoneal cavity was rinsed by cold PBS supplemented with 0.1% EDTA-Na2, and the lavage was centrifuged to collect cells. After removing erythrocytes by Red Cell Lysis Buffer, the macrophage was seeded into appropriate plated or dish and cultured in DMEM (High Glucose,10% FBS). Culture medium was refreshed after 30 min to remove unwanted cells.
Phagocytosis and bactericidal activity assessment
The macrophages were treated with S. pneumoniae D39 (MOI = 100), S. aureus 502A (MOI = 100) and P. aeruginosa PAK (MOI = 100) for 30 min. The extracellular bacteria were removed by Gentamycin (200 μg/mL) treatment for 30 min and washing. The cells were then lysed with ddH2O and scrapping, and the lysate was diluted and seeded on Columbia Blood Agar for CFU counting. For bactericidal activity assessment, the procedure is similar to the phagocytosis assay with 2 h of infection. The percent of bacteria killed by macrophage is calculated by the equation: Rkilled = 1-B2 h/B30 min, where Rkilled represents the ratio of bacteria eliminated by macrophage, B2 h represents the number of intracellular bacteria after 2-h infection and B30 min represents the number of intracellular bacteria after 30 min infection.
Immunoprecipitation
2 × 107 Primary murine macrophage was seeded in 9 cm dishes 1 day before PepO treatment. The macrophage was incubated with recombinant PepO (10 μg/mL) for 1 h to ensure the binding of PepO to TLR2 or TLR4. The culture medium was discarded, and macrophage was washed with cold PBS for 3 times. The macrophage was then incubated with cell lysis buffer for Western and IP (Beyotime) supplemented with Protease Inhibitor Cocktail (Bimake) for 1 min followed by collecting with cell scrapper and transferring into microcentrifuge tubes. The macrophage lysate was incubated with Protein-G coupled agarose beads (Millipore) to exclude unspecific interaction. Then, the lysate was incubated with anti-His-Tag antibody (Beyotime) and Protein-G coupled agarose beads at 4°C overnight with mild shaking. The beads were collected by centrifuging and then subjected to Western Blot.
Immunoblot
The cell lysate was subjected to appropriated SDS-PAGE. The separated protein was then transferred to PVDF membrane in ice bathing. The membrane was blocked by 5% BSA in TBS buffer (137 mM NaCl,20 mM Tris, pH = 7.6) and then incubated with corresponding antibodies at 4°C overnight with slow shaking. The membrane was then washed with TBS supplemented with 0.5% Tween-20 and incubated with appropriated HRP-conjugated second antibody. The image was obtained by ECL.
NOS assessment
The macrophages were treated with PepO or pre-treated with Bafilomycin A1 (Selleck) before PepO treatment, and then the cells were treated with NOS assay kit (Beyotime) according to the instruction of the manufacturer.
Preparation of recombinant PepO protein and its truncated mutant
CDS sequence of pepO in S. pneumoniae D39 was amplified by PCR and cloned to pet28a plasmid. The constructed plasmid is transformed into E. coli BL21 (DE3) after confirming by sequencing. The expression of PepO recombinant protein is induced by IPTG (0.4 mM) and PepO protein is collected and purified by affinity chromatography with Ni2+/NTA column. The purified protein is subjected to ToxinEraser™ Endotoxin Removal Kit (Genescript) to remove the residual endotoxin. The LytR, PepO 1–430, and PepO 431–630 mutant was constructed in the same manner. The primers used in this study are listed in the Supplementary materials.
Reagents
Antibodies against LC3 I/II, p70S6 K, ULK, and beclin 1 were purchased from Cell Signaling Technology. Antibodies against GAPDH and His-Tag were purchased from Beyotime. Antibodies against TLR2 and TLR4 were purchased from Abcam. Antibody against p-mTOR was purchased from Santa Cruz. Protein-G coupled agarose beads and RapidStep™ ECL Reagent were purchased from Millipore. Ni2+-NTA column was purchased from GE lifesciences. DMEM High glucose culture medium was purchased from Hyclone. Cell lysis buffer for Western and IP and Nitric Oxide Synthase Assay Kit was purchased from Beyotime. Protease Inhibitor Cocktail is purchased from Bimake. Red Cell Lysis Buffer was purchase from Tiangen. Rapamycin and Bafilomycin A1 were purchased from Selleck. Clodronate Liposome was purchased from Liposoma.
Statistical analysis
Data are presented as mean with SEM. Student’s t-test was employed. A p-value of less than 0.05 was considered significant.
