Abstract
Eosinophils and their products play an essential role in the pathogenesis of various reactive and neoplastic disorders. Depending on the underlying disease, molecular defect and involved cytokines, hypereosinophilia may develop and may lead to organ damage. In other patients, persistent eosinophilia is accompanied by typical clinical findings, but the causative role and impact of eosinophilia remain uncertain. For patients with eosinophil-mediated organ pathology, early therapeutic intervention with agents reducing eosinophil counts can be effective in limiting or preventing irreversible organ damage. Therefore, it is important to approach eosinophil disorders and related syndromes early by using established criteria, to perform all appropriate staging investigations, and to search for molecular targets of therapy. In this article, we review current concepts in the pathogenesis and evolution of eosinophilia and eosinophil-related organ damage in neoplastic and non-neoplastic conditions. In addition, we discuss classifications of eosinophil disorders and related syndromes as well as diagnostic algorithms and standard treatment for various eosinophil-related disorders.
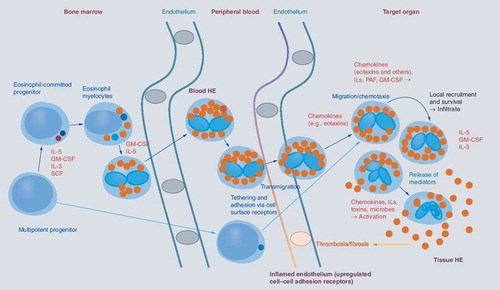
Eosinophils originate from multipotent and lineage-restricted hematopoietic progenitor cells. Eosinophil progenitors reside in the bone marrow but are also detectable in the peripheral blood. Eosinophil development is regulated by eosinophilopoietic cytokines (IL-3, GM-CSF and IL-5) and takes place primarily in the bone marrow. Cytokine-induced HE in the peripheral blood is often accompanied by tissue HE. Activation of eosinophils and activation of endothelial cells contribute to endothelial transmigration and infiltration of inflamed tissues. Eosinophil adhesion to endothelium and transmigration are mediated by certain homing receptors. Migration and accumulation of eosinophils in (inflamed) tissues are mediated by chemotactic peptides (chemokines), cytokines and other mediators. Eosinophil accumulation is also triggered by delayed eosinophil apoptosis, another cytokine-mediated phenomenon, in local tissue sites. Activation of eosinophils leads to degranulation and mediator secretion in tissues, with consequent organ damage, which may be accompanied by fibrosis and/or thrombosis and by deposition of eosinophil granule proteins. HE: Hypereosinophilia; GM-CSF: Granulocyte/macrophage colony-stimulating factor; IL: Interleukin; PAF: Platelet-activating factor; SCF: Stem cell factor.
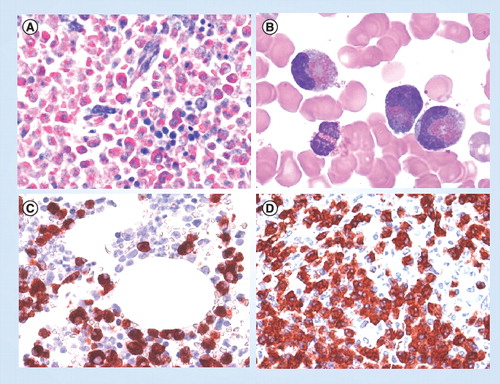
(A) Giemsa-stained bone marrow section in a patient with eosinophilic leukemia. Note the massive eosinophil infiltrate replacing the normal bone marrow. (B) Circulating immature eosinophils in a patient with FIP1L1/PDGFRA+ chronic eosinophilic leukemia. (C) & (D) Bone marrow biopsy sections obtained from a patient with FIP1L1/PDGFRA+ chronic eosinophilic leukemia. Sections were incubated with an antibody directed against eosinophil major basic protein and analyzed by indirect immunohistochemistry.
