
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objective
One of the difficulties in developing a novel drug for patients with amyotrophic lateral sclerosis (ALS) is the significant variation in the clinical course. To control this variation, a 12-week run-in period is used in some clinical trials. Based on the Amyotrophic Lateral Sclerosis Functional Rating Scale Revised (ALSFRS-R) change during the run-in period, only moderate progressors are selected in some clinical trials. Some reports showed that the ALSFRS-R progression rate was associated with survival. However, it is unclear whether the ALSFRS-R change in the run-in period is a useful prognostic factor of the ALSFRS-R change from baseline. In addition, we explore the inclusion criteria that could control the variability in ALS-function progression without setting a run-in period.
Methods
We utilized the Japanese and US ALS registry databases (JaCALS and PRO-ACT). Patients were classified into three populations (rapid, moderate, and slow progressors) based on the ALSFRS-R change prior to baseline. We also classified patients into three prognostic populations based on the ALSFRS-R change from baseline. We confirmed whether each of the three populations were matched with their respective three prognostic populations.
Results
Our data showed that the three groups classified by the ALSFRS-R change during the 12 weeks prior to baseline or by the rate of progression from onset to baseline did not accord with the three prognostic groups.
Conclusions
Our results showed that the ALSFRS-R change in the run-in period or from onset to baseline is not useful for stratifying subsequent progression of functional decline in clinical trials.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease in which both upper and lower motor neurons degenerate progressively (Citation1). Neurodegeneration leads to bulbar palsy, limb muscular atrophy, and death due to dysphagia or respiratory muscular palsy, with a median survival time of 2–4 years from onset (Citation1). It has been reported that different phenotypes of ALS might have different rates of progression (Citation2).
Treatment options for ALS are very limited, although riluzole (Citation3, Citation4), edaravone (Citation5, Citation6), sodium phenylbutyrate and taurursodiol (PB-TURSO) (Citation7–9), and tofersen (Citation10, Citation11) are available in the US. In the meantime, the degree of difficulty in developing new drugs is high as evidenced by the fact that more than 50 clinical trials have failed to find an effective form of treatment in the last 50 years (Citation12). However, due to the progress in ALS research, drug development for this condition has been active internationally, and the number of new clinical trials is increasing.
To monitor disease progression, the Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised (ALSFRS-R) is commonly used. It consists of 12 items to evaluate bulbar function, motor function, and respiratory function, and each item is evaluated from 0 (unable) to 4 (normal). The total points (0–48) also serve as the gold standard for primary efficacy outcomes in clinical trials (Citation13). Edaravone has successfully completed a phase 3 trial using the ALSFRS-R as the primary endpoint measure. The Phase 3 trial targeted early-stage patients and included subjects who had a 1- to 4-point worsening in the ALSFRS-R total score during the 12-week run-in period prior to the treatment period (Citation6). The variability in ALS progression is thought to have been controlled and the study appeared to be successful. Since then, a 12-week run-in period prior to the treatment period has been commonly used in clinical trials in Japan (Citation14–18). In the US and EU, a 12-week run-in period is not common in clinical studies because a run-in period of this duration is not considered suitable for ALS patients given their short life expectancy. Some reports showed that the ALSFRS-R progression rate was associated with survival (Citation19–22). Thakore et al reported that the run-in slope correlated with the post-baseline slope (Citation23). Although estimated the run-in slope is a predictor, another group reported the rate of change in the ALSFRS-R was frequently non-linear (Citation24). When the impact of predicting post-baseline slope using the run-in slope to set inclusion criteria is small, it would be better to intervene earlier rather than setting a run-in period. Therefore, it is important to evaluate the value of setting a run-in period.
