
Abstract
Objectives
This retrospective cohort study aims to provide a comprehensive account of death in Swedish patients with ALS, including clinical status preceding death, the death setting, as well as symptoms.
Methods
The study presents detailed information on a cohort of patients with ALS from Stockholm, Sweden, deceased in 2018–2020. In addition, selected information is presented on a larger complementary cohort of ALS patients from all regions of Sweden deceased in 2011–2020. Data were obtained from patient medical records, the Swedish Motor Neuron Disease Quality Registry, and the Swedish Quality Registry of Palliative Care.
Results
Ninety-three patients were included in the main cohort and 2224 patients in the complementary cohort. In the main cohort, there was a slow decline in weight and motor function during the 12 months preceding death. Most (93.4%) anticipated/prolonged deaths occurred in a palliative care unit, at home, or in an assisted living facility while 44.8% of precipitous deaths occurred in a hospital ward. Next of kin or health care staff were present at death for most patients (78.7%). In the final week of life, 41.1% experienced at least one symptom (either pain, anxiety, confusion, or dyspnea) that was only partially relieved or not at all.
Conclusion
The majority of patients died in their own homes or at a palliative unit in the presence of next of kin and most symptoms were adequately managed. This paper might be used in educating patients, next of kin as well as health professionals, decreasing uncertainty surrounding the end of life.
Keywords:
Introduction
Although the clinical course in Amyotrophic Lateral Sclerosis (ALS) is highly variable, the disease usually generalizes to widespread motor dysfunction eventually resulting in feeding difficulties, respiratory failure, and death (Citation1). There is currently no effective treatment for ALS and the expected survival from onset is short, ∼2–4 years (Citation2). Given the rapid progression and fatal outcome of the disease, a major preoccupation among patients is the circumstances surrounding the end of life (Citation3). Depression is common in the first year after diagnosis (Citation4), and up to half of the patient population consider hastening their death (Citation5,Citation6). Much of the patient's anxiety is driven by uncertainly about what to expect and many fear a distressing death. Common symptoms at the end of life are dyspnea, anxiety, pain, insomnia, and restlessness (Citation7–9), but research shows that many patients die peacefully (Citation7,Citation8). The majority of patients with ALS die in their own home or at a palliative unit (Citation7–9), which has been associated with increased well-being (Citation10–12), but a substantial proportion of patients [one in four in one study (Citation13)], more often male and/or unmarried, die in an acute care facility. The cause of death most frequently reported in medical records is respiratory failure (Citation7,Citation14), and at autopsy: aspiration/pneumonia, and heart failure (Citation15,Citation16).
The details surrounding death in patients with ALS differ greatly between countries and are poorly delineated for Swedish patients. This paper aims to provide a comprehensive account of various important aspects of death in a cohort of ALS patients from Stockholm, Sweden, including clinical status preceding death, the death setting (place of death, persons present at the death, etc.) as well as symptoms at the end of life.
Methods
Study population
We performed a retrospective study with detailed information in a cohort of 93 patients as well as information on selected variables of interest in a large complementary cohort (n = 2224). All patients had a diagnosis of probable, possible, or definite ALS, according to the El Escorial criteria (Citation17).
The main cohort includes 93 patients diagnosed in 2016 and forward at the ALS Clinical Research Center (ALS CRC) at the Karolinska University Hospital in Stockholm, Sweden, and deceased in 2018–2020. The ALS CRC provides multidisciplinary ALS care and is the only tertiary center for ALS patients in the Stockholm region with a population of ∼2 million inhabitants. The number of patients registered in the Swedish Motor Neuron Disease Quality Registry (SMNDR) fulfilling the inclusion criteria was 147. However, the main cohort only includes patients registered in both the SMNDR and the Swedish Quality Registry of Palliative Care (SRPC). Thus, since the SMNDR has almost complete coverage of Stockholm ALS patients, the coverage of this study is estimated to be 63% (93/147). A comparison of key characteristics in the main cohort and those only registered in the SMNDR showed no significant differences (Supplementary Table 1), and the main cohort was therefore considered representative of the Stockholm ALS population.
Table 1 Patient characteristics of the main cohort.