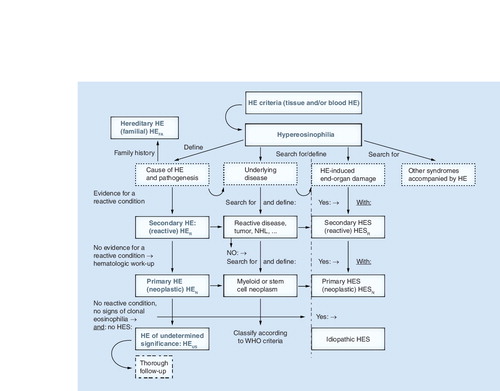
After having established that HE is present, the cause and clinical significance of HE need to be explored. With regard to the cause, the patient is examined for signs of a reactive process (helminth infection, drug allergy or others), evidence of a myeloid or stem cell neoplasm (where eosinophils usually are neoplastic cells), or of other malignancies. In rare cases, familial HE is diagnosed. When no underlying condition and no signs of overt organ damage are found, the (provisional) diagnosis HEUS is established, and the patient is carefully monitored. In the case of secondary (reactive or paraneoplastic) HE or clonal (neoplastic) HE, the final diagnosis is determined using the WHO criteria and other established criteria. When HE is accompanied by specific (eosinophil-induced) organ damage, diagnosis of HES is established. HES can occur in any type of HE and can present as secondary (reactive) HES, primary (neoplastic) HES, or idiopathic HES. Rare syndromes presenting with eosinophilia, such as the CCS, Gleich’s syndrome and other syndromes, must also be considered based on clinical findings. CCS: Churg–Strauss syndrome; HE: Hypereosinophilia; HEN: Clonal/neoplastic HE; HER: Reactive HE; HEUS: HE of undetermined significance; HES: Hypereosinophilic syndrome; HESN: Primary/neoplastic HES; HESR: Reactive HES; NHL: Non-Hodgkin´s lymphoma.
Eosinophils are highly specialized granulocytic effector cells that produce and store diverse biologically active molecules, including cytotoxic, cytostimulatory proteins, lipid mediators, chemo-tactic peptides and cytokines Citation[1–5]. Under various conditions, eosinophils can invade certain target organs after transendothelial migration and secrete their products into the surrounding tissue, thereby triggering local inflammation and tissue remodeling Citation[2–8]. When tissue and/or blood eosinophilia is marked and persistent, the term hypereosinophilia (HE) is appropriate. In patients with HE, eosinophil-derived substances may induce marked alterations in the microenvironment and thus chronic (and potentially irreversible) organ damage Citation[2–8]. In these patients, an array of signs and symptoms associated with eosinophilia may be present, and tissue inflammation is often accompanied by local (extracellular) deposition of eosinophil-derived proteins Citation[2–8]. In a subset of these patients, tissue fibrosis and/or thrombosis may develop Citation[2–8].
Blood and/or tissue HE is detectable in various inflammatory reactions, certain hematologic malignancies and sometimes in patients with solid tumors (Box 1) Citation[9–17]. Reactive eosinophilia is typically found in patients with helminth infections, toxic or allergic drug reactions and atopic disorders (Box 1). Hematopoietic malignancies typically (but not always) accompanied by eosinophilia are myeloproliferative neoplasms (MPN), certain variants of acute myeloid leukemia (AML), a smaller subset of patients with myelodysplastic syndromes (MDS), some MDS/MPN overlap disorders, several (mostly T-cell-derived) lymphoproliferative disorders and (advanced) systemic mastocytosis (SM) (Box 1) Citation[13,18–24]. These differential diagnoses should be considered in cases of unexplained HE, especially in the context of other blood count abnormalities. In such patients, a hematologic work-up, including bone marrow (BM) investigations with cytogenetics, FISH and molecular analyses, should be initiated Citation[18–24]. In patients with myeloid or stem cell-derived neoplasms, eosinophils usually belong to the malignant clone. In these patients, fusion genes involving PDGFRA, PDGFRB, FGFR1 or other tyrosine kinases may be present . This is of great importance given the fact that imatinib is highly effective in patients with PDGFRA or PDGFRB fusion genes, but not in neoplasms with FGFR1 fusion genes Citation[18–28]. In chronic eosinophilic leukemia (CEL), the FIP1L1-PDGFRA fusion gene (and the related cytogenetic surrogate CHIC2 deletion by FISH) is detected in approximately 10–20% of all cases, and is thus the most frequent recurrent aberration in CEL . Numerous other cytogenetic defects, such as loss of the Y chromosome, trisomy 8, trisomy 15, del(6q), del(20q) or i(17q), have also been reported Citation[13]. Although most of these defects are rare in patients with eosinophil neoplasms, they support the clonal nature of HE.
During the past two decades, our knowledge about eosinophils, their biology, their specific functions and the mechanisms underlying HE and HE-related organ damage has improved substantially. Moreover, several proposals for the classification of HE-related disorders have been presented Citation[17–24,29–33]. However, a number of open questions concerning the definition of HE and the classification of HE-related disorders remain. In this article, the authors review and discuss the recent literature and the current knowledge about eosinophils, eosinophil-induced organ damage and disorders presenting with HE. In addition, a diagnostic algorithm is proposed and related diagnostic parameters and treatment options are discussed.