In this study, we aimed to confirm whether the rate of deterioration in the 12 weeks before the treatment period was useful for controlling the heterogeneity of ALSFRS-R progression or not. In addition, we explored the inclusion criteria that could control the variability in ALS-symptom progression without setting a 12-week run-in period. In conducting the present research, we utilized Japanese and US ALS registry databases. The Japanese Consortium for Amyotrophic Lateral Sclerosis Research (JaCALS) has been utilized in the search for clinical markers, biomarkers, and modifier genes related to the course and prognosis of ALS (Citation2, Citation21, Citation22, Citation25–27). It is the largest ALS registry in Japan. The Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) database is a publicly available repository that merges data from existing publicly and privately conducted ALS clinical trials (Citation28). By using both databases, we planned to evaluate whether or not the change in ALSFRS-R during the 12 weeks prior to baseline is useful to control ALSFRS-R heterogeneity after baseline in clinical trials.
Materials and methods
Datasets
The JaCALS and PRO-ACT databases were used. For JaCALS database research, data were collected from 38 sites in Japan from January 2006 to the beginning of the analysis (24 August 2022). The protocol for our JaCALS research was approved by the School of Medicine Ethical Review Board of Aichi Medical University. This study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and in compliance with the protocol, and Ethical Guidelines for Medical and Biological Research Involving Human Subjects. PRO-ACT represents the largest aggregation of placebo and treatment-arm data from 29 Phase II/III clinical trials. As of 1 August 2022, 111,675 de-identified clinical patient records were included. We obtained the dataset in August 2022 via a web-based application (available at https://ncri1.partners.org/ProACT).
Eligibility criteria for JaCALS database research
All of the following criteria should be met; 1) Patients aged 18 years or older and less than 80 years at the time of onset, 2) Patients with an ALSFRS-R total score at baseline (baseline was defined in each patient as the time point closest to week 78 between weeks 52 and 104 after onset), 3) Definite, probable, laboratory-supported probable, or possible ALS according to the El Escorial criteria, 4) Patients with an ALSFRS-R total score at baseline and 12 weeks (±8 weeks) prior to baseline, 5) Patients who had at least one ALSFRS-R total score recorded during the 48 weeks after baseline. In addition, patients were not included if they met either of the following criteria: 1) Patients undergoing tracheostomy at baseline, 2) Patients who are considered unsuitable for analysis identified by data review.
Eligibility criteria for PRO-ACT database research
All of the following criteria should be met; 1) Patients aged 18 years or older and less than 80 years at the time of onset, 2) Patients with an ALSFRS-R total score at baseline (baseline was defined in each patient as the time point closest to week 78 between weeks 52 and 104 after onset), 3) Patients with an ALSFRS-R total score at baseline and 12 weeks (±8 weeks) prior to baseline, 4) Patients who had at least one ALSFRS-R total score recorded during the 48 weeks after baseline, 5) Patients in the placebo group. Patients were not included if they met either of the following criteria: 1) Patients undergoing tracheostomy at baseline, 2) Suspected ALS according to the El Escorial criteria.
Formula for calculating change/slope of ALSFRS-R total score
The change in ALSFRS-R total score during the 12 weeks prior to baseline was calculated using the ALSFRS-R total score closest to week 12, within the period from week 4 to week 20, prior to baseline, using the following equation:
The rate of progression of the ALSFRS-R total score from onset to baseline was calculated by the following equation:
Patients’ classification (rapid, moderate, and slow progression population)
Patients were classified into the following three populations based on the change in ALSFRS-R total score during the 12 weeks prior to baseline. This categorization was same as that used in the edaravone phase 3 studies (Citation5, Citation6).
Rapid progressors with ALSFRS-R total score change in the 12 weeks prior to baseline faster than -4
Moderate progressors with ALSFRS-R total score change of -1 to -4 during the 12 weeks prior to baseline
Slow progressors with ALSFRS-R total score change in the 12 weeks prior to baseline slower than -1
Patients were classified into the following three prognostic populations based on the change in ALSFRS-R total score from baseline at Weeks 24 and 48.
