
Abstract
Sickle cell disease (SCD) is a genetic disorder characterized by the production of abnormal hemoglobin that polymerizes at low oxygen concentrations, causing the erythrocyte to adopt a sickle-shaped morphology. SCD pathophysiology is extremely complex and can lead to numerous clinical complications, including painful vaso-occlusive crises (VOC), end-organ damage, and a shortened lifespan. An impressive number of investigational drugs are currently in early stages of clinical development with prospects for use either as chronic therapies to reduce VOC frequency and end-organ damage in SCD or for use at the time of VOC onset. Many of these agents have been developed using a pathophysiological-based approach to SCD, targeting one or more of the mechanisms that contribute to the disease process. It is plausible that a multi-drug approach to treating the disease will evolve in the coming years, whereby hydroxyurea (HU) (the only drug currently FDA-approved for SCD) is used in combination with drugs that amplify nitric oxide signaling and/or counteract hemolytic effects, platelet activation and inflammation.
1. Sickle cell disease
Sickle cell disease (SCD) is a class of genetic disorders, caused by point mutations in the gene encoding the hemoglobin β chain. The most common forms of SCD are homozygous sickle cell anemia (SCA; HbSS), where the presence of two βs globin alleles results in an almost exclusive production of hemoglobin S (HbS) instead of HbA in erythrocytes; HbSC disease, caused by dual mutations resulting in the production of both HbS and HbC, and HbS-beta thalassemia compound heterozygosity Citation[1]. The βs globin mutation is thought to have evolved in Africa, as heterozygosity for the gene confers resistance to malaria without significant manifestations. Consequently, the highest frequency of the HbS allele occurs in sub-Saharan Africa, but SCD is recognized as a global health problem Citation[2].
SCD pathogenesis is driven by the polymerization of HbS, under conditions of low oxygenation, causing the erythrocyte to become rigid and adopt a characteristic sickle-shaped morphology. The complex pathophysiology of SCD results in numerous clinical complications, including painful vaso-occlusive (VO) episodes, autoinfarction of the spleen, acute chest syndrome (ACS), stroke, pulmonary hypertension, organ damage, renal damage and a shortened lifespan Citation[1].
2. SCD pathophysiology
Vascular occlusion in SCD decreases organ perfusion, leading to tissue damage, recurrent pain and, potentially, end-organ damage. The VO process is complex and comprises multiple pathophysiological mechanisms, as summarized in (). HbS polymerization in red blood cells (RBC), under deoxygenated conditions, causes RBC sickling and makes cells more rigid and less deformable, contributing to occlusion of the vessels of the microcirculation. Furthermore, these fragile RBC are more susceptible to rupture, and resulting hemolysis has significant consequences in the blood vessel. Hemoglobin (Hb) released into the plasma results in local endothelium-derived nitric oxide (NO) depletion, facilitating vasoconstriction, and subsequent release of the toxic Hb product, hemin. RBC lysis also increases plasma arginase, consequently limiting the availability of L-arginine, the substrate for NO synthesis. Oxidative Hb reactions in the blood vessel, as well as reduced NO bioavailability and hemin production contribute to vascular oxidative stress and can activate the endothelial cells. In turn, activated endothelium produces inflammatory cytokines and expresses adhesion molecules on its surface, occasioning leukocyte capture to the vascular wall. Inflammatory mediator production, from activated platelets, leukocytes and endothelial cells, results in inflammatory processes and induces leukocyte and RBC adhesion to the endothelium, causing reduced blood flow, diminished oxygen concentrations, consequent RBC sickling and, eventually, vessel occlusion. Chronic vascular inflammation ensues with the occurrence of recurrent VO processes, leading to hypoxia/reperfusion injury and additional oxidative stress. Additionally, hypoxia can trigger the synthesis of pro-angiogenic factors and upregulation of angiogenesis in SCD with further endothelial activation and inflammation, possibly contributing to manifestations such as pulmonary hypertension and retinopathy.