To explore the representativeness of the findings to a larger population, as well as factors associated with symptoms at the end of life, a larger group of patients with ALS (n = 2224) that died during 2011–2020 in any region of Sweden were also included. Results from this cohort are only described in the section on symptoms at the end of life. Data on these patients were obtained only from the SRPC and were thus limited to a few variables: sex, age, symptoms at the end of life, place of death, and persons present at death. The distribution of these variables was compared to that of the main cohort and revealed significant differences only for the place of death (Supplementary Table 2).
Table 2 Cause of death, time to death, and persons present at death in ALS patients.
The study was approved by the ethics review board in Stockholm, Sweden (DNRs 2017/1895-31/1 and 2022-00306-02), and information was collected on an opt-out basis.
Data
Three data sources were used: the SMNDR, the SRPC, and patient medical records. The SMNDR was established in 2015 and recruits Swedish motor neuron disease (MND) patients at the time of diagnosis with prospective collection of information on clinical measures and quality of life outcomes. As of 2017, this registry had 99% coverage of all MND patients in Stockholm (Citation18). The SRPC contributes to research and development of palliative care in Sweden since 2005. The registry is based on a 30-item end-of-life questionnaire that covers the last week of life and is completed by the responsible staff after the death of a patient. The registry primarily includes anticipated deaths and was estimated to cover about 60% of all Swedish deaths in 2020 (Citation19). A large portion of the Swedish ALS patients is connected to palliative care services as the disease progresses (to assist in supportive care) and are thus included in the SRPC.
For the main cohort, all baseline variables had complete information, except for occupation (missing data in nine patients). For place of death and cause of death, there was missing data in three patients and in four patients for the variable persons present at death. For symptoms at the end of life, there was missing data in 14–17 patients depending on the specific symptom. For the large complementary cohort, data was complete except for persons present at death (missing data in 28 patients) and symptoms at the end of life (missing data in 233–428 patients depending on the specific symptom).
Measures and definitions
Comorbidity
Several comorbid conditions present at the time of ALS diagnosis were reported based on clinical relevance. Some conditions were reported separately, i.e. hypertension, diabetes mellitus, chronic obstructive pulmonary disease (COPD), and osteoarthritis. Others were grouped because of low prevalence or similar clinical characteristics, i.e. depression/anxiety, ischemic heart disease/heart failure. Patients were classified as multimorbid if having three or more documented conditions.
Clinical status
Functional impairment was assessed by the revised ALS Functional Rating Scale (ALSFRS-R) (Citation20), and nutritional status by body mass index (BMI).
Clinical phenotype at onset
Onset phenotype was defined based on the predominant symptom at the time of presentation: spinal (lower extremity or upper extremity) or bulbar onset.
Precipitous vs. anticipated death
A precipitous death was defined as an acute event (e.g. infection, sudden cardiac arrest, etc.) or a rapid (a few days) and unexpected clinical worsening resulting in death, as opposed to death as the culmination of a slow decline (the anticipated clinical course of ALS).
Symptoms at the end of life
Defined as symptoms reported in the week preceding death.
Statistical analyses
Categorical variables were summarized as proportions (percent) and bivariate analyses were performed using the χ2 test to assess for statistical significance of differences between groups. Continuous variables were reported as the median with interquartile range (IQR), or the 25th–75th percentile (in the event of skewed distribution), and the student’s t-test was used to assess for differences between groups. The statistical significance level was set to p < 0.05.
Patients with missing values were dropped from the analyses except for ALSFRS-R and BMI. Patients were scheduled for follow-up and evaluation of ALSFRS-R and BMI status every three months. Missing values for a specific month were extrapolated from values available at previous and later time points, e.g. 18 − ? − 20 → 18 − 19 − 20. However, when there was no neighboring value, missing data were left blank. The mean value from all registrations in each consecutive 3-month period (1–3, 4–6, 7–9, and 10–12) was first calculated individually for each patient (there were frequently multiple registrations during each period). We then reported the median values for the group.
Results
Results are primarily presented from the main cohort. Results from the complementary cohort are only presented in the section on symptoms at the end of life. In the sections describing cohort characteristics and clinical status preceding death, patients were grouped based on the clinical phenotype at onset: bulbar (n = 49, 52.7%) or spinal (n = 44, 47.3%). However, at the time of death, the disease tended to have generalized to involve the bulbar area as well as the extremities, making this grouping less relevant. Thus, for the sections describing the place of death, persons present at death, cause of death, and symptoms at the end of life, patients were instead grouped based on whether their death was anticipated (61 of 90 patients [67.8%]) or precipitous (29 of 90 patients [32.2%]).
