
1. Introduction
As molecular bases of pathophysiologies are being decoded, drug discovery efforts are also being increasingly focused on target-based approaches Citation[1]. Target identification is inevitably the first step in such a drug discovery pipeline. The target-centric approach reflects a major paradigm shift from the ligand-centric one. Target identification itself has not always taken advantage of rational systematic explorations. Many targets that enter the pipeline are first chosen, based on the knowledge of their roles in a given pathological process. Then they are typically taken through an in vitro and in vivo validation process which is laborious, time consuming and have high rates of attrition Citation[2]. Newer methods are undoubtedly required to select targets that are more likely to succeed. Toward this goal, computational approaches have a high potential to obtain first shortlists of targets Citation[3-6].
2. Properties preferred in drug targets
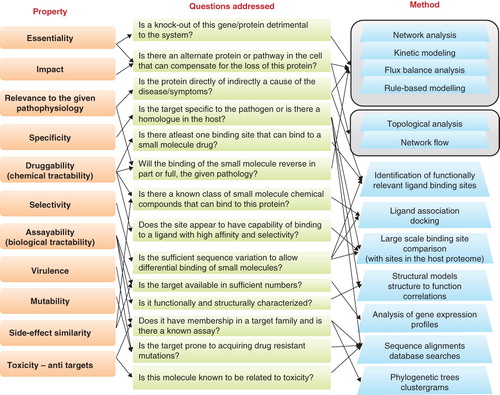
Now, let us attempt to define an ‘ideal target’. A primary property required of an ideal drug target is that the biological rationale of its use must be clearly evident. Put simply, one could define a drug target as a macromolecule, most often a protein, whose manipulation could lead to removing the causes or relieving the symptoms caused by the underlying pathophysiology. Manipulation is predominantly achieved by small molecules, although the use of biologicals including peptides and antibodies is catching up rapidly. Manipulation itself could be in the form of inhibition or augmentation of the natural function of the protein. However, though the biological relevance is an essential criterion, it is by itself insufficient to define a good drug target. Additional important criteria that need to be met by an ideal target are: i) essentiality – it should be essential to the system responsible for the pathophysiology, ii) process/condition specificity – it should be specific to the disease process or state, iii) species/family specificity – it should be specific to the pathogen species or family (where applicable), as detected by conservation of the protein in related organisms, iv) druggability or chemical tractability – it should reflect whether its function can be manipulated by an appropriate small molecule, v) biological tractability and assayability that reflects if the target is available in sufficient quantities in vivo and whether suitable methods are available to test the function of the protein and thereby study extent of inhibition or activation by candidate lead molecules Citation[1]. In addition, in the recent years, other criteria such as vi) virulence factors as novel strategies for therapeutic intervention Citation[7], vii) low mutability to lower chances of drug resistance Citation[7], viii) alterations in the quantitative profile of the target/reaction/pathway, ix) side effect similarity to a known drug/corresponding target Citation[8] and x) toxicity, are all being explored as additional criteria for target selection. Yet another important aspect being explored is the xi) combination of two or few targets from the same disease system, such that the impact will be synergistic. The following section describes how computational methods can be employed for screening entire genomes with each of these criteria ().
Figure 1. Properties preferred in drug targets and computational methods for identifying targets based on them. The nature of the method as well as the questions that need can be addressed while choosing a protein as a potential drug target are also indicated.
