
Abstract
Glioblastoma is a highly lethal cancer. Glioma stem cells (GSCs) are potentially an attractive therapeutic target and eradication of GSCs may impact tumor growth and sensitize tumors to conventional therapies. The brain is one of the most metabolically active organs with glucose representing the most important, but not the only, source of energy and carbon. Like all other cancers, glioblastoma requires a continuous source of energy and molecular resources for new cell production with a preferential use of aerobic glycolysis, recognized as the Warburg effect. As selected metabolic nodes are amenable to therapeutic targeting, we observed that the Warburg effect may causally contribute to glioma heterogeneity. This Editorial summarizes recent studies that examine the relationship between GSCs and metabolism and briefly provides our views for the future directions. The ultimate goal is to establish a new concept by incorporating both the cellular hierarchical theory and the cellular evolution theory to explain tumor heterogeneity. Such concept may better elucidate the mechanisms of how tumors gain cellular and molecular complexity and guide us develop novel and effective targeted therapies.
1. Clinical relevance of separating glioblastoma into subtypes
Despite aggressive multimodality therapy, glioblastomas (GBMs) remain universally fatal with current treatment offering only palliation Citation[1]. There is an urgent need to better understand GBM pathophysiology to develop effective therapies. Although it has been long recognized that cancers display aberrant metabolic reprogramming, recent studies of cancer genetics and metabolomics have just started to reveal the intrinsic crosstalk between oncogenic and oncometabolic pathways that facilitate tumor progression and therapeutic resistance Citation[2,3]. For example, whole exome sequencing of secondary GBM by the Vogelstein and Kinzler group discovered the genetic mutations of isocitrate dehydrogenases (IDH)1 Citation[4], which cause neomorphic enzymatic production of an oncometabolite 2-hydroxyglutarate (2-HG), resulting in global changes in DNA methylation and progression from low-grade tumor to high-grade one. However, the IDH1 mutation is uncommon in the vast majority of GBMs, suggesting that most GBMs dysregulate metabolism through alternative, most likely nongenetic, mechanisms Citation[4]. Based on the recent genome-wide transcriptome and DNA methylome analyses, presence of the four subtypes of adult GBM has been proposed: proneural (PN), classical, neural and mesenchymal (MES) Citation[5-7]. Given that distinct signaling pathways are activated in these individual GBM subtypes Citation[5-7], it is reasonable to believe that therapeutic strategies may be more effective if tailored to tumor subtypes instead of applying them to all GBM patients. Nonetheless, metabolic pathways in individual GBM subtypes with distinct genetic mutations remain poorly characterized. Also, clinical relevance of separation of GBM tumors into the four subtypes is still debatable, as patient prognoses of these subtypes are marginally different or contradictory between studies (unlike other cancer subtypes such as medulloblastoma and breast cancer). Despite this controversy, recent data points out that glioma may gain MES identity when they advance malignancy Citation[8]. Gene signature and DNA methylation pattern of classical GBM appear to overlap, at least partially, with those of PN or MES GBM Citation[7]. At least in the experimental GBM models, striking differential responses to individual targeted therapies are observed between PN and MES GBM cell cultures and/or xenografted tumors Citation[2,9]. To date, it remains unanswered whether separation of GBM into four (or more) subtypes has any clinically relevant significance, or gliomas including primary GBM arise from PN tumors, subsequently gaining MES identity during disease progression. We would speculate that the latter (PN to MES progression) takes a major, if not the only, role in GBM establishment (more details described later).
2. Clinical relevance of studying stem cell phenotype in GBM
Additional complexity in GBM heterogeneity is derived from recognition of cellular hierarchies in tumors with subpopulations of self-renewing, highly tumorigenic cells that activate stem cell pathways and give rise to the bulk of the tumor Citation[10-12]. Potential relevance of these stem-like cancer cells (glioma initiating/propagating cells or glioma stem cells [GSCs]) is derived from a number of experimental studies including ours demonstrating roles in therapeutic resistance, invasion into normal tissues and tumor angiogenesis Citation[10-12]. Targeting GSCs (or their dependent signals) may elucidate therapeutic windows that improve tumor-selective targeting with acceptable toxicity to somatic tissues. Collectively, intratumoral and intertumoral heterogeneity may be integrated into models of tumors as dynamic ecologies. Indeed, although convincing evidence exists to support presence of GBM cells with stem-like properties, it is still unclear whether these tumor cells represent a defined and irreversible population or they represent a ‘state’, where tumor cells can flux in and out this state. The evolution of tumors before, during and after treatment is likely created by a combination of cellular hierarchy and cellular evolution (). It remains undetermined, however, how cellular hierarchy and cellular evolution coexist to explain the complexity of tumor cells in single tumors.