Rapid progressors whose ALSFRS-R total score change from baseline at Week 48 is faster than −16 (ALSFRS-R total score change from baseline at Week 24 is faster than −8)
Moderate progressors whose ALSFRS-R total score change from baseline at Week 48 is -4 to -16 (ALSFRS-R total score change from baseline at Week 24 is − 2 to −8)
Slow progressors whose ALSFRS-R total score change from baseline at Week 48 is slower than –4 (ALSFRS-R total score change from baseline at Week 24 is slower than –2)
For calculating the slope from onset to baseline, ALSFRS-R total score at onset and baseline were used. In the event there was no score at onset, 48 points was adopted as the onset score.
Statistical analysis
Continuous variables were summarized using number of cases (n), mean, standard deviation (S.D.), and number of missing data cases. Discrete variables were presented as percentage of the number of cases and the number of cases included in the analysis as needed.
Cross tabulations were performed examine the distribution of the two different classifications of populations, as before and after the baseline. In order to evaluate whether the change in ALSFRS-R during the 12 weeks prior to baseline is not useful for stratifying subsequent progression, the consistency between three populations classified by ALSFRS-R total score change during 12 weeks prior to baseline and three prognostic populations at Week 48 was evaluated by the weighted kappa coefficient for ordinal scales. Quadratic weighting was used to calculate the weighted kappa coefficient. As a sensitivity analysis, the data up to Week 24 was analyzed in a same manner. In addition, for the exploration the inclusion criteria without setting a 12-week run-in period, the cutoff for rate of progression of ALSFRS-R total score from onset to baseline with the highest kappa coefficient is selected as the best control for the variability of ALS-functional progression. When ALSFRS-R total score at Week 48 (or Week 24) was missing, the change in ALSFRS-R total score from baseline at Week 48 (or Week24) was calculated based on the score at the time point closest to Week 48. Statistical analyses of JaCALS and PRO-ACT data were performed using SPSS Statistics 28.0 for Windows (IBM SPSS Statistics; Chicago, IL, USA) and SAS software, version 9.4 (SAS Institute; Cary, NC, USA), respectively.
Patients
Patient dispositions from the JaCALS and PRO-ACT databases are presented in . In terms of the JaCALS database, the analysis dataset included 246 ALS patients. For PRO-ACT database research, 585 ALS patients were used for the analysis. Patients were classified into three patient populations (rapid, moderate, and slow progressors) based on the change in ALSFRS-R total score during the 12 weeks prior to baseline. In JaCALS, the proportions of rapid, moderate, and slow progressors were 32.9% (81/246), 32.1% (79/246), and 35.0% (86/246). In PRO-ACT, the proportions of rapid, moderate, and slow progressors were 31.1% (182/585), 33.3% (195/585), and 35.6% (208/585). These proportions were well balanced between the two databases. The characteristics and baseline of the included patients are shown in . Male patients accounted for 58.5% in JaCALS and 62.6% in PRO-ACT. The mean age of all patients was 62.1 years and 56.5 years, respectively. The mean of the ALSFRS-R total score at baseline (± SD) was 32.6 ± 10.9 in JaCALS and 33.6 ± 8.0 in PRO-ACT. The main demographic and baseline data in JaCALS were similar to those in PRO-ACT.
Figure 1. Disposition. A: JaCALS; B: PRO-ACT. Abbreviations: ALS: amyotrophic lateral sclerosis; ALSFRS-R: amyotrophic lateral sclerosis functional rating scale - Revised.

Table 1. Patient characteristics and baseline.
Results
ALSFRS-R total score from onset to Week 48
showed the individual plots in the JaCALS and PRO-ACT databases. The ALSFRS-R total score for each population was analyzed up to 48 weeks from onset (). At Week 24, the percentage of patients who had ALSFRS-R data was 72.0% (177/246) in JaCALS and 72.8% (426/585) in PRO-ACT. At Week 48, the percentage of patients who had ALSFRS-R data was 52.8% (130/246) and 28.7% (168/585), respectively. The baseline score in the rapid progression group was lower than that of the slow progression group in JaCALS and PRO-ACT. However, the progression rate was not significantly different between either group at Week 24 and Week 48 in both databases.