3. Scope of computational approaches
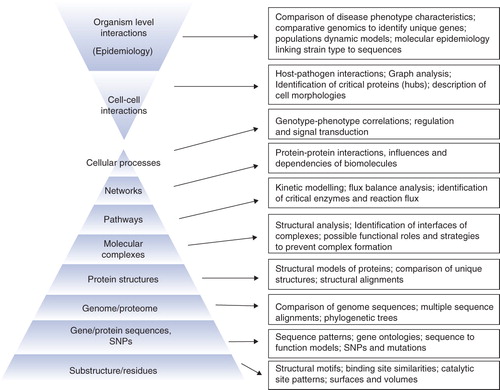
It is clear that biological information gathered in literature and databases span across various levels of hierarchy of biological organization since biology is studied in all these different levels Citation[9]. Clues for drug target identification can come from any of these levels shown in . For pathogenic diseases, comparisons of the sequence of the causative strain with those in the sequence databases readily identify the clade and the family the given genome belongs to. Comparisons with focused datasets of closely related sequences, lead to identification of those proteins that are either unique or at least sufficiently different from its counterparts from avirulent or less potent strains of the pathogen, thereby providing a first list of potential drug targets. Thus, specificity can be addressed easily through analyses of genome sequences. For designing broad-spectrum antibacterials, knowledge on clade specificity through phylogenetic profiling, will be extremely useful for identifying appropriate targets. Where three-dimensional structures or high-confidence prediction of structural models are available, specificity can be addressed at a higher resolution by comparing three-dimensional structures of the target protein(s) with other proteins in its own cell as well as any other relevant cell. For this purpose, sub-structure or functional site comparison is often more insightful Citation[10].
Figure 2. Various levels of hierarchy at which a system can be studied to identify drug targets. Some are already being used widely, but some have the potential to be explored in the coming years for target discovery. The lower pyramid illustrates the different levels of organization in the cell, while the upper pyramid illustrates high order interactions, at cellular levels as well as organismal levels.
Besides providing insights into specificity, structural models can be used to analyze another important parameter, that is, druggability Citation[11]. Molecular recognition events that enable specific binding of the drug to the target are fundamental to all aspects of drug action. Knowledge of the structure of the target macromolecule helps us in estimating whether it can bind specifically to a small molecule and whether its function can be manipulated with suitable small molecule ligands. For this, the binding site(s) are identified and the nature of the site characterized. Those that have deep grooves with sufficient cavity convexity and interaction potential and located strategically to influence the function of the protein would make the targets druggable Citation[12]. Structural bioinformatics methods including ligand docking can be valuable in estimating ligand binding potential Citation[13,14]. A study of Mycobacterium tuberculosis (Mtb) proteome indicates the usefulness of computational methods for this purpose Citation[15]. Using proteome-wide structural models of both host cell and pathogen, possible pockets were first detected by binding site prediction algorithms, and then compared with each other by all-versus all-site matching, thereby identifying unique pockets in Mtb. By combining this with residue-wise conservation analyses, a druggability check is automatically performed. A similar concept reported subsequently, termed as a chemical systematic biology approach, identifies off-targets through its ligand-binding sites Citation[16].
Essentiality is inherently a systems' property and cannot be addressed by studying proteins individually. Systems approaches are needed to study such issues. This can be achieved through different approaches. Kinetic modeling is one of the best approaches so far, to get a quantitative appreciation, but can generally be used at the level of individual metabolic or signaling pathways Citation[17]. Since parameters required for kinetic modeling are not available in most cases, especially at a genome scale, several constraint-based methods have been in use. Flux balance analysis is one such method, in which genome-scale metabolic models can be analyzed through systematic perturbations to identify lethal gene deletions. This method has been used to study metabolism in many bacteria Citation[18]. The set of proteins, without which no growth is observed in the models are considered as essential and hence as potential drug targets. Networks capturing structural or functional protein–protein interactions can be analyzed to identify the set of proteins that are important for maintaining network structure Citation[19]. Topological analyses using graph theoretical methods can be used to identify hubs and choke-points Citation[20], useful for target identification. Resilience of network topologies can also be probed by deletion of nodes or edges Citation[21]. Conditional specificity such as the relevance of a certain protein for a given state of the disease or disease-causing organism can be studied through networks by integrating genomics data from gene-expression profiling or similar studies Citation[22].
Emergence of drug resistance is a major problem confronting us for many diseases, particularly with antibacterial and antiviral agents. Suggested strategies to counter this include use of proteins that are less mutable and hence less prone to resistance Citation[7]. Virulence factors in bacteria are believed to come under this category and could serve as possible targets. Using network analyses it has been hypothesized that certain proteins may serve as hub nodes in triggering the emergence of resistance and could be co-targeted along with the primary target, so as to inhibit the resistance mechanism itself Citation[23].