Cohort characteristics
Overall, sex distribution was essentially equal, but male patients predominated in the bulbar onset group (57.1%) (). Most patients were retired (75.3%) and a third (32.3%) were reported to have multimorbidity. The median age at death was 72 (66–76, 25th–75th percentile). Differences in characteristics between bulbar and spinal onset patients were non-significant apart from the prevalence of inactive malignancy.
Clinical status preceding death (the last 12 months)
shows a steady decline in ALSFRS-R during the 12 months preceding death. In both the bulbar and spinal onset groups, the median score was ∼20 in the last three months of life, but bulbar onset patients tended to have lower scores throughout the preceding 12 months (starting out at a median of 28 vs. 33 in the spinal onset group). The figures also show the gradual increase of patients on regular non-invasive ventilation (NIV), reaching about 50% at the time of death in both groups.
Figure 1 (A–D) ALSFRS-R and BMI 12 months preceding death in ALS. The box plots display the longitudinal decline in ALSFRS-R (A,B) and BMI (C,D) per three months during the 12 months preceding death in ALS patients with bulbar (A,C) vs. spinal (B,D) onset. The right axis shows the percentage of patients on non-invasive ventilation (NIV) or with gastrostomy as indicated by the red line.
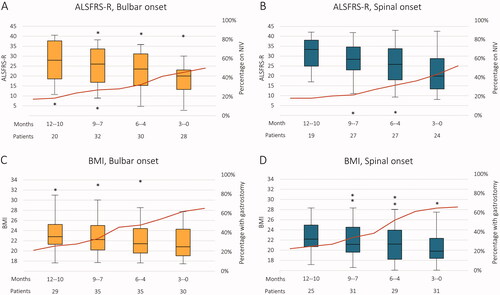
Also, during the 12 months preceding death, patients steadily lost weight as shown in . However, the change was relatively modest at a group level: from a median BMI slightly above 22 to ∼20 in 12 months. There was little difference between groups, although patients with bulbar onset had a slightly higher BMI overall. The percentage of patients with gastrostomy increased to ∼65% at the time of death in both groups.
Place of death
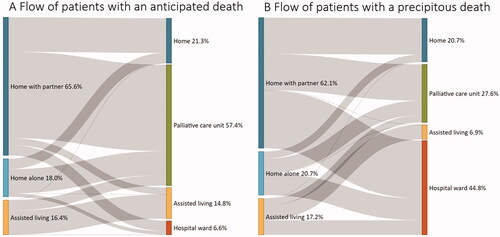
As for the permanent place of residency, most patients (58 [64.4%]) were living at home with a partner or another relative, 17 patients (18.9%) were living at home alone, whereas 15 patients (16.7%) were in assisted living. However, in the immediate period preceding death, there was a substantial flow of patients from their permanent place of residency to other locations (their place of death) which is illustrated in . In the anticipated death group, most patients, 57.4%, died in a palliative care unit which compares to only 27.6% of patients with a precipitous death. Conversely, death in a hospital ward was more common among patients with a precipitous death compared to those with an anticipated death: 44.8% vs. 6.6% (18.9% in total). However, the proportion of deaths in their own home was similar between the two groups: 21.3% in those with an anticipated death and 20.7% in those with a precipitous death.
Figure 2 (A,B) Flow of ALS patients from their permanent place of residency to the place of death. Shown separately for patients with an anticipated (A) vs. precipitous (B) death.
Persons present at death
Next of kin were present at death for most patients: 51 (57.3%) (). In 19 patients (21.3%) only health care staff were present. Nineteen patients (21.3%) died alone, and this proportion was larger for those with a precipitous death: 32.1% compared to 16.4% among those with an anticipated death.