Figure 2. ALSFRS-R individual plot. A: JaCALS; B: PRO-ACT. Abbreviations: ALSFRS-R: amyotrophic lateral sclerosis functional rating scale - Revised.
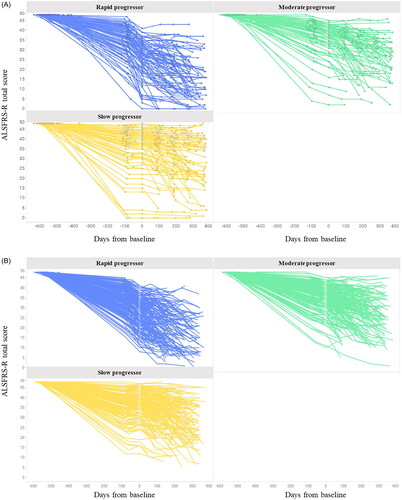
Table 2. ALSFRS-R total score from onset to Week 48.
Prognosis of the populations classified by ALSFRS-R total score change during the 12 weeks prior to baseline
showed that three groups classified by the change during the 12 weeks prior to baseline were not in concordance with the three prognostic groups in JaCALS (Kappa coefficient 0.126). Of the patients in the moderate group, 49.4% (39/79) showed a moderate decline (-4 to −16 points for 48 weeks) after baseline. The other 50.6% (40/79) showed a slow (less than −4 points for 48 weeks) or rapid (more than −16 points for 48 weeks) progression. The analysis of PRO-ACT produced similar results (Kappa coefficient: 0.180). Of the patients in the moderate group, 48.2% (94/195) showed a moderate decline (-4 to −16 points decrease for 48 weeks). In the total population without categorization based on the ALSFRS-R change prior to baseline, 45.9% (113/246) of JaCALS patients and 48.2% (282/585) of PRO-ACT patients in the moderate group showed a decline of −4 to −16 points over 48 weeks. As a sensitivity analysis, the data up to Week 24 were analyzed in the same manner. The results were consistent with those obtained using the data up to Week 48 (Supplementary Table 1).
Table 3. Prognosis of the populations classified by changes during 12 weeks prior to baseline.
Prognosis of the populations classified by ALSFRS-R total score from onset to baseline
Cutoffs for the rate of progression of the ALSFRS-R total score from onset to baseline similar to the three classifications based on the change in the ALSFRS-R total score during the 12 weeks prior to baseline (rapid, moderate, slow progression populations) were explored. The cutoffs with the highest Kappa coefficients were Rapid (change per month < −1.20), Moderate (−1.20 ≦change per month ≦–0.45), and Slow (change per month > −0.45) in JaCALS (Supplementary Table 2). However, the concordance between the three populations classified by the rate of progression from ALS onset to baseline and the three prognostic populations (those with a change from baseline to 48 weeks faster than −16, ranging from −16 to −4, and slower than −4, respectively) was low (Kappa factor 0.036) (). The same analysis was conducted using the PRO-ACT data. The cutoffs with the highest Kappa coefficients were Rapid (change per month <–1.10), Moderate (–1.10≦change per month ≦–0.50), and Slow (change per month > −0.50) in PRO-ACT (Supplementary Table 2). The concordance between the three populations classified by the rate of progression from onset to baseline and the three prognostic populations was low ().
Table 4. Prognosis of the population classified by ALSFRS-R total score from onset to baseline.