Traditionally, drug safety has been addressed by modification of the drug molecule itself, but a careful choice of the target molecule can be helpful in addressing safety right at the beginning of the drug discovery process. To address the issue of toxicity, similarity to the gut flora proteins can be computed. For this, the potential set of targets can be compared by sequence alignment methods to the millions of proteins in the meta-genome of the gut flora containing about 100 different organisms. Those that have close homologues in any of the gut flora can be eliminated from the target discovery pipeline Citation[15]. This is done so that we do not ultimately design a drug that will unwittingly inhibit proteins in the gut flora, which are now well recognized to be required for the maintenance of our normal health. Side effects of many existing antibiotics are linked to this phenomenon Citation[8]. Another important issue that is addressed through genome sequence comparisons is the emergence of toxicity due to unintentional binding to anti-targets. Care could be taken to choose a target such that it is sufficiently different from the host proteins, belonging to the class of anti-targets. Proteins such as the transporters and pumps, which modify the bio-availability of a drug by their efflux action, or those proteins that trigger hazardous side effects, such as the hERG protein are termed as ‘anti-targets’ Citation[24]. By considering these aspects early in the drug discovery pipeline, the risk of failure of the drug candidates in the later stages of drug discovery can be minimized.
It has been suggested that a more useful approach for target discovery would be to identify protein combinations that perturb the robustness often seen in disease-causing phenotypes, rather than the conventional approach of hitting one target at a time Citation[25]. A fact that lends support to this approach is the wide clinical practice of prescribing a combination of drugs for many diseases. Systems level analyses are useful in identifying optimal combinations as possible drug targets Citation[26]. Databases capturing a variety of these aspects are proving to be useful resources for scientists in the field. TDR database which contains druggability predictions for potential drug targets in tropical disease pathogens and compound desirability information is one such example Citation[27].
4. Expert opinion
Drug discovery has witnessed a paradigm shift from the traditional medicinal chemistry-based ligand-oriented approaches to rational drug target identification and target-driven lead discovery, by targeting the molecular mechanisms of disease. However, there is some concern that target-based discovery has not shown increase in productivity over the traditional physiology-based approach Citation[28,29]. A solution to this could be in the careful choice of targets by considering multiple aspects that influence the outcome of various steps of the drug discovery pipeline, so that target identification becomes more prudent and less prone to failures.
Availability of high throughput ‘omics’-scale data is influencing every sphere of biology research, pervading to drug discovery as well. The scale of the data and the inherent complexity of the underlying system, necessitates the use of computational approaches. The field has many examples of overstated anticipations, disappointments as well as successes. It is important to recognize that different approaches come with their own advantages and limitations. Hence integration of different approaches in an appropriate manner is possibly the key to success. Computational approaches should be viewed as providing a strong focus to designing specific experiments so that only a handful of possibilities can be tested rather than having to search for a needle in a haystack.
Computational methods of the type discussed here have several distinct advantages: i) model building is based on precise descriptions of both the genotype and phenotype, making correlations more meaningful, ii) such models enable the dissection of precise roles of each component through systematic perturbations, iii) models are amenable to simulations under a wide variety of conditions, some of which may be impractical to study experimentally and iv) they can be carried out at low cost and high speed, yet integrating much of the knowledge gained on that component over the years. However, it is important to note that a model is only as good as our understanding of what constitutes a system. Both the resolution of individual components as well as the abstraction levels of the models need to be, therefore, borne in mind to draw conclusions more meaningfully.
Many of the criteria required for defining a good target, can be analyzed using computational methods. Computational models span a wide range of levels, covering sequence, structural and recently also systems levels. The first two provide functional clues, and ligand recognition properties, for evaluating target feasibility. The conventional method of focusing on a single protein at a time, however, important the protein may be, would mean losing perspective of its larger context. Broader insights about the appropriateness of a potential target can be obtained by considering pathways and whole-system models relevant to that disease. Although systems thinking is not new to biology, the current practice of systems biology attempts to reconstruct the system, brick by brick and hence facilitates an understanding of why and how an event takes place, automatically leading to ‘what if’ type of questions, and hence predictions, rather than the conventional approach of merely recording a systems' output from a ‘black-box’ without knowing why or how, such an output results. Systems models allow us to address important aspects in target discovery such as essentiality, safety, combination targets and even polypharmacology.