3. Currently available treatment options for SCD
The only drug approved at present by the FDA for use as a therapy for SCD is Hydroxyurea (HU), or hydroxycarbamide. HU is a cytostatic agent and may also exert some of its effects via NO generation in vivo Citation[3,4]. The drug acts principally by inducing the production of fetal hemoglobin (HbF) in RBC, but also reduces leukocyte counts amongst other effects. HbF is generally synthesized during intrauterine life, but production switches to HbA or HbS after birth; however, the induction of HbF production, even at relatively low levels (15 – 25% of total hemoglobin), can inhibit the polymerization of HbS and significantly improve the disease’s clinical course, ameliorating mortality rates and reducing hospitalization for vaso-occlusive crises (VOC), the incidence of ACS, as well as the necessity for transfusions Citation[3,4]. Although HU has recognizably successful benefits in SCD, the search continues for agents that control other aspects of the pathophysiology of the disease for use in combination with, or as an alternative to, HU.
Other current therapeutic approaches for SCD include the use of transfusion therapy to reduce HbS concentrations, particularly in children identified as at risk for stroke Citation[5]; however, transfusion therapy can have long-term consequences such as alloimmunization and iron overload. Hematopoietic stem cell transplantation (HSCT) is a curative option for treating SCD, but is not widely available and only usually offered to a very small proportion of patients with HLA-matched sibling donors and with infrequent, but serious, transplant-associated complications that include death, graft versus host disease and engraftment difficulty Citation[5]. Recent advances in non-myeloablative conditioning regimens and the use of HLA-haploidentical donors may make HSCT transplantation available to more individuals with SCD in the future, although a high rate of graft failure remains an obstacle to be overcome Citation[6].
At present there are no agents available for use specifically during VOC, and the current treatment of these extremely painful and debilitating crises, which usually require hospitalization, is purely symptomatic, relying on rest, strong analgesics and hydration.
4. Investigational therapies in development for SCD
An impressive number of drug candidates are currently at various stages of clinical development, either as potential therapies for SCD or for use at the time of the onset of VOC. Many of these agents have been developed using a pathophysiological-based approach to SCD, targeting one or more of the mechanisms that contribute to the disease process (; ). Some drugs, previously developed for other clinical indications, are also under investigation for use in SCD, based on evidence that these drugs could ameliorate some of these same pathophysiological aspects ().
Figure 1. Pathophysiology of Sickle Cell Disease (SCD). SCD is characterized by the production of hemoglobin S (HbS) in red blood cells (RBC). Hb S polymerizes when deoxygenated, causing the cells to become rigid and adopt a sickled state, as well as making cells more fragile and susceptible to rupture (hemolysis). Increasing levels of fetal hemoglobin (HbF) in RBC can reduce hemoglobin polymerization and therefore RBC sickling and hemolysis. Intravascular hemolysis has significant consequences; hemoglobin (Hb) released into the plasma results in endothelium-derived nitric oxide (NO) depletion in the blood vessel and the release of the toxic Hb product, hemin. Red cell lysis also increases plasma arginase, consequently limiting the availability of the substrate for NO synthesis, L-arginine. Oxidative Hb reactions, as well as reduced NO bioavailability and hemin production, contribute significantly to vascular oxidative stress and endothelial cell activation. Activated endothelium produces inflammatory cytokines and expresses adhesion molecules on its surface, resulting in leukocyte capture, and RBC and platelet adhesion to the vascular wall. Inflammatory mediator production from activated platelets, leukocytes and endothelial cells results in the inflammatory state that is associated with SCD and that drives recurrent vaso-occlusive processes, which result from leukocyte and RBC adhesion to the endothelium, occasioning reduced blood flow, diminished oxygen concentrations, consequent red cell sickling and eventually the occlusion of vessels. In a vicious cycle, chronic vascular inflammation and recurrent vaso-occlusive processes occur, causing hypoxia/reperfusion injury and resulting in further oxidative stress. Additionally, hypoxia triggers the synthesis of pro-angiogenic factors that results in the upregulation of angiogenesis and further endothelial activation and inflammation. Currently early investigational therapies under development for SCD use a pathophysiological-based approach to abolish or reduce one or more of the mechanisms that contribute to this disease’s complex pathophysiology.
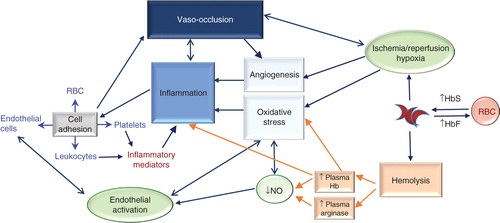
Table 1. Early investigational therapies for sickle cell disease and their pathophysiological targets.