Figure 1. Schema showing the combined concept of cellular hierarchy model and cellular evolution model. Association with glucose metabolism is also described.
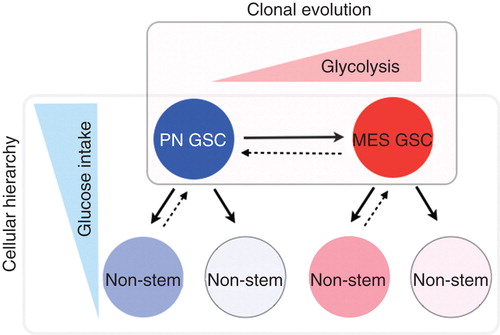
GSCs are not distributed uniformly within tumors but rather mimic the organization of the bone marrow with two functional niches: perivascular and hypoxic. Microenvironment of these two niches for GSCs is quite distinct: perivascular areas in tumors are enriched with oxygen and glucose provided by tumor vessels, while hypoxic areas have very limited nutrition including glucose. In addition, GSCs in perivascular and hypoxic niches are regulated by distinct molecular mechanisms including VEGF and hypoxia-inducible factors (HIFs), respectively Citation[13]. HIFs are critical to GSC maintenance and chronic hypoxic and/or hypo-glucose condition triggers stem cell features in cancer cells. Limited oxygen and nutrition induces stress to challenge tumor cells to survive. It remains elusive, however, whether GSCs in these two niches have different gene signatures and phenotypes. The predominant source of VEGF secretion for tumor cells is vascular endothelial cells. Given that the brain is one of the most vascular-rich organs, premalignant cells that arise and propagate GBM are presumably surrounded with enriched vascular endothelial cells, at least in the initial stage (favorable condition for PN tumor cells). On the other hand, when a tumor expands rapidly, vascularization may not catch up with requirement of nutrition for individual tumor cells, thereby creating a large central necrosis (favorable microenvironment for MES tumor cells). It is possible that GBM arises from PN subtype that eventually shifts its phenotype to MES tumor cells. Future studies will likely tell us the answer.
3. Glucose metabolism in GSCs as a therapeutic target
Under limited energy sources within a tumor mass, glucose uptake and its intracellular metabolism may determine whether individual tumor cells can preferentially survive and grow. We recently reported that restricted glucose conditions lead to increased GSC frequency through a combination of preferential GSC survival and acquisition of stem-like features in non-GSCs Citation[3]. GSCs competed to absorb glucose by co-opting high-affinity glucose transporters normally expressed by neurons and embryonic stem cells glucose transporter 3 (Glut3) Citation[3]. Targeting Glut3 expression in GSCs disrupted stem cell self-renewal and tumor growth, and high Glut3 expression was associated with increased tumor grade and poorer patient survival Citation[3]. Collectively, in the harsh microenvironment, GSCs may instruct tumor heterogeneity because of their enhanced survival potential over nonstem tumor cells.
In a variety of cancers, reprogramming of cellular metabolism is well recognized to be a shared feature of malignancy Citation[14-17]. Almost 90 years ago, Otto Warburg demonstrated that cancer cells preferentially obtain energy from aerobic glycolysis, which produces high amounts of lactate from pyruvate – termed the Warburg effect. The conversion of glucose to lactate in the presence of oxygen is a critical aerobic pathway that allows cancer cells to proliferate rapidly and the amount of lactate produced is correlated well with tumor malignancy Citation[14-17]. In the MES, but not PN, subtype of GSCs, glycolysis pathway is highly upregulated creating heterogeneous cellular populations Citation[2]. Elevated radioresistance of GSCs is likely associated with MES signature and activated glycolysis pathway and disruption of glycolysis-derived MES identity increased aggressiveness and radiosensitivity of tumor cells Citation[2]. Collectively, these data suggest that the Warburg effect is not a consequence of a malignant phenotype but rather a causative metabolic paradigm, leading to tumor development and propagation to create tumor heterogeneity at both the cellular and the global levels. Given these observations, elimination of the glycolysis-induced metabolic reprogramming is likely a promising approach for the treatment of various cancers including GBM Citation[18-21]. Recently, a small-scale first-in-human study was conducted with five GBM patients using the oral glycolysis inhibitor, dichloroacetate (DCA), for up to 15 months together with the oral methylator, temozolomide, with some promising results Citation[22]. Out of five patients, three patients showed radiographical regression of tumors and four patients were clinically stable for up to 15 months. Matched immunohistochemistry analysis with three tumors before and after treatment exhibited that DCA normalizes mitochondrial functions, inhibits the target gatekeeper enzyme of DCA, pyruvate dehydrogenase kinase II and promotes cellular apoptosis, resulting in antitumor effects. In preclinical studies, an IDH1 inhibitor delayed growth and induced differentiation of mutant glioma cells Citation[23]. These early studies suggest that targeting metabolism may offer benefit in GBM treatment.