Results
PepO triggers autophagy in macrophage
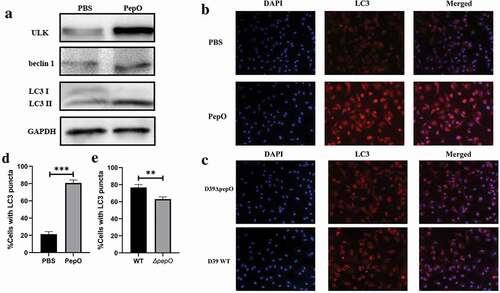
We previously reported that PepO enhances macrophage phagocytosis and manipulated the phosphorylation of Akt, a major signal molecule in autophagy regulation. Since autophagy participates in the internalization of pathogen in phagocytes [Citation16], we assumed that PepO may be involved in macrophage autophagy. After PepO treatment, the autophagy marker LC3 showed a significant turnover of LC3I to LC3II, while autophagy-related protein marker ULK-1 and beclin 1 increased consistently ()). To rule out the effect of impurity in the recombined protein, e.g. LPS from E. coli, we treated macrophage with recombined LytR, a S. pneumoniae protein which produced in the same process, and detected no significant alteration in LC3 expression (SFigure 2). The formation of autophagosome was monitored by immunofluorescent microscopy. PepO significantly increased the number of LC3 puncta, indicating the formation of autophagosome (). To further confirm the autophagy induced by PepO, we constructed a ΔpepO mutant by replacing pepO gene with an erythromycin-resistant coding sequence. WT S. pneumoniae D39 infected peritoneal derived macrophages (PDMs) showed oblivious aggregation of LC3 puncta, while D39ΔpepO::Pcerm infected PDM showed significantly lower amount of LC3 puncta (), taken together, these results confirmed that PepO induces macrophage autophagy.
Figure 1. PepO triggers macrophage autophagy. (a) PepO induced the elevation of autophagy marker ULK and beclin 1 and the turnover of LC3 I to LC3 II after 6 h of PepO treatment (10 μg/mL). (b) WT macrophage was differently treated and the LC3 was monitored by immunofluorescent microscopy. Recombinant PepO protein (10 μg/mL) induced obviously LC3 puncta aggregation comparing to PBS treated macrophage. (c) WT D39 infected macrophage showed significantly higher amounts of LC3 puncta than D39ΔpepO infected macrophage after 6 h of infection (MOI = 100). (d) and (e) The cells with numerous LC3 puncta of differently treated macrophage were counted (n = 100) in three independent experiments. Images were representative of three independent experiments. (d) and (e) were the statistical analyzation of (b) and (c), respectively. Data were shown as mean + SEM (n = 3) and are representative of three independent experiments. **p < 0.01; ***p < 0.001. Student’s t-test was employed for statistical analysis
PepO-induced autophagy contributes to the elimination of bacteria
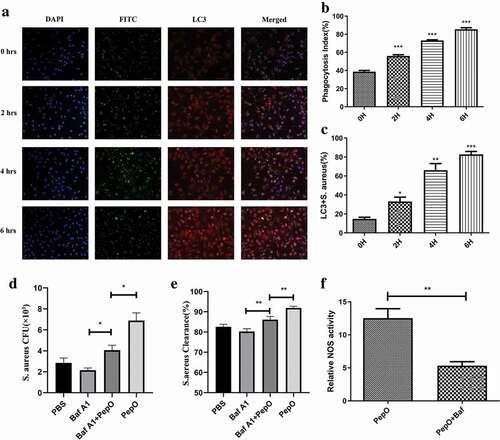
Autophagy is known as a self-defense process, and multiple researches had revealed the relationship between macrophage autophagy and bacteria clearance [Citation16]. Since the formation of LC3-associated structure is critical for autophagic clearance of bacteria, we then investigated the spatial relationship of internalized bacteria and LC3 puncta by immunofluorescent microscopy to verified if PepO-induced autophagy is related with phagocytosis. We found that the longer PepO treated the macrophage, the more S. aureus was internalized into macrophage (), while the partial of LC3-co-localized bacteria is increased after PepO treatment , suggesting that PepO-induced autophagy is related to the enhanced phagocytosis. Meanwhile, the ratio of bacteria-colocalized LC3 puncta to total LC3 puncta is significantly increased with longer treatment time (4 h or 6 h) (SFigure 4), suggesting the internalized bacteria are likely to be eliminated by process related with PepO-induced LC3 puncta formation. Thus, we assume that PepO-induced autophagy contributes to the elimination of bacteria by macrophage. For further confirming, we inhibited macrophage autophagy by Bafilomycin A1 before PepO treatment and S. aureus infection. As shown in ,e), Bafilomycin A1 significantly decreased PepO-induced phagocytosis and bactericidal activity enhancement. Nitric oxide synthase (NOS) is an enzyme which controls the production of nitro oxide, a bactericidal-related small molecule effector. Consistently, the autophagy inhibition reduced the activity of NOS ()). We noticed that PepO still promoted macrophage bacterial clearance after Bafilomycin A1 blocked autophagy, indicating there is other mechanisms involved in this process.
Figure 2. PepO-induced macrophage autophagy is related to the enhanced phagocytosis and bactericidal activity. (a) The co-localization of S. aureus and LC3 puncta was monitored by immunofluorescent microscopy. The longer PepO treated macrophage, the more co-localization of FITC-labeled S. aureus (green) and LC3 (red) was observed. (b) The phagocytosis index was calculated (n = 100) in three independent experiments. (c) S. aureus co-localized with LC3 puncta was counted (n = 100). (d) The macrophages were treated with Bafilomycin A1 (100 nM) for 1 h before PepO (10 μg/mL) treatment for 6 h, and the phagocytosis against S. aureus was assessed. (e) The bactericidal activity against S. aureus of Bafilomycin A1 and PepO-treated macrophages was assessed. (f) The relative NOS activity of different macrophages treated with PepO (10 μg/mL, 6 h) or with Bafilomycin A1 (100 nM, 1 h) before PepO treatment was assessed. Images were representative of three independent experiments. Data were shown as mean + SEM (n = 3) and are representative of three independent experiments. ns: no significance; *p < 0.05; **p < 0.01; ***p < 0.001. Student’s t-test was employed for statistical analysis
PepO-induced autophagy relies on both TLR2 and TLR4
We reported in our earlier research that PepO-elicited innate immune response is related to both TLR2 and TLR4 [Citation13], thus we confirmed if PepO induces autophagy via TLR2/TLR4. By treating WT or deficient macrophages respectively with PepO, we found that regardless of single or double deficiency of tlr2 or tlr4, the autophagy level is significantly lower than that of WT macrophage (), indicating that PepO-induced autophagy is dependent on both TLR2 and TLR4.