The predictive power provided by data-driven computation has long been a critical component in product development and safety testing in engineering. It is only logical to expect that biology too, and in particular drug discovery, will increasingly benefit from large-scale informatics, modeling and simulations in the coming years.
Declaration of interest
The author states no conflict of interest and has received no payment in preparation of this manuscript.
Bibliography
- Lindsay MA. Target discovery. Nat Rev Drug Discov 2003;6:831-8
- van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov 2003;2:192-204
- Knowles J, Gromo G. A guide to drug discovery: target selection in drug discovery. Nat Rev Drug Discov 2003;2:63-9
- Keiser MJ, Setola V, Irwin JJ, Predicting new molecular targets for known drugs. Nature 2009;462:175-81
- Yang Y, Adelstein SJ, Kassis AI. Target discovery from data mining approaches. Drug Discov Today 2009;14:147-54
- Scheiber J, Chen B, Milik M, Gaining insight into off-target mediated effects of drug candidates with a comprehensive systems chemical biology analysis. J Chem Inf Model 2009;49:308-17
- Tan Y-T, Tillett DJ, McKay IA. Molecular strategies for overcoming antibiotic resistance in bacteria. Mol Med Today 2000;6:309-14
- Levy J. The effects of antibiotic use on gastrointestinal function. Am J Gastroenterol 2000;95:8-10
- Chandra N. Computational systems approach for drug target discovery. Expert Opin Drug Discov 2009;4:1221-36
- Blundell TL, Sibanda BL, Montalvao RW, Structural biology and bioinformatics in drug design: opportunities and challenges for target identification and lead discovery. Philos Trans R Soc Lond B Biol Sci 2006;361:413-23
- Al-Lazikani B, Gaulton A, Paolini G, The Molecular basis of predicting. Wiley-VCH Verlag GmbH; Druggability: 2008
- Perot S, Sperandio O, Miteva MA, Druggable pockets and binding site centric chemical space: a paradigm shift in drug discovery. Drug Discov Today 2010;15:656-67
- Chandra N, Anand P, Yeturu K. Structural bioinformatics: deriving biological insights from protein structures. Interdiscip Sci 2010;2:347-66
- Gao Z, Li H, Zhang H, PDTD: a web-accessible protein database for drug target identification. BMC Bioinformatics 2008;9:104
- Raman K, Yeturu K. Chandra N. targetTB: a target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst Biol 2008;2:109
- Xie L, Xie L, Bourne PE. Structure-based systems biology for analyzing off-target binding. Curr Opin Struct Biol 2011;21:189-99
- Voit EO. Computational analysis of biochemical systems. Cambridge University Press; Cambridge, UK; 2000
- Oberhardt MA, Palsson BO, Papin JA. Applications of genome-scale metabolic reconstructions. Mol Syst Biol 2009;5:320
- Zhao S, Li S. Network-based relating pharmacological and genomic spaces for drug target identification. PLoS One 2010;5:e11764
- Hasan S, Daugelat S, Rao PSS, Prioritizing genomic drug targets in pathogens: application to mycobacterium tuberculosis. PLoS Comput Biol 2006;2:e61
- Strong M, Eisenberg D. The protein network as a tool for finding novel drug targets. Prog Drug Res 2007;64:193-215
- Forst CV. Network genomics – A novel approach for the analysis of biological systems in the post-genomic era. Mol Biol Rep 2002;29:265-80
- Raman K, Chandra N. Mycobacterium tuberculosis interactome analysis unravels potential pathways to drug resistance. BMC Microbiol 2008;8:234
- Vaz RJ, Klabunde T. Antitargets: prediction and prevention of drug side effects. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany; 2008
- Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 2008;4:682-90
- Raman K, Vashisht R, Chandra N. Strategies for efficient disruption of metabolism in Mycobacterium tuberculosis from network analysis. Mol Biosyst 2009;5:1740-51
- Aguero F, Al-Lazikani B, Aslett M, Genomic-scale prioritization of drug targets: the TDR Targets database. Nat Rev Drug Discov 2008;7:900-7
- Sams-Dodd F. Target-based drug discovery: is something wrong? Drug Discov Today 2005;10:139-47
- Brown D. Unfinished business: target-based drug discovery. Drug Discov Today 2007;12:1007-12