Increasing levels of HbF in RBC has been known for some time to reduce HbS polymerization and, whereas HU therapy can successfully elevated HbF in most patients, other drugs that may be able to induce HbF production even more efficiently are also under investigation. The immunomodulating thalidomide derivative, pomalidomide, is currently in Phase I/II trials for use in SCD and is reported to be well tolerated and increase HbF efficiently at higher doses Citation[7]. Other agents, such as the thalidomide-hybrid compounds 4c and 4d, and lenalidomide have been shown to elevate HbF and have also demonstrated anti-inflammatory effects in pre-clinical studies Citation[8,9]. Histone deacetylase inhibitors, such as sodium butyrate, azacitidine and decitabine can induce γ-globin gene expression and, therefore, HbF production and are also at the Phase I/II trial stage to evaluate safety and efficacy in SCD Citation[9]. Additionally, allosteric effectors of Hb are also under study, such agents include 5-hydroxymethylfurfural (5-HMF; Aes-103), which reduces HbS sickling by increasing its affinity for oxygen and stabilizing Hb in its R state; preliminary clinical studies indicate 5-HMF to be well tolerated in steady-state SCD Citation[10].
The use of L-glutamine for improving the redox potential of sickle RBC and, therefore, countering oxidative stress has been in development for some time and data reported from a recent Phase III study indicate that supplementation may reduce the frequency of VOC and hospitalization Citation[11]. Furthermore, Gardos channel inhibitors, such as senicapoc (ICA-17043), have been administered to patients with a view to reducing RBC dehydration and improving hemolysis, but no significant improvement in the rate of sickle cell painful crises was observed in patients during a Phase III study Citation[12].
In line with our growing understanding of VO processes in SCD, drugs currently in early investigational stages generally target specific mechanisms of SCD pathophysiology. Much effort has been devoted to developing agents that inhibit the adhesion of blood cells to the vascular endothelium. Animal models of SCD indicate that leukocyte adhesion to the activated endothelium may trigger VO Citation[13]; the pan-selectin antagonist, rivipansel (GMI-1070), was developed with a view to decreasing leukocyte/endothelial interactions, as well as the formation of heterocellular leukocyte aggregates, and for use at the onset of VOC. Phase II studies have shown that GMI-1070 improves clinical outcomes such as time to resolution of crisis, time to discharge, and I.V. opioid use Citation[14,15]. The SelG1 humanized anti-P selectin mAb has been developed for use via periodical intravenous infusion with the aim of inhibiting RBC adhesion to the endothelium and has the potential to reduce heterocellular platelet interactions; a Phase II clinical trial to evaluate efficacy in reducing the frequency of VOC crises is currently underway (NCT01895361). The use of low-molecular-weight heparins that block P-selectin (expressed on activated endothelial cells and platelets) and L-selectin (expressed on leukocytes) at sub-anti-coagulant doses during steady-state SCD is also under investigation; selectin-targeted therapies in development for SCD have recently been reviewed in this journal Citation[16]. Other potential approaches for reducing adhesive interactions in SCD include the use of intravenous gamma globulin (IVIG) at the time of admission for VOC, which downregulates the neutrophil Mac-1-integrin function Citation[17], and has been shown to be well tolerated in patients during VOC in a recent Phase I study, whereas Poloxamer 188 (MST-188) is a non-ionic surfactant that reduces blood viscosity and RBC adhesion that has already reached Phase III trials and may hold promise for use during VOC and for the management of ACS (Citation[18,19] NCT01737814).
Inhibition of endothelial activation and suppression of chronic inflammation in SCD are targets for a number of investigational drugs. Exploitation of the pleiotropic anti-inflammatory effects of statins constitutes a potential approach for SCD. A Phase I/II trial demonstrated simvastatin to be well tolerated during steady-state SCD Citation[20]. NKTT120 is a humanized mAb that depletes invariant natural killer T cells (iNKT), reducing inflammatory molecule release and is well tolerated in steady-state SCD adults at low dosages Citation[21]. Similarly, the adenosine A2A receptor agonist ragadenoson has been shown to decrease iNKT activation and inflammation in SCD and was well-tolerated in individuals during steady-state and following 24h-infusion during VOC; a larger Phase II trial is underway to evaluate the efficacy of ragadenoson administration during VOC Citation[22] (NCT 01788631). Additionally, pre-clinical studies have shown that etanercept, a TNF-α-antagonist, reduces inflammation and VO processes in a mouse model of SCD Citation[23].