Origin, development, migration & activation of eosinophils: role of cytokines and peptide mediators
Eosinophils are constantly replenished from a pool of pluripotent and lineage-committed hematopoietic progenitor cells in the BM Citation[34–36]. Bipotent precursor cells for eosinophils and basophils (CFU-eo/ba) are detectable in the BM and peripheral blood in healthy subjects as well as in patients suffering from allergic or other inflammatory reactions Citation[34–38]. The production of eosinophils from multipotent and lineage-restricted progenitors is tightly controlled by a network of transcription factors (GATA-1, PU.1, c/EBPs and others) Citation[39], growth factors and growth-inhibitory cytokines. Major growth factors for eosinophils are IL-5, granulocyte/macrophage colony-stimulating factor (GM-CSF) and IL-3 Citation[40–42]. These growth regulators are produced and secreted by (activated) immune cells such as T cells and mast cells, stromal cells and by eosinophils themselves Citation[43–47]. Cell surface receptors for IL-3, IL-5 and GM-CSF are detectable on immature multipotent eosinophil progenitor cells, eosinophil-committed precursors and on mature eosinophils Citation[48–50]. In line with this observation, these cytokines trigger not only proliferation of eosinophil progenitor cells, but also migration, adherence, cytokine production, activation and survival (by delayed apoptosis) of mature eosinophils & ) Citation[48–51]. The mobilization of eosinophils from the BM into the blood is regulated predominantly by IL-5 and eotaxin.
In addition to classical eosinophilic growth factors, other cytokines such as platelet-derived growth factor or NGF may play a role in the regulation of eosinophil development and function Citation[52,53]. Although constitutively activated receptors for these cytokines may be involved in the development of HE, the effects of platelet-derived growth factor and NGF on eosinophil growth and function remain largely unknown Other cytokines and chemokine ligands (CCL), such as IL-16, vascular endothelial growth factor, CCL5 (RANTES), CCL11 (eotaxin), CCL24 (eotaxin-2), CCL26 (eotaxin-3) and platelet-activating factor, induce eosinophil migration and chemotaxis Citation[54–68]. Most of these peptide mediators also trigger eosinophil activation, and some of them alter the adhesive properties of eosinophils . Eosinophil-activating cytokines, such as IL-3 or IL-5, have also been described to enhance responsiveness of eosinophils to other peptides and cytokines Citation[69]. Another related observation is that eosinophils isolated from inflamed tissue show enhanced migratory responses to various ligands compared with eosinophils in healthy tissue, and thus are in an activated (primed) state Citation[70].
Finally, growth, survival and function of blood eosinophils are controlled by various ‘negative-regulators’ such as Siglec-8 and other cell surface inhibitory receptors Citation[71,72]. It has also been described that TGF-β, IFN-α and IFN-γ suppress cytokine-induced growth and differentiation of human eosinophils in vitro Citation[73–75]. In addition, TGF-β induces apoptosis in mature eosinophils Citation[76]. Moreover, such soluble ‘negative-regulators’, like IFN-γ, reportedly counteract cytokine-induced migration of eosinophils Citation[77]. All of these cytokine effects are considered to be mediated via specific receptors expressed on eosinophils. Eosinophils also possess receptors for glucocorticosteroids (GC), which inhibit eosinophil growth and function, and the number of GC receptors detectable in eosinophils correlates with the responses of these cells to GCs Citation[78,79].
Expression of pathogenetically important cell surface receptors & other target antigens on eosinophils
Adhesion, migration, activation and survival of eosinophils are regulated by a network of cytokines, chemokines, other biologically active ligands and their receptors . Indeed, eosinophils express a unique composition of cell surface receptors relevant to adhesion, homing and migration Citation[58,59,61–65,80–82]. Several of these receptors are involved in the transmigration of eosinophils through endothelial monolayers Citation[82–93], and thus in homing to tissues, which is critical for the development of a local eosinophil infiltrate typically seen in patients with HE-related organ damage . Eosinophils have also been reported to express L-selectin as well as E- and P-selectin ligands, various integrins, sialyl Lewis x, intercellular-adhesion molecule 1, the C3bi receptor (CD11b/CD18) and leukocyte-function antigen-1 (CD11a/CD18) Citation[83–95]. In addition, eosinophils express leukosialin (CD43) and the leukocyte invasion receptor CD44. Whereas selectins are considered to mediate rolling and tethering of eosinophils on endothelial cell membranes and thus marginalization of eosinophils in the blood stream, integrins and other adhesion receptors mediate firm binding of eosinophils to endothelium before transmigration.
A number of proinflammatory cytokines and peptides promote the expression of adhesion molecules on eosinophils. Proinflammatory cytokines, such as IL-1, also promote the expression of ‘leukocyte-binding’ adhesion molecules on endothelial cells Citation[85–97]. In line with this observation, eosinophils derived from inflamed tissues express higher levels of certain cell surface- adhesion receptors. Conversely, GCs and other anti-inflammatory agents inhibit cytokine-induced expression of adhesion molecules on eosinophils and endothelial cells and thus eosinophil adhesion and transendothelial migration Citation[96,97].