Cause of death
In the anticipated death group, 32 patients (52.5%) died of respiratory failure and 29 patients (47.5%) died of multiorgan failure whereas in the precipitous death group, over a third of patients (11 [37.9%]) died of unknown causes and the majority (16 [55.2%]) died of respiratory failure (). Out of all deaths from respiratory failure, 11 out of 48 cases were associated with pneumonia. In those with a precipitous death, this ratio was seven out of 16 cases.
Time to death
Overall, the median time from disease onset to death was 28 (IQR 17) months: 32 months (IQR 23) for the anticipated death group and 24 months (IQR 18) for the precipitous death group, although this difference was not statistically significant (). A “no resuscitation order” was placed in 85 patients (94.4%), at a median of 3.4 months (IQR 6.7) before death.
Symptoms at the end of life
Many patients suffered from various symptoms in the week before death. For most patients, these could be managed effectively but in a substantial proportion, the symptoms were only partially relieved or not at all (). For those with an anticipated death (in the main cohort), this proportion was 28.6% for anxiety, 29.8% for dyspnea, 6.9% for pain, 22.4% for excessive bronchial secretions, and 8.8% for confusion. There was a trend toward less symptoms in patients with a precipitous death, but the only statistically significant difference was seen for the number of patients experiencing pain. In total, 41.1% of the main cohort patients experienced at least one symptom (either pain, anxiety, confusion, or dyspnea) that was only partially relieved or not at all.
Figure 3 (A–C) Symptoms at the end of life in ALS patients. End of life is defined as the week preceding death. Shown separately for patients of the main cohort with an anticipated (A) vs. precipitous (B) death, as well as for patients from a large complementary cohort with either an anticipated or a precipitous death (unknown status) (C). EBS: excessive bronchial secretions.
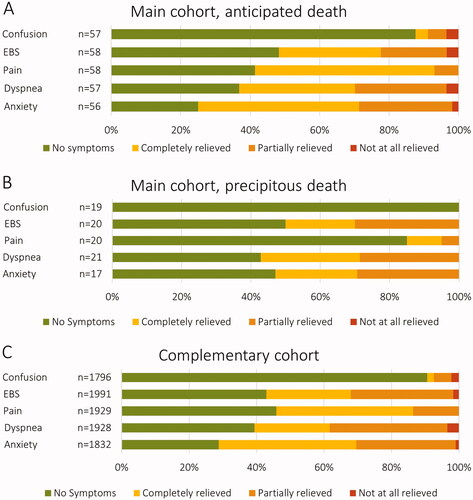
To further explore the extent of symptoms at the end of life and associated factors, these were also examined in a large complementary cohort (n = 2224). The overall distribution of symptoms was similar as compared to the main cohort (). When comparing patients with symptoms (either dyspnea, anxiety, pain, or confusion) that were only partially relieved or not at all (n = 785 [49.3%]) to those with no symptoms or symptoms that were completely relieved (n = 807 [50.7%]), there were significant differences for sex and place of death (). The high symptom burden group had a larger proportion of males (54.0% vs. 48.4%) and a larger proportion dying in a hospital ward (26.6% vs. 17.0%). There were no statistically significant differences for persons present at death.
Table 3 Comparison of key variables in patients with low vs. high symptom burden in a complementary cohort of 2224 patients with ALS.
Since the study period coincided with the outbreak of the COVID-19 pandemic we compared the place of death, cause of death, and persons present at death in the years 2018, 2019, and 2020 (Supplementary Table 3). There were no statistically significant differences.
Discussion
In ALS, a major preoccupation for both patients and next of kin early on is the circumstances surrounding the end of life (Citation3), but previous research shows that many patients die peacefully (Citation7,Citation8). Indeed, in our main cohort, for each of the specific symptoms reported in the last week in life, over 70% of patients either had no symptoms or symptoms that were completely relieved. The results were confirmed in a larger complementary cohort, where these proportions were well over 60%. There was a trend toward more symptoms in those with a prolonged death, but statistically significant only for the number of patients experiencing pain. The lower symptom burden in patients with a precipitous death might be explained by them being in a better general clinical condition in the week preceding death (which is the period when symptoms were reported in this study).
Most patients died in their own homes or in a palliative care unit in the presence of next of kin. The proportion of deaths in a hospital ward, which is preferably avoided, was 18.9% in total in our main cohort (44.8% of those with a precipitous death and 6.6% of those with a prolonged death), compared to 39% in the general population dying of any cause (Citation19). There was a greater symptom burden in those dying in a hospital ward. However, an increase in symptoms might be the reason for admission and not the result of differences in treatment.