Discussion
Our database research is the first study to evaluate whether the ALSFRS-R change during the 12 weeks prior to baseline is useful to control ALSFRS-R heterogeneity in clinical trials. We conducted our study using the JaCALS and PRO-ACT databases. The main demographic and baseline data were similar in both databases in our analysis population. Our data showed that three groups classified by the change during the 12 weeks prior to baseline were not in concordance with the three prognostic groups in JaCALS (Kappa coefficient 0.126). The analysis using 24-week data was conducted to minimize the impact of missing data. The results obtained using 24-week data were similar to those using 48-week data. In addition, the results from the PRO-ACT database research were robustly consistent with those of JaCALS. In the JaCALS database research, 49.4% (39/79) of patients in the moderate group showed moderate progression after baseline. However, 45.9% (113/246) of all subjects also showed moderate decline without classification based on the change in the 12 weeks prior to baseline. Given the above, ALSFRS-R change during the 12 weeks prior to baseline would not be a key factor to stratify subsequent decline of ALSFRS-R in the setting of clinical trials although a previous research reported that estimated the run-in slope correlated with the post-baseline slope (Citation23). We focused on the likely target population in clinical trial and directly evaluated the usefulness of the run-in slope as a single factor. It might be a potential reason for the divergent conclusions. Our research also showed that 40-50% of slow progressors and rapid progressors classified by the ALSFRS-R change in the 12 weeks prior to baseline became moderate progressors after baseline. Thus, patients might lose the opportunity to participate clinical trials due to the eligibility criteria based on the ALSFRS-R change in the run-in period.
PB-TURSO was approved by the FDA based on the positive ALSFRS-R results (Citation7). In the trial, early ALS patients who were within 18 months of symptom onset were enrolled without a run-in period. In Japan, an ultrahigh-dose methylcobalamin phase 3 study showed the effect of treatment on ALSFRS-R (Citation14). In the phase 3 study, patients within 12 months of symptom onset were enrolled. In addition, post-hoc analysis of dexpramipexole and edaravone studies showed that targeting early ALS patients is important to detect the therapeutic effects of treatment (Citation29, Citation30). Given these results, intervention at the early stage of ALS appears to be a critical factor. A post-hoc survival analysis of the PB-TURSO phase 2 study (CENTAUR) also clarified the advantage of early intervention (Citation8, Citation9). In CENTAUR, patients with ALS were randomized 2:1 to PB-TURSO or placebo. CENTAUR consisted of a 24-week double-blind randomized phase and an open-label extension in which all subjects were administered PB-TURSO. A survival benefit was observed among participants originally randomized to PB-TURSO compared to those originally randomized to placebo. This indicates that 6-month early intervention is important to detect a survival benefit and highlights the fact that a 12-week run-in period delays intervention by 12 weeks. This could have the effect of impairing the results of ALS clinical trials.
The alternative eligibility criteria using the slope from onset to baseline have been used in several clinical trials (Citation11, Citation31, Citation32). Our study showed that the concordance between the three populations classified by the rate of progression from ALS onset to baseline and the three prognostic populations (those with a change from baseline to 48 weeks faster than −16, those with a change ranging from −16 to −4, and those with a change slower than −4) was low based on the JaCALS and PRO-ACT database research. Given these results, eligibility criteria using the slope from onset to baseline does not appear to be a key factor to stratify subsequent progression of ALSFRS-R after baseline. Post-hoc analyses also showed that the correlation coefficients between the change during 12 weeks prior to baseline and the change from baseline at Week 48 were low (Pearson: 0.014, Spearman: 0.118) in JaCALS. The correlation was also low (Pearson: −0.103, Spearman: 0.006) for the rate of progression from onset to baseline. Results in PRO-ACT were similar to those in JaCALS (Supplementary Table 3). A previous report showed that the rate of change in the ALSFRS-R was frequently non-linear from onset (Citation24). The non-linearity was observed in our research as well although the research focus was different. The previous work did not mimic the setting of recent clinical trials. On the other hand, we set a similar baseline point to other clinical trials and evaluated the slope between pre- and post-baseline. Therefore, our results showed the impact of non-linearity on the clinical trial design. In the setting of clinical trials, a 12-week run-in period, which requires extra time, would not have an additional value in the setting of clinical trials compared to the index (the rate of progression from onset to baseline) obtained in a single baseline assessment.