Eosinophils also express a number of other biologically active cell surface molecules, including complement receptors, Toll-like receptors, Fc receptors, gangliosides and various structural glycoproteins Citation[79–83,98]. In addition, eosinophils express various lineage-restricted myeloid (target) antigens, such as Siglec-3 (CD33), and inhibitory receptors, including CD300a, EP4, SIRPα/CD172a and Siglec-8 Citation[71,99,100]. CD300a and Siglec-8 are also expressed on basophils and mast cells, but are not expressed on other leukocytes Citation[100,101]. Crosslinking of Siglec-8 on eosinophils is associated with apoptosis Citation[102,103], whereas crosslinking of other inhibitory receptors described to date appears to downmodulate eosinophil responses to activation signals.
Altogether, a wide variety of surface molecules, including cytokine receptors, adhesion receptors, peptide (chemokine) receptors and siglec molecules, have been considered as potential targets of therapy in eosinophil-related disorders Citation[101–106]. Whether such receptors can indeed be used as targets of drug therapy, and whether suitable targeted drugs can be developed, remains to be determined in preclinical studies and future clinical trials.
Eosinophil-derived mediators & their potential role in the pathogenesis of eosinophil-induced organ damage
Eosinophils are a rich source of proinflammatory mediators and cytokines, including hematopoietic growth factors, chemotactic and leukocyte-activating peptides, vasoactive mediators and profibrotic and/or angiogenic molecules Citation[107–113]. Eosinophils also synthesize various lipid mediators, including cysteinyl leukotrienes and prostaglandins, which play a role in eosinophil biology, as eosinophils serve as de novo producers and target cells of these compounds Citation[2,5,6]. In addition, eosinophils express several, more or less cell-specific basic proteins, including eosinophil cationic protein, eosinophil major basic proteins (MBP1 and MBP2), eosinophil peroxidase (EPO) and eosinophil-derived neurotoxin (EDN) Citation[109,113–116]. The eosinophil granule proteins possess numerous biological properties, including direct toxicity to cells and microorganisms and the ability to activate cells and platelets.
With regard to HE-related organ damage, little is known about the pathogenetic role of eosinophil-derived mediators and cytokines in specific disease states. Based on known biological activities, several eosinophil-derived mediators and cytokines may contribute to local inflammation and recruitment of other leukocytes . Other eosinophil-derived compounds may display cytotoxic properties in local tissue sites, assist in microbe killing (basic proteins, extracellular DNA traps and others) Citation[117–119], counteract or degrade vasoactive molecules such as histamine (by eosinophil-derived histaminase), regulate lymphocyte function Citation[110–113,120] or facilitate the development of fibrosis or thrombosis Citation[107–113]. Of note, eosinophil products have been shown to promote fibrosis and thrombosis both by activating (and possibly damaging) endothelial cells and/or platelets and through anti-fibrinolytic or ‘prothrombotic’ actions mediated by expression and release of plasminogen activator inhibitor-2 Citation[121] and other compounds. In fact, activated eosinophils and neoplastic eosinophils are a particularly rich source of proinflammatory, angiogenic and fibrogenic cytokines Citation[45,107–114]. These eosinophil-derived mediators and cytokines may all act together to cause tissue damage in patients with HE.
Definition & classification of HE & HE-related organ damage (hypereosinophilic syndromes)
The normal eosinophil count in the peripheral blood ranges from 50 to 500 × 109/l. Blood eosinophilia can be divided into mild eosinophilia (up to 1500 × 109/l) and marked eosinophilia (>1500 × 109/l). The term HE should be used when marked blood eosinophilia has been documented and is persistent, with or without an additional marked tissue eosinophilia Citation[33]. The defining features of ‘tissue HE’ have not been well characterized. Physiologically, substantial numbers of eosinophils are observed in the BM and in lymphatic organs as well as in the mucosal linings of the intestinal tract, especially the stomach, small intestine and colon Citation[122]. In most other tissues and organs, however, even low numbers of eosinophils can be regarded as ‘eosinophilia’. True tissue HE is thus characterized by a local marked increase in eosinophils and/or marked deposition of eosinophil-derived proteins such as MBP Citation[122–124]. Sometimes, the deposition of MBP is a predominant finding even in the absence of marked eosinophil accumulation Citation[122–124]. Based on underlying conditions and etiology, several variant forms of HE exist for which the following terminology has recently been proposed Citation[33]: a hereditary (familial) form (HEF), HE of undetermined (clinical) significance (HEUS), primary (neoplastic) HE (HEN) where eosinophils are considered to be clonal cells and secondary (reactive) HE (HER) where eosinophils are considered to be nonclonal cells expanded (and activated) by a reactive process Citation[33]. Rarely, HER is triggered by a clonal process such as Hodgkin’s lymphoma or lung adenocarcinoma. In most cases, HER may be caused by eosinophilopoietic cytokines such as IL-5.