Although the majority of patients in our study did not seem to suffer from many distressing symptoms, a large proportion (41.1% in the main cohort and 49.7% in the complementary cohort) experienced at least one symptom (either pain, anxiety, confusion, or dyspnea) that was only partially relieved or not at all. Moreover, a substantial proportion (21.3% in the main cohort and 13.1% in the complementary cohort) died alone. This might indicate an unmet need for support. Health care professionals have an important role in supporting patients at all stages of the disease, and the multidisciplinary team has been shown to extend survival and improve quality of life (Citation21). Eventually, terminal care in a palliative care unit or at home (with health care support) is recommended as it has been shown to improve quality of life (Citation10–12). An essential part of the support is educating patients and next of kin on the expected disease course and since the end of life is a major preoccupation for many, this topic must be addressed early to decrease anxiety and uncertainty. Importantly, clinical worsening might happen unexpectedly, further empathizing the importance of an early discussion of end-of-life expectations and wishes.
Finally, it is important to note that death is a very subjective experience that is not well-captured by quantitative measures at a group level, which may not truly reflect the experiences of patients and next of kin. Thus, using our data to make any statement regarding the “quality of death” for the individual patient is problematic.
Strengths and limitations
This study is a comprehensive description of death in ALS, including rich information on patient characteristics, longitudinal clinical data as well as details on the death setting and symptoms at the end of life. Although limited by a small cohort size (as frequently occurs in rare diseases such as ALS), a major strength is the inclusion of a complementary cohort to verify the results in a much larger sample.
There are several important limitations regarding the data on symptoms. (1) Symptom burden was reported by health care professionals and not the patients themselves or their next of kin. (2) The SRPC does not specify whether the symptoms persisted despite maximal treatment or if they were inadequately treated. (3) Variations in reported symptom burden might be the result of varying oversight/documentation by health care providers in different settings.
Generalizability
The description of the main cohort of 93 patients was based on data from two high-quality registers: the SMNDR and the SRPC, and the coverage was estimated to be 63%. Since the SRPC mostly includes anticipated deaths, the number of precipitous deaths was probably underestimated. However, a comparison of key characteristics in the main cohort versus those only registered in the SMNDR showed no significant differences (Supplementary Table 1). This indicates that our study population and findings are representative of the Stockholm region. Also, we performed a comparison of the main cohort and the large complementary cohort of ALS patients from all regions of Sweden (Supplementary Table 2). This analysis showed significant differences only for the place of death, which would suggest that the main cohort is also relatively representative of Sweden as a whole. However, the differing years of inclusion (2018–2020 vs. 2011–2020) and a small set of variables makes it difficult to draw any conclusions. Whether the results are generalizable to countries other than Sweden remains to be determined.
Selection bias
Only patients diagnosed in 2016 forward were included in the SMNDR and thus in the main cohort. Therefore, some patients with long survival might have been excluded. Previous research has shown that the median time from onset to diagnosis is ∼1 year (Citation22,Citation23). To be included in our study, this effectively limits the longest survival time to about 3 years from diagnosis for patients deceased in early 2018. This should only affect a small number of individuals, as survival in ALS patients is 2–4 years on average from onset (Citation2) and the annual number of deaths was relatively evenly distributed in 2018–2020. However, this might have skewed the cohort to more rapid progressors, explaining the higher-than-expected proportion of bulbar patients [who are more likely to have a rapid disease progression (Citation2)].
Conclusions
Most patients experienced few distressing symptoms in the week preceding death, although, in a substantial proportion, symptoms were not adequately managed. This emphasizes the importance of providing support at all stages of ALS and particularly at the end of life. An essential component is preparing the patients and next of kin for what death might be like and what to expect, preferably early in the disease course, and we hope that this paper might prove helpful in this communication. Also, we would like to encourage more education of health professionals in addressing these topics and more research exploring various aspects of end of life in ALS.