Figure 3. PepO-induced macrophage autophagy is dependent on TLR2 and TLR4. (a) WT, tlr2−/-, tlr4,−/- and tlr2/tlr4−/- macrophage were treated with PepO (10 μg/mL, 6 h) respectively, and the LC3 was monitored by immunofluorescent microscopy. (b) The cells with numerous LC3 puncta of PepO-treated different macrophages were counted (n = 100) in three independent experiments. (c) PepO inhibited the phosphorylation of mTOR and downstream signaling molecule p70S6 K, while inducing the turnover of LC3 I to LC3 II in WT macrophage. (d) PepO did not affect the mTOR signaling and autophagy in tlr2/tlr4−/- macrophage. Images were representative of three independent experiments. (e) PepO (10 μg/mL) induced a 2-phase alteration of mTOR phosphorylation, while AMPK phosphorylation is upregulated at the initial phase and decreased in longer time of treatment. The ULK-1 and LC3 II level is upregulated after PepO treatment. **p < 0.01; ***p < 0.001. Student’s t-test was employed for statistical analysis
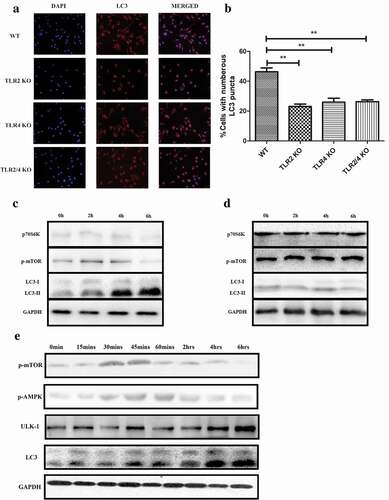
As reported before, PepO manipulates Akt signaling in a TLR2/TLR4-dependent manner [Citation13], thus we confirmed if PepO induces autophagy via mTOR. We treated WT and tlr2/tlr4 double deficient macrophage respectively with PepO for different time, and it is found that PepO suppressed mTOR and its downstream signaling molecule p70SK6 K phosphorylation in WT macrophage ()), while the phosphorylation of these two-signal molecules is unaffected in tlr2/tlr4−/- macrophage ()). Consistent with signaling alteration, PepO failed to induce autophagy in tlr2/tlr4−/- macrophage, indicating that PepO-induced autophagy relies on TLR2 and TLR4. We found that the initiation of autophagy has not occurred at the same time as mTOR dephosphorylation; thus, we assumed that there could be other mechanisms involved in this process. We found that PepO manipulates mTOR phosphorylation in a 2-phase manner ()). In the initial phase of PepO stimulation, PepO upregulates mTOR phosphorylation, however, in the later phase PepO inhibited the phosphorylation of mTOR. Thus, we assume that there may be other mechanisms involved in PepO inducing autophagy. It was reported that ULK-1 is regulated by AMPK, and we observed the upregulation of phosphorylation of AMPK by PepO in the initial phase of PepO stimulation. The time that ULK-1 elevation is consistent with AMPK phosphorylation. Besides, AMPK also regulates mTOR activation, thus we assume that AMPK activation is the key factor in autophagy induction while mTOR is more essential in the later phase.
TLR2 and TLR4 both contribute to PepO-induced macrophage function enhancement
Since PepO induces macrophage autophagy in a TLR2/TLR4 dependent manner, we confirmed the interaction of PepO with TLR2 or TLR4 by co-immunoprecipitation ()). To identify the binding site of TLR2 and TLR4 in PepO, we constructed two truncated mutants of PepO, PepO 1–430, and PepO 431–630 according to the conserved domain prediction (SFigure5), and the Co-IP result showed that both TLR2 and TLR4 bind to PepO in the M13_N domain of PepO ()). The phagocytosis of WT or gene-deficient macrophage was also assessed. tlr2−/- and tlr4−/- macrophage still reacted to PepO stimulation, though higher dose was required to promote macrophage phagocytosis (). However, the tlr2/tlr4 double deficiency abrogated the ability of macrophage responding to PepO completely ()), indicating that TLR2 and TLR4 both contribute to PepO promoting phagocytosis. Next, we identified the role of TLR2 and TLR4 in this process in vivo by gene deficiency mice model. As shown in ), PepO failed to decrease the lung bacteria load in tlr2/tlr4 double deficient mice while PepO-treated wild mice bore significant lower bacteria load than PBS treated wild mice ()), indicating that the enhancement of bacteria clearance induced by PepO is TLR2 and TLR4 dependent.