To counter hypoxia, pegylated Hb carbon monoxide (CO) carriers are under investigation; SanguinateTM releases CO while also providing oxygen delivery to hypoxic tissues and recruitment for Phase I trials to evaluate safety, efficacy and pharmacokinetics in SCD is currently underway (NCT01848925; NCT01374165). Release of CO by another pegylated-Hb, MP4CO, has significant anti-inflammatory and cytoprotective effects and a Phase Ib single dose escalation study demonstrated the molecule to be well tolerated in steady-state SCD Citation[24].
The effects of reducing platelet activation by inhibiting αIIbβ3-integrin function on the platelet surface has been investigated using the synthetic peptide inhibitor, eptifibatide; however, whereas a preliminary Phase I/II study showed the drug to be apparently safe it failed to significantly reduce opioid use or time until hospital discharge when administered during VOC Citation[25]. On the other hand, prasugrel, a P2Y12 ADP receptor antagonist, has been found to be well tolerated when administered over a 30-day period to steady-state SCD patients, diminishing platelet activation and demonstrating a trend towards decreased pain Citation[26].
Vascular hemolysis may be central to SCD pathophysiology and preclinical studies indicate that scavengers of Hb, or its products, may abrogate the inflammatory and oxidative effects of cell-free Hb. Vaso-occlusion and ACS can be prevented by the infusion of plasma-purified haptoglobin (a Hb-binding protein found in plasma) or recombinant hemopexin (a plasma protein that sequesters the Hb product, hemin) in SCD mouse models Citation[27,28]. Other Hb-binding proteins have been identified and synthesized, such as α1-microglobulin (A1M) and hE-Hb-B10 Citation[29,30], respectively, and further pre-clinical and clinical trials will be necessary to evaluate the efficacy and safety of such Hb/hemin-scavenging therapeutics for use in SCD.
Finally, reposition of vascular NO has been advocated for SCD, following evidence that hemolysis and cell-free Hb release decreases NO bioavailability in SCD. Whereas inhalation of NO during VOC was not found to significantly decrease pain or improve the time until crisis resolution Citation[31], therapy with L-arginine (the substrate for NO production) was found to significantly reduce opioid use when administered to children hospitalized for VOC Citation[32]. HU, the only currently approved therapy for SCD, exerts some of its effects via in vivo donation of NO Citation[33]; preclinical studies indicate that amplification of NO-dependent signaling, either in combination with HU, or not, may be of therapeutic benefit in SCD. Phosphodiesterase inhibitors decrease the degradation of cGMP and can demonstrate tissue specificity. An inhibitor of phosphodiesterase 9, expressed in hematopoietic cells, has been shown to reduce VO processes in an SCD-mouse model Citation[33] and a Phase I study to investigate the safety and tolerability of PF-04447943, a PDE9 inhibitor, in steady-state SCD individuals, with and without co-administration of HU, is planned (NCT 02114203).
5. Conclusion
Considerable progress in our understanding of the pathophysiology of SCD has been made over the past decade or so, allowing the identification of a number of molecular and mechanistic targets for SCD. These findings have led to the development of the significant number of drugs that are currently in the early stages of clinical investigation for the disease; however, extensive clinical studies are still required to confirm the full potential of these different approaches for SCD therapy.
Given the multi-systemic nature of SCD manifestations, advances in other specialties of medicine should be monitored with a view to applying such approaches to SCD. For instance, potential applications for anti-VEGF drugs, hypoxia-inducible factor-1 antagonists, in current development for treating ocular neovascularization, as well as β-adrenoreceptor inhibitors, should be investigated for countering disorganized angiogenesis in SCD Citation[34-36]. Novel approaches under investigation for nephropathy, liver fibrosis and pulmonary hypertension, include the use of nuclear erythroid 2-related factor 2 (Nrf2; an oxidative stress-mediated induced transcription factor) agonists, palmitoylethanolamide (PEA; a lipid with anti-inflammatory actions), and endothelin receptor and endothelin synthesis inhibitors Citation[37-39], all of which could present potential for use in SCD.