It is important to note that some patients with unexplained HE are asymptomatic at presentation and may not develop clinical manifestations for many years. These patients would initially be classified as HEUS. In patients with HEUS, no underlying (neoplastic or non-neoplastic) disease and no organ damage (or dysfunction) is detected. Some of these patients may develop signs and symptoms of eosinophil-induced organ damage in the follow-up. When organ damage accompanies HE (of any type), the term ‘hypereosinophilic syndrome’ (HES) is appropriate, provided that organ involvement is clinically relevant and can be attributed to HE Citation[33]. This formulation closely matches to the traditional definition of HES Citation[125–128] and remains a useful concept in clinical practice.
Similar to HE, HES should be divided into variants based on the underlying etiology Citation[33]: idiopathic HES (with unknown etiology), primary (neoplastic) HES with an underlying clonal myeloid or stem cell disorder and secondary (reactive) HES where an underlying non-neoplastic or paraneoplastic condition is detected and is responsible for the expansion of nonclonal eosinophils. The lymphoid variant of HES (HE) was recognized as special situation. Although diagnostic criteria are not well established, this condition exhibits several features of reactive HES. Typically, lymphocytes from patients with lymphoid-variant HES show an aberrant phenotype, most often CD3−/CD4+, by flow cytometry. Monoclonality of T cells as demonstrated by a positive (monoclonal) T-cell receptor rearrangement may also be detected. The clinical course tends to be indolent, with rare cases of evolution to T-cell lymphoma.
Although several different organ systems may be affected in HES, organ damage is typically restricted to certain organ systems, including the heart, lungs, skin, spleen, GI tract and central nervous system Citation[125–129]. Endomyocardial fibrosis, development of intracavitary thrombi and occurrence of intravascular thrombosis represent serious cardiovascular complications of HE Citation[123,129–135]. Endomyocardial fibrosis and thrombus formation are frequently seen in (untreated) patients with eosinophilic leukemias carrying the FIP1L1-PDGFRA fusion gene, but may also develop in other patients with HES. Patients with FIP1L1-PDGFRA+ eosinophilic leukemia usually respond well to imatinib, and early treatment prevents irreversible organ damage in most patients Citation[25–27,136–138]. Therefore, it is of great importance to start staging and genetic investigations early in order to document or exclude overt or imminent organ damage and to identify patients with a myeloid neoplasm likely to respond to imatinib therapy.
Approach to the patient with HE & staging investigations
A detailed medical history should be elicited from all patients with HE, including complete drug/medication and travel histories. Testing for potential helminth infections, including stool examination and serologies, should be guided by the exposure history. Nonessential drugs should be discontinued, and the chronology of events between exposure and the development of HE should be determined for the remaining medications. If no inciting drug or infection is identified, a thorough investigation for allergic/atopic or autoimmune disorders, blood cell disorders and other neoplastic conditions should be initiated Citation[139,140]. This is usually performed in a stepwise approach. Important initial parameters that can be obtained from routine laboratory testing include a complete blood count with differential counts (confirmed by microscopy), routine chemistries (including tests of hepatic and renal function), levels of inflammatory markers and autoantibodies, serum IgE, vitamin B12 and tryptase levels, and a molecular test for FIP1L1-PDGFRA. BM investigation is warranted in all patients in whom HE remains unexplained or if a hematopoietic neoplasm is suspected. In patients with eosinophilic leukemia or other myeloid neoplasms, eosinophils are often (not always) immature and may display atypical morphology on Wright-Giemsa or May-Grunwald-Giemsa stained aspirate smears . Eosinophil granule abnormalities, including reduced granule size, leading to hypodense eosinophils, have also been described in patients with HEN Citation[141]. In some patients, the BM smear shows an increase in blast cells. However, HEN can also occur with morphologically normal eosinophils, and in most patients, blast counts are normal at diagnosis.
BM investigations should also include a core biopsy with histology and immunohistochemistry . Ideally, an immunohistochemical marker panel including CD34, CD117, tryptase and CD25 should be applied Citation[142]. In addition, cytogenetics, FISH and molecular analyses (seeking PDGFRA-, PDGFRB and FGFR1 fusion genes, BCR-ABL, JAK2 V617F, KIT D816V as well as clonal TcR rearrangement) should be performed Citation[23,33]. Many, but not all, of the molecular analyses are equally sensitive using peripheral blood cells. BM is usually required for cytogenetic examinations, although studies on peripheral blood can be successful if precursor cells circulate in the peripheral blood, which can be the case in HEN and HESN. Finally, lymphocyte (T cell) phenotyping (by flow cytometry) should be performed in patients with HE to identify aberrant populations, most commonly CD3−/CD4+ T cells, which have been associated with eosinophilopoietic cytokine production Citation[143,144].