Figure 4. TLR2 and TLR4 both participate in PepO inducing macrophage function enhancement. (a) The PDMs were incubated with PepO, PepO 1–430, and PepO 431–630, respectively, the cell lysates were subjected to His-Tag IP and the interaction of different proteins with TLR2 and TLR4 were identified by TLR2 or TLR4 immunoblot. GAPDH was used as loading control. (b) PepO enhanced the phagocytosis of tlr2−/- macrophage, though higher dose was required. (c) PepO enhanced the phagocytosis of tlr4−/- macrophage. (d) tlr2/tlr4−/- macrophage did not respond to PepO stimulation. (e) PepO failed to reduce the bacterial load of S. pneumoniae in tlr2/tlr4−/- mice (n = 6 mice/group). (f) The macrophages were treated with Bafilomycin A1 before PepO treatment, and the phagocytosis against S. aureus was assessed. (g) The bactericidal activity against S. aureus of Bafilomycin A1 and PepO-treated macrophages was assessed. (h, i, and j) The relative NOS activity of different macrophages treated with PepO or with Bafilomycin A1 before PepO treatment was assessed. Data are shown as mean + SEM (n = 3) and are representative of three independent experiments. ns: not significant; **p < 0.01. Student’s t-test was employed for statistical analysis
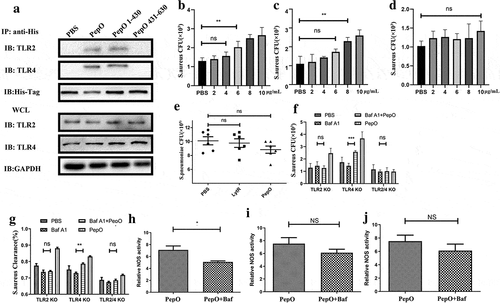
To further elucidate the correlation between TLR2/TLR4 and autophagy, we blocked autophagy with Bafilomycin A1 in gene-deficient macrophage. After inhibition of autophagy, PepO failed to promote phagocytosis in tlr2−/- and tlr2/tlr4−/- macrophage, while tlr4 deficiency did not completely abolish the reaction of macrophage to PepO ()), suggesting that there is other TLR2-involved mechanisms, distinguished from autophagy, regulating PepO-induced phagocytosis, e.g. miR155 mediated phagocytosis enhancement. Consistent with phagocytosis, in autophagy blocked macrophage, PepO enhanced bacteria killing in tlr4−/- macrophage, while tlr2−/- and tlr2/tlr4−/- macrophage did not react to PepO stimulation ()). PepO-induced NOS activity elevation was not compromised by the blockade of in tlr2/tlr4−/- macrophage ()). However, out of expectation, autophagy blockade impaired PepO-induced NOS activation in tlr2−/- macrophage ()) but not in tlr4−/- ()) and tlr2/tlr4−/-macrophage ()). It was reported that TLR4/IFNR2 signaling is involved in NOS production [Citation28]; thus, we assume that the tlr4 deficiency inhibited PepO-induced NOS production, while tlr2−/- macrophages react to the PepO stimulation.
PepO promotes macrophage unspecific phagocytosis and bactericidal activity in a dose-dependent manner
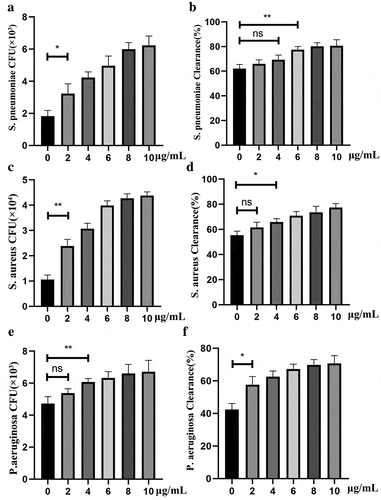
Since PepO-induced macrophage autophagy is an intrinsic process without the contribution of adaptive immunity, we assume that PepO enhanced phagocytosis and bactericidal activity in an unspecific manner. Thus, we invested the reaction of PepO-treated macrophage against different bacteria. When treated with PepO, the phagocytosis of murine peritoneal derived macrophage (PDM) is significantly elevated compared to PBS treated macrophage in a dose-dependent manner ()). Although no significant difference was found in lower dose (2–4 μg/mL) PepO-treated PDM, higher dose PepO (over 6 μg/mL) enhanced the bactericidal activity of PDM ()). Consistent with S. pneumoniae, PepO also promoted macrophage phagocytosis and bacteria killing of S. aureus () and P. aeruginosa (). These results indicated that PepO enhances macrophage phagocytosis and bactericidal activity in a species-independent manner, for it altered the intrinsic response pattern of macrophage.