6. Expert opinion
Goals for the treatment of SCD include reducing end-organ damage, decreasing VOC frequency and ultimately increasing patient survival, as well as improving quality of life. To this end, chronic therapeutic approaches to ameliorate the clinical course of the disease are still being sought. Whereas HU is proven to be an effective drug for SCD, reducing patient mortality and morbidity, this drug does not successfully elevate HbF in all patients and complete protection against organ damage has not yet been evidenced Citation[4]. Our understanding of the pathophysiology of SCD has now improved greatly and it is plausible that a multi-drug approach to treating the disease will evolve in the coming years. Exploration of the use of drugs in combination with HU to amplify NO signaling and counteract any residual hemolytic effects, as well as platelet activation and inflammation may prove important for improving the long-term benefits of HU.
Furthermore, at present, no acute therapy currently exists for treating painful SCD VOC and procuring drugs that can relieve VOC remains a priority for investigators. Many of the drugs under current investigation for use during VOC have been developed with the aim of reducing pain, accelerating crisis resolution and diminishing severe complications such as ACS. However, clinical investigators face enormous difficulties in pinpointing workable clinical end points to allow adequate statistical power for measuring efficacy in VOC. With the ever-increasing number of drugs undergoing early clinical investigation for use in SCD VOC, it is hoped that the growing experience acquired by investigators in such studies and the identification of biomarkers for efficiently monitoring the progress of VOC may attenuate such limitations.
Declaration of interest
N Conran is a São Paulo State government employee at the University of Camipinas, Brazil. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Notes
Bibliography
- Stuart MJ, Nagel RL. Sickle-cell disease. Lancet 2004;364(9442):1343-60
- Piel FB, Tatem AJ, Huang Z, et al. Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000. Lancet Glob Health 2014;2(2):e80-9
- Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med 1995;332(20):1317-22
- McGann PT, Ware RE. Hydroxyurea for sickle cell anemia: what have we learned and what questions still remain? Curr Opin Hematol 2011;18(3):158-65
- Kassim AA, DeBaun MR. The case for and against initiating either hydroxyurea therapy, blood transfusion therapy or hematopoietic stem cell transplant in asymptomatic children with sickle cell disease. Expert Opin Pharmacother 2014;15(3):325-36
- Bolanos-Meade J, Fuchs EJ, Luznik L, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood 2012;120(22):4285-91
- Kutlar A, Swerdlow PS, Meiler SE, et al. Pomalidomide in sickle cell disease: Phase I study of a novel anti-switching agent. Blood 2013;122:abstract 777
- dos Santos JL, Lanaro C, Lima LM, et al. Design, synthesis, and pharmacological evaluation of novel hybrid compounds to treat sickle cell disease symptoms. J Med Chem 2011;54(16):5811-19
- Fard AD, Hosseini SA, Shahjahani M, et al. Evaluation of novel fetal hemoglobin inducer drugs in treatment of beta-hemoglobinopathy disorders. Int J Hematol Oncol Stem Cell Res 2013;7(3):47-54
- Safo MK, Kato GJ. Therapeutic strategies to alter the oxygen affinity of sickle hemoglobin. Hematol Oncol Clin North Am 2014;28(2):217-31
- Niihara Y, Koh HA, Tran L, et al. A phase III study of L-glutamine therapy for Sickle cell anemia and sickle ß0-thalassemia. Blood (ASH) 2014;124(21):abstract:86
- Ataga KI, Reid M, Ballas SK, et al. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a Phase III randomized, placebo-controlled, double-blind study of the gardos channel blocker senicapoc (ICA-17043). Br J Haematol 2011;153(1):92-104
- Chang J, Patton JT, Sarkar A, et al. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 2010;116(10):1779-86
- Wun T, Styles L, DeCastro L, et al. Phase I study of the E-selectin inhibitor GMI 1070 in patients with sickle cell anemia. PLoS One 2014;9(7):e101301
- Wun T, Telen MJ, Krishnamurti L, et al. Pan-selectin antagonist rivipansel (GMI-1070) reduces soluble E-selectin levels while improving clinical outcomes in SCD vaso-occlusive crisis. Blood (ASH) 2014;124:abstract:2704