Concomitant with the evaluation described above, directed at identifying the etiology of the eosinophilia and depending in part on the signs and symptoms in the individual patient, the extent of eosinophil-mediated organ damage should be assessed. In all patients, these investigations should include a physical examination with thorough skin examination (by a dermatologist), detailed cardiologic evaluations including assays of serum troponins and an echocardiogram (and MRI if echocardiographic findings are difficult to interpret), assessment of pulmonary function, chest x-ray, abdominal imaging, and gastrointestinal examinations (with endoscopy if indicated).
Differential diagnoses, underlying disorders & diagnostic algorithm
As mentioned above, a number of different underlying conditions and disorders may induce HE with or without organ damage (HES). It is important to search for such disorders in patients with unexplained HE. When no underlying disease, family history, organ damage or other related disease (rare syndrome associated with eosinophilia) is discovered, the (provisional) diagnosis is HEUS Citation[33].
A number of reactive conditions and disorders can cause HE and may produce a clinical picture resembling HES (Box 1). Reactive hypereosinophilia (HER) may develop in the setting of an underlying parasitic infection, drug allergy or neoplasm. Solid tumors, such as adenocarcinomas occurring in the lung, cervix or digestive tract, may produce paraneoplastic HE as a result of ectopic production of IL-5 or other cytokines by malignant cells. Similarly, there are hematological malignancies where eosinophilia is reactive, such as in Hodgkin’s lymphoma, T-cell lymphomas or B-cell acute lymphoblastic leukemia. On the other hand, HE may also develop as a clonal (neoplastic) process (HEN) in the setting of a hematopoietic malignancy .
Hematopoietic neoplasms that typically (but not always) present with HE include Ph+ chronic myeloid leukemia, certain variants of AML and some forms of advanced SM (Box 1) Citation[12,13,23,24,33]. In addition, eosinophilia (but rarely HE) may occur in patients with MDS, Ph- MPN and MDS/MPN overlap disorders. All of these malignancies should be classified according to WHO criteria Citation[21,30–32]. The WHO classification of hematopoietic neoplasms has also defined specific categories (subvariants) for eosinophilic leukemias and other neoplasms in which abnormalities (mutant forms) of PDGFRA, PDGFRB or FGFR1 are detected Citation[31,32]. However, although clearly relevant as driver mutants and potential therapeutic targets, it remains uncertain whether these fusion gene products can be recommended as major criteria of eosinophil-related neoplasms, or whether they should rather serve as minor or cocriteria in a revised classification of eosinophil neoplasms Citation[23,33]. In fact, unlike chronic myeloid leukemia, where BCR/ABLp210 is almost invariably associated with the disease, abnormalities in PDGFRA, PDGFRB or FGFR1 may alternatively be found in a wide variety of myelogenous malignancies, including MPN, MDS/MPN overlap neoplasms and acute leukemias, often without HE Citation[31–33].
The WHO classification has also included the category ‘chronic eosinophilic leukemia – not otherwise specified’ (CEL NOS) Citation[30,31]. Again, the current recommendation is to diagnose CEL NOS according to this classification and the proposed WHO criteria Citation[31]. However, for the future, it may be advisable to revise this classification, and to adjust these criteria on the basis of novel markers and histomorphological criteria Citation[33].
Finally, in other hematopoietic neoplasms, such as advanced mastocytosis, acute leukemias or stem cell-derived neoplasms, eosinophils may represent a mixture of clonal and nonclonal cells. The dilemma is that it is impossible to confirm (or exclude) clonality of eosinophils in these patients in daily practice.
Additional rare syndromes with specific organ pathologies can produce HE. These syndromes have been separated from the classical variants of HES because, although it is tempting to surmise that abundant eosinophils contribute to pathogenesis, the exact relationships between HE and the observed pathologies are often uncertain. It is important to be aware of these potential diagnoses when approaching a patient with HE. These disorders include, among others, episodic angioedema and eosinophilia (Gleich’s syndrome), Churg–Strauss syndrome (CSS), Eosinophilia Myalgia syndrome, Omenn syndrome and the Hyper-IgE syndrome . Gleich’s syndrome is characterized by cyclic, recurrent angioedema, associated with peripheral blood HE and elevated polyclonal IgM Citation[145,146]. In some cases, lymphocyte phenotyping reveals abnormal (activated) T-cell subsets (CD4+ T cells with decreased or absent expression of surface CD3). Therefore, Gleich’s syndrome is also regarded as a special manifestation of the lymphoid variant of HES Citation[140,144,147]. CSS is characterized by necrotizing vasculitis often accompanied by asthma and eosinophilia Citation[148–150]. Antineutrophil cytoplasmic antibodies are detectable in a subset (less than 50%) of CSS patients Citation[148–151]. Eosinophilia Myalgia syndrome is defined by severe myalgia and is often accompanied by neurologic symptoms and skin changes. Epidemic cases were associated with l-tryptophan exposure and with the toxic oil syndrome Citation[151–153]. Omenn syndrome and the Hyper-IgE syndrome are rare inherited immunodeficiency syndromes accompanied by eosinophilia . There are other clinical conditions and syndromes characterized by the presence of eosinophilia, sometimes resembling HES. In many (if not most) of these conditions, eosinophilia is likely triggered by aberrant lymphocyte production of eosinophilopoietic cytokines such as IL-5 Citation[143,144].