Figure 5. PepO enhanced unspecific phagocytosis and bactericidal activity of macrophage. Murine primary macrophages were treated with PepO and then infected with S. pneumoniae (MOI = 100), S. aureus (MOI = 100), and P. aeruginosa, respectively (MOI = 50). (a, c and e) PepO enhanced macrophage phagocytosis against S. pneumoniae, S. aureus, and P. aeruginosa, respectively, in a dose-dependent manner. (b, d, and f) PepO enhanced macrophage bactericidal activity against the three species referred above. Data were shown as mean with SEM (n = 3) and are representative of three independent experiments. ns: not significant; *p < 0.05; **p < 0.01. Statistically significant differences were carried out by Student’s t-test
PepO promotes bacteria clearance in pulmonary infection
Pulmonary infection is a major cause of death worldwide, especially in children and people in developing countries [Citation29]. Macrophage is the first immunocyte that participates in bacteria clearance in pulmonary infection [Citation30]. Since PepO enhanced the phagocytosis and bactericidal activity of macrophage, we next investigated if PepO protects against pulmonary bacterial infection. PepO was administrated to C57/BL6 mice intranasally 6 h prior to bacterial infection. To rule out the unspecific effects of microorganism protein, the recombinant S. pneumoniae protein LytR prepared in the same manner was set as control. We first infected the mice with S. pneumoniae D39 strain intranasally. Comparing to PBS or LytR, the bacteria load in the lung of PepO-treated mice significantly decreased at 24-h post-infection ()), indicating that PepO strongly enhances host anti-bacteria defense. Consistently, when infected with another typical gram-negative bacteria, P. aeruginosa, PepO-treated mice bore lower bacteria load comparing to PBS or LytR treated mice ()). In particular, we used a multidrug-resistant clinical isolated P. aeruginosa strain in this experiment, the satisfying result suggested the potential of PepO for MDR bacterial infection treatment.
Figure 6. PepO promotes pulmonary bacterial clearance in C57/BL6 mice. Mice were treated with 10 μg rPepO 6 h before bacterial infection. PBS and LytR were employed as control. (a) PepO significantly reduced the bacteria load of S. pneumoniae D39-infected mice. (b) PepO significantly reduced the bacteria load of an MDR P. aeruginosa-infected mice. (c) In macrophage depleted mice, PepO failed in reducing the bacteria load of D39 infected mice. Data were expressed as mean with SEM (n = 6 mice/group). ns: not significant; *p < 0.05; **p < 0.01; ***p < 0.001. Statistically significant differences were carried out by Student’s t-test
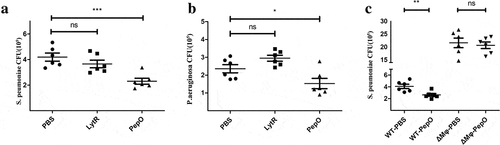
Since PepO significantly enhances macrophage phagocytosis and bactericidal activity in vivo, and macrophage is a key effector cell in the pulmonary tract, we wondered if the main effector cell in this process is macrophage. By eliminating macrophage in C57/BL6 mice with Clodronate Liposomes before PepO treatment [Citation31], we confirmed this hypothesis. Unsurprisingly, the bacteria load in macrophage deficient mice is significantly higher than WT mice. What is more important is that macrophage deficiency completely abrogated the ability of PepO enhancing host defense ()), suggesting that PepO promotes bacteria clearance by regulating macrophage activity.
Discussion/conclusion
Due to the widespread of multidrug resistance bacteria and novel found pathogens, existing antibiotics are already very hard to meet the need of clinical practice. World Health Organization listed antimicrobial resistance as one of the ten threats to global health in 2019 [Citation32]. Thus, new agents to solve this problem is urgently required. However, what concerning is that the development of novel antibiotics is far from satisfying clinicians for this moment. The only two novel classes of antibiotics discovered in the past two decades, lipopeptides and oxazolidinones, cannot cover gram-negative bacteria. Up to 2017, among the 44 novel antibiotic agents under clinical trial, only 15 is effective against gram-negative bacteria, and only 5 derived from existing antibiotics in the 15 had entered phase 3 clinical trial [Citation33]. While the detection rate of gram-negative bacteria is rising horribly fast [Citation34] and gram-positive bacteria causes mass loss [Citation35], it is not wise to put all the odds on the development of novel antimicrobial agents. Thus, we should look for a new strategy to meet the challenge of bacterial infection.
Immunomodulators against bacterial infection are a potential strategy [Citation1,Citation2,Citation12]. By avoiding drug resistance risk, these agents may have advantage in some way comparing to antibiotics. However, most of these agents are small molecules, which development has come to a bottleneck period [Citation36]. In the past decades, the rate of increase of novel small molecule drugs had slowed down, indicating that their development is restrained. The development of small molecular agents relies on the establishment of molecular library and large-scale screening [Citation37], and the relatively low specificity of small molecular agents is a limitation hard to overcome. Benefit from the development of gene engineering, the advantage of protein drugs had shown their advantage. The function and activity of a certain protein are usually known, and its high specificity decreased the risk of uncertain side effect [Citation38]. Moreover, protein drug is relatively economical efficient in production. These advantages made the protein agent a new strategy in drug development. However, there is few protein immunomodulator existing.
In this research, we reported a novel identified TLR2/TLR4 bi-ligand protein, PepO, which promotes host anti-bacterial defense by promoting macrophage function. The species-independent effect of PepO on macrophage provides a broad-spectrum anti-bacterial effect, indicating the therapeutic potential of PepO. In vivo, PepO reduced the bacteria load and protected C57/BL6 mice against two major causes of pulmonary infection, S. pneumoniae and P. aeruginosa, representing gram-positive and gram-negative bacteria, respectively. It is noticeable that the P. aeruginosa strain we used in this research is an MDR strain, and the satisfying results indicated the possibility of PepO as an immunotherapy agent against pulmonary bacterial infection.