4. Known unknown
In cancer cells, glucose provides acetyl-CoA that condenses with oxaloacetate to produce citrate and 2-HG production Citation[24,25]. As a critical metabolite, α-ketoglutarate (α-KG) is actively carboxylated by the NADPH-linked mitochondrial and cytosolic IDH1/2 to form citrate and 2-HG, respectively Citation[25,26]. Subsequently, citrate is required for facilitating cancer cell proliferation and 2-HG is an oncometabolite synthesized by carboxylation of α-KG. Increased α-KG production and carboxylation appear to be essential for accelerating cancer cell growth. Nonetheless, despite the accelerated cancer cell growth by α-KG, IDH-mutant tumors are more indolent and less aggressive compared with wild-type tumors. It is possible that citrate promotes lipid synthesis as a critical intermediate, which may serve as the common mechanism for aggressiveness of GBM that do not have IDH mutations. Collectively, these data indicate the complexity of distinct dependence of GBM tumors in various other, yet undetermined, oncometabolic pathways. While α-KG phenocopies malignant transformation of human fibroblasts driven by Rho family GEF oncogenic Dbl, DCA treatment of cultured GBM cells paradoxically increases α-KG expression Citation[27]. The physiological roles of α-KG as glycolysis mediator are still controversial and require more investigation.
Significance of elucidating the molecular mechanisms of tumor metabolism in GBM has no question, and it is critical to translate this knowledge into the development of effective GBM-targeted therapeutic approaches. Successful achievement will likely elucidate GSC-specific signaling pathways and effective targeted therapies for GBM. In addition, to study the molecular mechanisms by which glucose uptake and glycolysis regulate GSCs can be applicable to other stem cells and/or cancers.
Declaration of interest
The author has no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Acknowledgement
The American Cancer Society MRSG-08-108-01, NIH/NCI P01 CA163205, R21 CA175875, NIH/NINDS R01 NS083767 and R01 NS087913.
Notes
Bibliography
- Jarzabek MA, Sweeney KJ, Evans RL, et al. Molecular imaging in the. development of a novel treatment paradigm for glioblastoma (GBM): an integrated multidisciplinary commentary. Drug Discov Today 2013;18:1052-66
- Mao P, Joshi K, Li J, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci USA 2013;110:8644-9
- Flavahan WA, Wu Q, Hitomi M, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci 2013;16:1373-82
- Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765-73
- Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006;9:157-73
- Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17:98-110
- Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012;22:425-37
- Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010;463:318-25
- Bhat KP, Balasubramaniyan V, Vaillant B, et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013;24:331-46
- Gilbert CA, Ross AH. Cancer stem cells: cell culture, markers, and targets for new therapies. J Cell Biochem 2009;108:1031-8
- Heddleston JM, Hitomi M, Venere M, et al. Glioma stem cell maintenance: the role of the microenvironment. Curr Pharm Des 2011;17:2386-401
- Ji J, Black KL, Yu JS. Glioma stem cell research for the development of immunotherapy. Neurosurg Clin N Am 2010;21:159-66
- Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009;15:501-13
- Clendening JW, Penn LZ. Targeting tumor cell metabolism with statins. Oncogene 2012;31:4967-78
- Romero-Garcia S, Lopez-Gonzalez JS, Baez-Viveros JL, et al. Tumor cell metabolism: an integral view. Cancer Biol Ther 2011;12:939-48
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 2009;23:537-48
- Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 2008;13:472-82
- Shuch B, Linehan WM, Srinivasan R. Aerobic glycolysis: a novel target in kidney cancer. Expert Rev Anticancer Ther 2013;13:711-19
- Hagenbuchner J, Kuznetsov AV, Obexer P, Ausserlechner MJ. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene 2013;32(40):4748-57
- Lu W, Hu Y, Chen G, et al. Novel role of NOX in supporting aerobic glycolysis in cancer cells with mitochondrial dysfunction and as a potential target for cancer therapy. PLoS Biol 2012;10:e1001326
- Sun Q, Chen X, Ma J, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA 2011;108:4129-34
- Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010;2:31ra34
- Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013;340:626-30
- Kim M, Le H, McInerney MJ, Buckel W. Identification and characterization of re-citrate synthase in Syntrophus aciditrophicus. J Bacteriol 2013;195:1689-96
- Wise DR, Ward PS, Shay JE, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci USA 2011;108:19611-16
- Leonardi R, Subramanian C, Jackowski S, Rock CO. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J Biol Chem 2012;287:14615-20
- Wang JB, Erickson JW, Fuji R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010;18:207-19