The present study has some limitations. First, this was a retrospective study. Second, data were insufficient to confirm robustness in certain subgroups such as a subgroup population with forced vital capacity of more than 80%. A run-in period might be useful in these subgroup populations. Third, the baseline was set arbitrarily in our study because there is no baseline timepoint in JaCALS’s real-world data. The baseline was defined in each patient as the time point closest to week 78 between weeks 52 and 104 after onset given the mean of ALS duration in the edaravone phase 3 studies (Citation5, Citation6). This could be a potential source of bias. Fourth, in our research, the total ALSFRS-R score at baseline was relatively severe than other clinical trials (Citation5–7).
In conclusion, our research results showed that the ALSFRS-R change during the 12 weeks prior to baseline or from onset to baseline is not useful to for stratifying subsequent progression of functional decline in clinical trials. It would be better to intervene early rather than setting a run-in period.
Supplemental Material
Download MS Word (161.9 KB)Acknowledgements
We would like to thank the doctors and staff as well as all the patients with ALS who participated in JaCALS and PRO-ACT. We would also like to extend our thanks to Ariko Takanashi, Shinichi Kanazawa, Yoshinori Imokawa and Takahiro Higuchi from A2 Healthcare Corporation who supported the analysis of the data from JaCALS.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
PRO-ACT data are openly available at https://ncri1.partners.org/ProACT. The participants of JaCALS did not give written consent for their data to be shared publicly, so due to the sensitive nature of the research supporting data is not available.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (http://dx.doi.org/10.1080/21678421.2024.2320987).
Additional information
Funding
References
- Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, et al. Amyotrophic lateral sclerosis. Lancet. 2022;400:1363–80.
- Watanabe H, Atsuta N, Hirakawa A, Nakamura R, Nakatochi M, Ishigaki S, et al. A rapid functional decline type of amyotrophic lateral sclerosis is linked to low expression of TTN. J Neurol Neurosurg Psychiatry. 2016;87:851–8.
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole study group. N Engl J Med. 1994; 330:585–91.
- Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis/Riluzole Study Group II. Lancet. 1996; 347:1425–31.
- Abe K, Itoyama Y, Sobue G, Tsuji S, Aoki M, Doyu M, et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014; 15:610–7.
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2017; 16:505–12.
- Paganoni S, Macklin EA, Hendrix S, Berry JD, Elliott MA, Maiser S, et al. Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020; 383:919–30.
- Paganoni S, Hendrix S, Dickson SP, Knowlton N, Macklin EA, Berry JD, et al. Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve. 2021; 63:31–9.
- Paganoni S, Hendrix S, Dickson SP, Knowlton N, Berry JD, Elliott MA, et al. Effect of sodium phenylbutyrate/taurursodiol on tracheostomy/ventilation-free survival and hospitalisation in amyotrophic lateral sclerosis: long-term results from the CENTAUR trial. J Neurol Neurosurg Psychiatry. 2022; 93:871–5.
- Miller T, Cudkowicz M, Shaw PJ, Andersen PM, Atassi N, Bucelli RC, et al. Phase 1-2 Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2020; 383:109–19.
- Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC, et al. Trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2022; 387:1099–110.
- Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol. 2014; 13:1127–38.
- van Eijk RPA, de Jongh AD, Nikolakopoulos S, McDermott CJ, Eijkemans MJC, Roes KCB, et al. An old friend who has overstayed their welcome: the ALSFRS-R total score as primary endpoint for ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2021; 22:300–7.
- Oki R, Izumi Y, Fujita K, Miyamoto R, Nodera H, Sato Y, et al. Efficacy and safety of ultrahigh-dose methylcobalamin in early-stage amyotrophic lateral Sclerosis: a randomized clinical trial. JAMA Neurol. 2022; 79:575–83.
- Aizawa H, Kato H, Oba K, et al. Randomized phase 2 study of perampanel for sporadic amyotrophic lateral sclerosis. J Neurol. 2022; 269:885–96.
- Aoki M, Warita H, Kato M, et al. Application of hepatocyte growth factor for amyotrophic lateral sclerosis. Brain Nerve 2019; 71:1253–60.