TLR4 is a cell surface receptor critical for phagocytosis of macrophage. TLR4 ligand, e.g. LPS and E6020, promotes macrophage phagocytosis by multiple mechanisms [Citation39,Citation40]. ΔA146Ply [Citation41] and GAPDH [Citation42] are also TLR4 interacting proteins; however, they failed in promoting macrophage phagocytosis [Citation14]. Though TLR4 is the first functionally characterized and the best-characterized TLRs member, the function of TLR4 is mostly described in a model of LPS stimulation. TLR4 recognizes and triggers downstream signal transduction requires a complex consist of other three proteins: LPS-binding protein (LBP), CD14, and MD-2, referred as the lipopolysaccharide-multireceptor complex [Citation3]. They act subsequently to treat LPS and participate in endosome generation. Different TLR4 interacting molecules, including exogenous (e.g. LPS and several proteins derived from different viruses [Citation43,Citation44]) and endogenous (e.g. HMGB1 [Citation45], β-defensins [Citation46] and LDL [Citation47]), employ these accessory molecules (all or in part) to induce the signal transduction. TLR4 signals were conducted from both cell membrane and endosomes, mediated by TIRAP/MyD88 and TRAM/TRIF, respectively, and led to NF-κB and IRF3 activation [Citation3]. However, most of these events are described in LPS-stimulation, since different TLR4 ligand interacting with LPS-multireceptor complex differently, it is important to clarify how a certain ligand bind to this complex and triggered downstream signal transduction. As a TLR2/TLR4 bi-ligand, how PepO interacts with these receptors and activates intracellular signaling to promote phagocytosis requires further investigation.
Autophagy has been shown to be essential for innate immunity by degrading internalized microorganism. Accumulating evidence had shown that autophagy contributes to bacterial clearance [Citation48]. Investigations into L. monocytogenes [Citation49,Citation50], M. tuberculosis [Citation51] and S. typhimurium [Citation21,Citation52] highlighted the critical role of autophagy in restricting bacterial intracellular replication and controlling infection. Besides these traditional intracellular bacteria, the activation of autophagy also enhances the internalization and elimination of extracellular bacteria, including S. aureus [Citation53], P. aeruginosa [Citation54,Citation55] and E. coli [Citation50]. In macrophages, xenophagy is the most detailed described autophagy process associated with bacteria clearance, targeting internalized microbes in macroautophagy pathway [Citation17,Citation56,Citation57]. Recently, researchers found that a noncanonical autophagy process named LC3-associated phagocytosis (LAP) also contributes to the clearance of bacteria by macrophage [Citation17,Citation56,Citation58]. Though there is still overlap in the molecular mechanism with macroautophagy, LAP is characterized by some special features, including the difference in membrane structure, the requirement for ROS and the formation time of LC3-associated vesicles [Citation59,Citation60]. LC3 on the LAPosome can be detected as early as 10 min and it usually reaches a peak point at 1 h, while macrophage autophagosome requires several hours to form. We found in our results that the co-localization of LC3 puncta and bacteria reaches a peak point at 4 h (SFigure 4), which is consistent with canonical macroautophagy rather than LAP. In addition, since the recombinant PepO is sufficient to induce macrophage autophagy, it is more likely to induce nonspecific autophagy which is beneficial for the bacteria clearance rather than selective bacteria-specific one, but further investigation is required for confirmation.
By contributing in antigen processing and presentation of antigen-presenting cells including macrophages and dendritic cells, autophagy linked innate immunity to adaptive immunity [Citation61]. Therefore, autophagy is redeemed as a potential target for anti-infection treatment. Several studies had demonstrated that manipulating autophagy could enhance host anti-microorganism defense and treat the infection. Levine et al. identified an autophagy inducing peptide, Tat-beclin 1, which promotes the clearance of multiple pathogens including bacteria and viruses [Citation62]. Carbamazepine has been reported to protect against M. tuberculosis infection by enhancing autophagic killing [Citation63]. On the other hand, researches have revealed that autophagy prevents over-inflammation, which is the biggest obstacle of TLR agonists application [Citation64]. Autophagic elimination of damaged mitochondria caused by invading bacteria limits the inflammation injury in vivo and in vitro [Citation65], and the deficiency in atg7 results in the dysregulation of IL-1β and pyroptosis [Citation66]. As an autophagy inductive protein, PepO may be more safely in use than the existing agents, but further investigation is required for confirming the advantage in the safety of PepO.
In conclusion, we identified PepO as an immunomodulator that interacts with both TLR2 and TLR4. By initiating autophagy, PepO promotes macrophage phagocytosis and bactericidal activity, leading to enhanced pulmonary bacteria clearance and anti-bacterial immunity. In this study, we provided evidence for PepO’s or TLR agonists’ therapeutic potential and a new strategy for anti-bacterial infection treatment.
Author contributions
Z.S., Y.Y., and X.Z. conceived and designed the study; Z.S., J.Y., J.Z., and S.L. conducted the experiment; J.Y., H.W., and Y.L. provided important insights in experimental design and performing; Z.S., H.Z., and Y.L. analyzed the data; Z.S. and H.Z. wrote the paper; Y.Y. and X.Z. revised the paper.