Management & therapy of patients with HE & HE-related disorders
In patients with HEUS and HEF, it seems appropriate to follow-up the patient without treatment provided that there are no signs or symptoms of eosinophil-related organ damage despite careful medical monitoring. Notably, both HEUS and HEF must be regarded as provisional diagnoses, since in both conditions, organ damage may develop over time, or a hematologic or other underlying disease may eventually be detected.
The reactive form of HE is best managed by treating the underlying disease. In many cases, symptomatic therapy is sufficient to control problems related to eosinophil activation in these patients. However, it is not appropriate to treat HE per se in these patients, such as when no eosinophil-related organ damage (HES) is present. If the underlying disease is accompanied by HES and cannot be managed with or is resistant to conventional (symptomatic) therapy, the eosinophil count can usually be suppressed with glucocorticosteroids Citation[140,154–157]. These drugs act directly and indirectly to reduce eosinophilia by inducing apoptosis in eosinophils and eosinophil precursors, and by inhibiting production of eosinophilopoietic cytokines by T lymphocytes .
For patients presenting with HES in the absence of a detectable underlying cause (so-called idiopathic HES) or with lymphoid variant HES, glucocorticoids are also indicated, but side effects and poor tolerance are often dose-limiting. The most commonly used corticosteroid-sparing agents in this setting are hydroxyurea and IFN-α Citation[140,154–157]. Mepolizumab, an anti-IL-5 antibody, has also been shown to be a safe and effective steroid-sparing agent for patients with FIP1L1/PDGFRA-negative HES (idiopathic and lymphoid variants) but is not commercially available Citation[158–161].
In contrast to patients with idiopathic HES and reactive HES, those with HES in the setting of a clonal (neoplastic) BM disease (HESN) do not typically respond to glucocorticoid therapy. Although IFN-α and hydroxyurea have been used with some success, the mortality in this subgroup of patients with HES remained high until the recent development of therapeutic agents targeting specific molecular defects in CEL and other hematopoietic neoplasms with eosinophilia. Notably, a number of molecular targets related to CEL or other hematopoietic malignancies presenting with eosinophilia have been identified in the past decade Citation[13,18–24,162–165]. It is essential to define any molecular defects (and thereby therapeutic targets), the exact histomorphological diagnosis and extent of organ involvement (organ damage) by appropriate staging investigations in order to develop an optimal treatment plan.
The most frequent fusion oncoprotein and target detectable in patients with CEL is FIP1L1/PDGFRA Citation[18–24]. This fusion gene product is a target of imatinib and also sensitive to other tyrosine kinase inhibitors (TKIs) such as nilotinib or dasatinib Citation[18–24, 166–168]. A number of other fusion gene-related oncoproteins that serve as targets of imatinib have also been described (less commonly) in eosinophil disorders Citation[18–24]. Most of these fusion genes involve PDGFRA or PDGFRB, and are sensitive to imatinib, whereas FGFR1 mutants are resistant Citation[25–28].
Based on clinical trials, imatinib is considered standard firstline therapy in patients with FIP1L1/PDGFRA+ eosinophilia Citation[25–27,137,169,170]. Although most patients show a long-lasting response to a standard start dose of 100 mg daily or less, some patients require a dose of 400 mg daily Citation[25–27,137,169,170]. Very few patients may develop resistance, which is mainly caused by rare FIP1L1/PDGFRA point mutations associated with decreased (or loss of) drug-binding capacity Citation[26,170–172]. For these patients, novel TKIs such as sorafenib, nilotinib or midostaurin may be considered (in clinical trials) Citation[173–176]. Another approach is to apply alternative cytoreductive agents, such as IFN-α, hydroxyurea or experimental drugs, including new antileukemic, targeted small-molecule-type drugs or targeted antibodies Citation[177,178]. In patients who progress to acute leukemia, high-dose chemotherapy and hematopoietic stem cell transplantation must be considered.