Supplemental Material
Download MS Word (21.7 KB)Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Data availability statement
The study is based on data from the Swedish Motor Neuron Disease Quality Registry and the Swedish Quality Registry of Palliative Care. Having obtained the appropriate approval from a research ethics board, these data can be accessed from the registry offices upon request. Data obtained from medical records can be accessed from the principal author upon request after obtaining the appropriate ethics board approval.
Additional information
Funding
References
- Brown RH Jr., Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:1602.
- del Aguila MA, Longstreth WT Jr., McGuire V, Koepsell TD, van Belle G. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60:813–9.
- Averill AJ, Kasarskis EJ, Segerstrom SC. Psychological health in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2007;8:243–54.
- Roos E, Mariosa D, Ingre C, Lundholm C, Wirdefeldt K, Roos PM, et al. Depression in amyotrophic lateral sclerosis. Neurology. 2016;86:2271–7.
- Ganzini L, Johnston WS, McFarland BH, Tolle SW, Lee MA. Attitudes of patients with amyotrophic lateral sclerosis and their care givers toward assisted suicide. N Engl J Med. 1998;339:967–73.
- Stutzki R, Weber M, Reiter-Theil S, Simmen U, Borasio GD, Jox RJ. Attitudes towards hastened death in ALS: a prospective study of patients and family caregivers. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:68–76.
- Neudert C, Oliver D, Wasner M, Borasio GD. The course of the terminal phase in patients with amyotrophic lateral sclerosis. J Neurol. 2001;248:612–6.
- Mandler RN, Anderson FA, Miller RG, Clawson L, Cudkowicz M, Del Bene M, et al. The ALS Patient Care Database: insights into end-of-life care in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2:203–8.
- Ganzini L, Johnston WS, Silveira MJ. The final month of life in patients with ALS. Neurology. 2002;59:428–31.
- Bede P, Oliver D, Stodart J, van den Berg L, Simmons Z, O Brannagáin D, et al. Palliative care in amyotrophic lateral sclerosis: a review of current international guidelines and initiatives. J Neurol Neurosurg Psychiatry. 2011;82:413–8.
- Teno JM, Clarridge BR, Casey V, et al. Family perspectives on end-of-life care at the last place of care. JAMA. 2004;291:88–93.
- Oliver D. The quality of care and symptom control–the effects on the terminal phase of ALS/MND. J Neurol Sci. 1996;139 Suppl:134–6.
- Goutman SA, Nowacek DG, Burke JF, Kerber KA, Skolarus LE, Callaghan BC. Minorities, men, and unmarried amyotrophic lateral sclerosis patients are more likely to die in an acute care facility. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:440–3.
- Gil J, Funalot B, Verschueren A, Danel-Brunaud V, Camu W, Vandenberghe N, et al. Causes of death amongst French patients with amyotrophic lateral sclerosis: a prospective study. Eur J Neurol. 2008;15:1245–51.
- Kurian KM, Forbes RB, Colville S, Swingler RJ. Cause of death and clinical grading criteria in a cohort of amyotrophic lateral sclerosis cases undergoing autopsy from the Scottish Motor Neurone Disease Register. J Neurol Neurosurg Psychiatry. 2009;80:84–7.
- Corcia P, Pradat P-F, Salachas F, Bruneteau G, Forestier N, Seilhean D, et al. Causes of death in a post-mortem series of ALS patients. Amyotroph Lateral Scler. 2008;9:59–62.
- Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9.
- Longinetti E, Regodón Wallin A, Samuelsson K, Press R, Zachau A, Ronnevi L-O, et al. The Swedish motor neuron disease quality registry. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:528–37.
- Swedish registry of palliative care. Year report of 2020. 2021. Available at: www.Palliativregistret.se.
- Cedarbaum JM, Stambler N. Performance of the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) in multicenter clinical trials. J Neurol Sci. 1997;152 Suppl 1: S1–S9.
- Hogden A, Foley G, Henderson RD, James N, Aoun SM. Amyotrophic lateral sclerosis: improving care with a multidisciplinary approach. J Multidiscip Healthc. 2017;10:205–15.
- Paganoni S, Macklin EA, Lee A, Murphy A, Chang J, Zipf A, et al. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:453–6.
- Nzwalo H, de Abreu D, Swash M, Pinto S, de Carvalho M. Delayed diagnosis in ALS: the problem continues. J Neurol Sci. 2014;343:173–5.