- Morimoto S, Takahashi S, Fukushima K, Saya H, Suzuki N, Aoki M, et al. Ropinirole hydrochloride remedy for amyotrophic lateral sclerosis - Protocol for a randomized, double-blind, placebo-controlled, single-center, and open-label continuation phase I/IIa clinical trial (ROPALS trial). Regen Ther. 2019; 11:143–66.
- Imamura K, Izumi Y, Nagai M, Nishiyama K, Watanabe Y, Hanajima R, et al. Safety and tolerability of bosutinib in patients with amyotrophic lateral sclerosis (iDReAM study): A multicentre, open-label, dose-escalation phase 1 trial. EClinicalMedicine 2022; 53:101707.
- Kimura F, Fujimura C, Ishida S, Nakajima H, Furutama D, Uehara H, et al. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006; 66:265–7.
- Kollewe K, Mauss U, Krampfl K, Petri S, Dengler R, Mohammadi B, et al. ALSFRS-R score and its ratio: a useful predictor for ALS-progression. J Neurol Sci. 2008; 275:69–73.
- Watanabe H, Atsuta N, Nakamura R, Hirakawa A, Watanabe H, Ito M, et al. Factors affecting longitudinal functional decline and survival in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2015; 16:230–6.
- Nakamura R, Atsuta N, Watanabe H, Hirakawa A, Watanabe H, Ito M, et al. Neck weakness is a potent prognostic factor in sporadic amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry. 2013; 84:1365–71.
- Thakore NJ, Lapin BR, Pioro EP,. Trajectories of impairment in amyotrophic lateral sclerosis: Insights from the pooled resource open-access ALS clinical trials cohort. Muscle Nerve. 2018; 57:937–45.
- Ramamoorthy D, Severson K, Ghosh S, Sachs K, Glass JD, Fournier CN, et al. Identifying patterns in amyotrophic lateral sclerosis progression from sparse longitudinal data. Nat Comput Sci. 2022; 2:605–16.
- Yokoi D, Atsuta N, Watanabe H, Nakamura R, Hirakawa A, Ito M, et al. Age of onset differentially influences the progression of regional dysfunction in sporadic amyotrophic lateral sclerosis. J Neurol. 2016; 263:1129–36.
- Hayashi N, Atsuta N, Yokoi D, Nakamura R, Nakatochi M, Katsuno M, et al. Prognosis of amyotrophic lateral sclerosis patients undergoing tracheostomy invasive ventilation therapy in Japan. J Neurol Neurosurg Psychiatry. 2020; 91:285–90.
- Nakamura R, Tohnai G, Nakatochi M, Atsuta N, Watanabe H, Ito D, et al. Genetic factors affecting survival in Japanese patients with sporadic amyotrophic lateral sclerosis: a genome-wide association study and verification in iPSC-derived motor neurons from patients. J Neurol Neurosurg Psychiatry. 2023;94:816–24. jnnp-2022-330851. Epub ahead of print.
- Atassi N, Berry J, Shui A, Zach N, Sherman A, Sinani E, et al. The PRO-ACT database: design, initial analyses, and predictive features. Neurology 2014; 83:1719–25.
- Bozik ME, Mitsumoto H, Brooks BR, Rudnicki SA, Moore DH, Zhang B, et al. A post hoc analysis of subgroup outcomes and creatinine in the phase III clinical trial (EMPOWER) of dexpramipexole in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014; 15:406–13.
- EDARAVONE (MCI-186) ALS 16 STUDY GROUP A post-hoc subgroup analysis of outcomes in the first phase III clinical study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2017; 18:11–9.
- Mora JS, Genge A, Chio A, Estol CJ, Chaverri D, Hernández M, et al. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized clinical trial. Amyotroph Lateral Scler Frontotemporal Degener. 2020; 21:5–14.
- Haji S, Fujita K, Oki R, Osaki Y, Miyamoto R, Morino H, et al. An exploratory trial of EPI-589 in amyotrophic lateral sclerosis (EPIC-ALS): protocol for a multicenter, open-labeled, 24-week, single-group study. JMIR Res Protoc. 2023;12:e42032.