Statement of ethics
All experiments related to animal in this study were approved by the Animal Ethics Committee of Chongqing Medical University.
Supplemental Material
Download MS Word (2 MB)Disclosure statement
The authors have no conflicts of interest to declare.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Schaad UB. OM-85 BV, an immunostimulant in pediatric recurrent respiratory tract infections: a systematic review. World J Pediatr. 2010;6(1):5–12.
- Steurer-Stey C, Bachmann LM, Steurer J, et al. Oral purified bacterial extracts in chronic bronchitis and COPD: systematic review. Chest. 2004;126(5):1645–1655.
- Brubaker SW, Bonham KS, Zanoni I, et al. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290.
- Moresco EM, LaVine D, Beutler B. Toll-like receptors. Curr Biol. 2011;21(13):R488–93.
- Hofmann U, Ertl G, Frantz S. Toll-like receptors as potential therapeutic targets in cardiac dysfunction. Expert Opin Ther Targets. 2011;15(6):753–765.
- Erard F, Ryffel B. Toll like receptor - potential drug targets in infectious disease. Infect Disord Drug Targets. 2008;8(4):221–231.
- Feng Y, Mu R, Wang Z, et al. A toll-like receptor agonist mimicking microbial signal to generate tumor-suppressive macrophages. Nat Commun. 2019;10(1):2272.
- Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3(2):196–200.
- Abdul-Cader MS, De Silva Senapathi U, Nagy E, et al. Antiviral response elicited against avian influenza virus infection following activation of toll-like receptor (TLR)7 signaling pathway is attributable to interleukin (IL)-1beta production. BMC Res Notes. 2018;11(1):859.
- Cluff CW, Baldridge JR, Stover AG, et al. Synthetic toll-like receptor 4 agonists stimulate innate resistance to infectious challenge. Infect Immun. 2005;73(5):3044–3052.
- Reppe K, Radunzel P, Dietert K, et al. Pulmonary immunostimulation with MALP-2 in influenza virus-infected mice increases survival after pneumococcal superinfection. Infect Immun. 2015;83(12):4617–4629.
- Kerber-Momot T, Leemhuis D, Luhrmann A, et al. Beneficial effects of TLR-2/6 ligation in pulmonary bacterial infection and immunization with Pseudomonas aeruginosa. Inflammation. 2010;33(1):58–64.
- Zhang H, Kang L, Yao H, et al. Streptococcus pneumoniae endopeptidase O (PepO) elicits a strong innate immune response in mice via TLR2 and TLR4 signaling pathways. Front Cell Infect Microbiol. 2016;6:23.
- Yao H, Zhang H, Lan K, et al. Purified streptococcus pneumoniae endopeptidase O (PepO) enhances particle uptake by macrophages in a Toll-like receptor 2- and miR-155-dependent manner. Infect Immun. 2017;85:4.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335.
- Bah A, Vergne I. Macrophage autophagy and bacterial infections. Front Immunol. 2017;8:1483.
- Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. 2015;264(1):182–203.
- Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253–1257.
- Delgado MA, Elmaoued RA, Davis AS, et al. Toll-like receptors control autophagy. Embo J. 2008;27(7):1110–1121.
- Liu W, Zhuang J, Jiang Y, et al. Toll-like receptor signalling cross-activates the autophagic pathway to restrict Salmonella Typhimurium growth in macrophages. Cell Microbiol. 2019;21(12):e13095.
- Li X, He S, Zhou X, et al. Lyn Delivers Bacteria to Lysosomes for Eradication through TLR2-Initiated Autophagy Related Phagocytosis. PLoS Pathog. 2016;12(1):e1005363.
- Tao L, Cao F, Xu G, et al. Mogroside IIIE attenuates LPS-induced acute lung injury in mice partly through regulation of the TLR4/MAPK/NF-kappaB axis via AMPK activation. Phytother Res. 2017;31(7):1097–1106.
- Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008;283(48):33175–33182.
- Chuang SY, Yang CH, Chou CC, et al. TLR-induced PAI-2 expression suppresses IL-1beta processing via increasing autophagy and NLRP3 degradation. Proc Natl Acad Sci U S A. 2013;110(40):16079–16084.
- Ye W, Zhang J, Shu Z, et al. Pneumococcal lytr protein is required for the surface attachment of both capsular polysaccharide and teichoic acids: essential for pneumococcal virulence. Front Microbiol. 2018;9:1199.
- Wu K, Zhang X, Shi J, et al. Immunization with a combination of three pneumococcal proteins confers additive and broad protection against streptococcus pneumoniae infections in Mice. Infect Immun. 2010;78(3):1276–1283.
- Lamrani M, Sassi N, Paul C, et al. TLR4/IFNgamma pathways induce tumor regression via NOS II-dependent NO and ROS production in murine breast cancer models. Oncoimmunology. 2016;5(5):e1123369.
- Everard ML. Paediatric respiratory infections. Eur Respir Rev. 2016;25(139):36–40.
- Byrne AJ, Mathie SA, Gregory LG, et al. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 2015;70(12):1189–1196.