- Okpala I. Investigational selectin-targeted therapy of sickle cell disease. Expert Opin Investig Drugs 2015;24(2):229-38
- Manwani DM, Chen C, Carullo V, et al. Vaso-occlusion-promoting neutrophil mac-1 integrin activation in human sickle cell crises is stabilized by a single dose of intravenous gammaglobulin. Blood (ASH) 2014;124:abstract:4089
- Cheung AT, Chan MS, Ramanujam S, et al. Effects of poloxamer 188 treatment on sickle cell vaso-occlusive crisis: computer-assisted intravital microscopy study. J Investig Med 2004;52(6):402-6
- Ballas SK, Files B, Luchtman-Jones L, et al. Safety of purified poloxamer 188 in sickle cell disease: Phase I study of a non-ionic surfactant in the management of acute chest syndrome. Hemoglobin 2004;28(2):85-102
- Hoppe C, Kuypers F, Larkin S, et al. A pilot study of the short-term use of simvastatin in sickle cell disease: effects on markers of vascular dysfunction. Br J Haematol 2011;153(5):655-63
- Field JJ, Ataga KI, Majerus EM, et al. A Phase I single ascending dose study of NKTT120 in stable adult sickle cell patients. Blood 2013;122(21):977
- Field JJ, Nathan DG, Linden J. The role of adenosine signaling in sickle cell therapeutics. Hematol Oncol Clin North Am 2014;28(2):287-99
- Solovey A, Somani A, Chen CS, et al. Interference with TNF alpha using long-term etanercept in S+S-Antilles sickle transgenic mice ameliorates abnormal endothelial activation, vasoocclusion, and pulmonary hypertension including its pulmonary arterial wall remodeling. Blood 2013;122(21):728
- Howard J, Thein SL, Galacteros F, et al. Safety and tolerability of MP4CO: a dose escalation study in stable patients with sickle cell disease. Blood 2013;122(21):2205
- Desai PC, Brittain JE, Jones SK, et al. A pilot study of eptifibatide for treatment of acute pain episodes in sickle cell disease. Thromb Res 2013;132(3):341-5
- Wun T, Soulieres D, Frelinger AL, et al. A double-blind, randomized, multicenter Phase II study of prasugrel versus placebo in adult patients with sickle cell disease. J Hematol Oncol 2013;6:17
- Belcher JD, Chen C, Nguyen J, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014;123(3):377-90
- Ghosh S, Adisa OA, Chappa P, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest 2013;123(11):4809-20
- Hanson MS, Xu H, Flewelen TC, et al. A novel hemoglobin-binding peptide reduces cell-free hemoglobin in murine hemolytic anemia. Am J Physiol Heart Circ Physiol 2013;304(2):H328-36
- Olsson MG, Allhorn M, Bulow L, et al. Pathological conditions involving extracellular hemoglobin: molecular mechanisms, clinical significance, and novel therapeutic opportunities for alpha(1)-microglobulin. Antioxid Redox Signal 2012;17(5):813-46
- Gladwin MT, Kato GJ, Weiner D, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA 2011;305(9):893-902
- Morris CR, Kuypers FA, Lavrisha L, et al. A randomized, placebo-controlled trial of arginine therapy for the treatment of children with sickle cell disease hospitalized with vaso-occlusive pain episodes. Haematologica 2013;98(9):1375-82
- Almeida CB, Scheiermann C, Jang JE, et al. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood 2012;120(14):2879-88
- De Falco S. Antiangiogenesis therapy: an update after the first decade. Korean J Intern Med 2014;29(1):1-11
- Iwase T, Fu J, Yoshida T, et al. Sustained delivery of a HIF-1 antagonist for ocular neovascularization. J Control Release 2013;172(3):625-33
- Casini G, Dal Monte M, Fornaciari I, et al. The beta-adrenergic system as a possible new target for pharmacologic treatment of neovascular retinal diseases. Prog Retin Eye Res 2014;42:103-29
- Impellizzeri D, Esposito E, Attley J, et al. Targeting inflammation: new therapeutic approaches in chronic kidney disease (CKD). Pharmacol Res 2014;81:91-102
- Maguire JJ, Davenport AP. Endothelin@25 - new agonists, antagonists, inhibitors and emerging research frontiers: IUPHAR Review 12. Br J Pharmacol 2014;171(24):5555-72
- Yang JJ, Tao H, Huang C, et al. Nuclear erythroid 2-related factor 2: a novel potential therapeutic target for liver fibrosis. Food Chem Toxicol 2013;59:421-7
- Nakagawa A, Lui FE, Wassaf D, et al. Identification of a small molecule that increases hemoglobin oxygen affinity and reduces ss erythrocyte sickling. ACS Chem Biol 2014;9(10):2318-25