- Nakamura T, Abu-Dahab R, Menger MD. et al. Depletion of alveolar macrophages by clodronate-liposomes aggravates ischemia-reperfusion injury of the lung. J Heart Lung Transplant. 2005;24(1):38–45.
- Organization WH. Ten threats to global health in 2019 2019. Available from: https://www.who.int/emergencies/ten-threats-to-global-health-in-2019.
- Tacconelli E, Carrara E, Savoldi A, et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18(3):318–327.
- Vasoo S, Barreto JN, Tosh PK. Emerging issues in gram-negative bacterial resistance: an update for the practicing clinician. Mayo Clin Proc. 2015;90(3):395–403.
- Watkins RR, Holubar M, David MZ. Antimicrobial resistance in methicillin-resistant Staphylococcus aureus to newer antimicrobial agents. Antimicrobial agents and chemotherapy. 2019;63(12):e01216–19.
- Tuffery P. Accessing external innovation in drug discovery and development. Expert Opin Drug Discov. 2015;10(6):579–589.
- Wu B, Zhang Z, Noberini R, et al. HTS by NMR of combinatorial libraries: a fragment-based approach to ligand discovery. Chem Biol. 2013;20(1):19–33.
- Dimitrov DS. Therapeutic proteins. Methods Mol Biol. 2012;899:1–26.
- Boivin A, Pineau I, Barrette B, et al. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J Neurosci. 2007;27(46):12565–12576.
- Church JS, Milich LM, Lerch JK, et al. E6020, a synthetic TLR4 agonist, accelerates myelin debris clearance, Schwann cell infiltration, and remyelination in the rat spinal cord. Glia. 2017;65(6):883–899.
- Su Y, Li D, Xing Y, et al. Subcutaneous immunization with fusion protein DnaJ-DeltaA146Ply without additional adjuvants induces both humoral and cellular immunity against pneumococcal infection partially depending on TLR4. Front Immunol. 2017;8:686.
- Sun X, Wang J, Zhou J, et al. Subcutaneous immunization with Streptococcus pneumoniae GAPDH confers effective protection in mice via TLR2 and TLR4. Mol Immunol. 2017;83:1–12.
- Kurt-Jones EA, Popova L, Kwinn L, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1(5):398–401.
- Rassa JC, Meyers JL, Zhang Y, et al. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc Natl Acad Sci U S A. 2002;99(4):2281–2286.
- Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201(7):1135–1143.
- Biragyn A, Ruffini PA, Leifer CA, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298(5595):1025–1029.
- Miller YI, Viriyakosol S, Worrall DS, et al. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. 2005;25(6):1213–1219.
- Deretic V, Kimura T, Timmins G, et al. Immunologic manifestations of autophagy. J Clin Invest. 2015;125(1):75–84.
- Mitchell G, Cheng MI, Chen C, et al. Listeria monocytogenes triggers noncanonical autophagy upon phagocytosis, but avoids subsequent growth-restricting xenophagy. Proc Natl Acad Sci U S A. 2018;115(2):E210–e7.
- Sorbara MT, Foerster EG, Tsalikis J, et al. Complement C3 Drives Autophagy-Dependent Restriction of Cyto-invasive Bacteria. Cell Host Microbe. 2018;23(5):644–52.e5.
- Franco LH, Nair VR, Scharn CR, et al. The ubiquitin ligase Smurf1 functions in selective autophagy of mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe. 2017;22(3):421–423.
- Liu W, Jiang Y, Sun J, et al. Activation of TGF-beta-activated kinase 1 (TAK1) restricts Salmonella Typhimurium growth by inducing AMPK activation and autophagy. Cell Death Dis. 2018;9(5):570.
- Maurer K, Torres VJ, Cadwell K. Autophagy is a key tolerance mechanism during Staphylococcus aureus infection. Autophagy. 2015;11(7):1184–1186.
- Krokowski S, Lobato-Marquez D, Chastanet A, et al. Septins recognize and entrap dividing bacterial cells for delivery to lysosomes. Cell Host Microbe. 2018;24(6):866–74.e4.
- Liu C, Qi J, Shan B, et al. Pretreatment with cathelicidin-BF ameliorates Pseudomonas aeruginosa pneumonia in mice by enhancing NETosis and the autophagy of recruited neutrophils and macrophages. Int Immunopharmacol. 2018;65:382–391.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222.
- Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev. 2011;240(1):92–104.
- Martinez J, Almendinger J, Oberst A, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108(42):17396–17401.
- Wong SW, Sil P, Rubicon: MJ. LC3-associated phagocytosis and beyond. Febs J. 2018;285(8):1379–1388.
- Heckmann BL, Boada-Romero E, Cunha LD, et al. LC3-Associated Phagocytosis and Inflammation. J Mol Biol. 2017;429(23):3561–3576.
- Valecka J, Almeida CR, Su B, et al. Autophagy and MHC-restricted antigen presentation. Mol Immunol. 2018;99:163–170.
- Shoji-Kawata S, Sumpter R, Leveno M, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206.
- Schiebler M, Brown K, Hegyi K, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med. 2015;7(2):127–139.
- Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14(2):243–251.
- Jabir MS, Hopkins L, Ritchie ND, et al. Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy. 2015;11(1):166–182.
- Pu Q, Gan C, Li R, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in pseudomonas sepsis. J Immunol. 2017;198(8):3205–3213.