Rarely, the FIP1L1/PDGFRA fusion gene is detected in untreated patients with a longstanding apparently stable course of HE without evidence of organ damage. Although a polymorphism in the IL-5RA gene has been associated with disease severity Citation[179], there are as yet no predictive markers that can define the exact risk of progression or development of organ damage in these patients. Moreover, imatinib is a very well-tolerated drug without documented long-term side effects or additional mutagenic effects of the drug in leukemic patients (except in pregnant patients). Therefore, the current recommendation is to treat even asymptomatic patients with CEL early with imatinib to prevent the development of irreversible complications, should disease progression occur.
In patients with FGFR1 mutants, imatinib is not effective and thus is not recommended. In many of these patients, the condition behaves as an aggressive stem cell disease or a leukemia/lymphoma syndrome (also known as 8p11 syndrome). For these patients, chemotherapy plus allogeneic stem cell transplantation or novel alternative targeted drugs Citation[180–182] are potential treatment options. In other patients with myeloid neoplasms associated with HE, the treatment plan should be based on the underlying disease and the availability of relevant therapeutic agents.
Concluding remarks & expert commentary
HE is an important diagnostic checkpoint in various hematologic and nonhematologic disorders. In patients with HE, it is important to document or exclude an underlying neoplastic or non-neoplastic disease and to document or exclude the presence of definitive or imminent HE-related organ damage (HES). Several immunological, serological and molecular/cytogenetic markers are available to delineate variants of HE, HES and underlying conditions. Independent of the underlying disease, patients with overt or imminent organ damage (HES) are candidates for early therapeutic intervention. In those with secondary HES, treatment of the underlying disease is usually effective. In some patients with HES, IL-5-targeting antibodies may work. In patients who have mutated variants of PDGFRA or PDGFRB, imatinib or other PDGFR-targeting tyrosine kinase inhibitors are usually effective. Our increasing knowledge about eosinophilia, the development of more specific predictive biomarkers, markers of eosinophil clonality and gene chip profiling-based evaluations, as well as the development of new therapeutic approaches should improve diagnosis and prognosis of eosinophil disorders in the future.
Five-year view
During the next 5 years, in addition to recently identified markers, novel markers that distinguish varied eosinophil diseases and their therapeutic targets will be identified. Using such new parameters and molecular targets, the proposed definitions, criteria and classification of eosinophil diseases may be updated and refined. With regard to the diversity of eosinophilic diseases, we contribute our collective classification proposals and their bases. We would welcome the WHO in creating a new multidisciplinary consensus classification of eosinophilic diseases that includes clinical, histomorphological, cytogenetic and molecular parameters.
Table 1. Gene abnormalities and cytogenetic defects in myeloid and stem cell neoplasms associated with eosinophilia.
Table 2. Cytokines, peptides and receptors regulating growth and function of eosinophils.
Table 3. Major eosinophil products and their potential role in the development of hypereosinophilia.
Table 4. Examples of rare syndromes accompanied by eosinophilia.
Table 5. Selected treatment options in patients with eosinophil disorders.
Box 1. Major causes of hypereosinophilia.
• Non-neoplastic reactive conditions (secondary/reactive HE)Footnote†
– Helminth infections
– Scabies, other infestations
– Allergic bronchopulmonary aspergillosis
– Drug reactions (allergic or toxic)
– Other allergic reactions
– Atopic diseases
– Chronic graft-versus-host disease
– Chronic inflammatory disorders (e.g., IBD)
– Autoimmune diseases
– L-HES
• Neoplastic conditions with secondary/reactive HE (paraneoplastic)Footnote†
– Hodgkin´s disease
– B- or T-cell lymphoma/leukemia
– Langerhans cell histiocytosis
– Solid tumors/malignancy
• Myeloid neoplasms and stem cell neoplasms (primary HE)Footnote‡
– Chronic eosinophilic leukemia – NOS
– Hematopoietic neoplasms with eosinophilia and abnormalities in PDGFRA
– Hematopoietic neoplasms with eosinophilia and abnormalities in PDGFRB
– Hematopoietic neoplasms with eosinophilia and abnormalities in FGFR1
– CML with eosinophilia
– AML with inv(16) and eosinophilia (AML-M4-eo)
– JAK2 V617F+ MPN with eosinophilia
– ASM with eosinophiliaFootnote§
– MDS with eosinophilia
– MPN/MDS overlap syndromes with eosinophilia
• Idiopathic: HEUS
Key issues
• Eosinophils represent a distinct hematopoietic cell lineage with unique features. Chronic (hyper)eosinophilia may be accompanied by a characteristic pattern of organ damage.
• Hypereosinophilia (HE) is an important checkpoint in the diagnostic algorithm.
• Independent of the underlying disease, HE-associated organ damage is called hypereosinophilic syndrome (HES).
• Depending on the underlying disease, HE and HES can be further subclassified.
• In hematologic neoplasms with HE, eosinophils are usually clonal cells, although direct proof of eosinophil clonality is rarely obtained.
• Close clinical follow-up of HE of undetermined significance (HEUS) is necessary to monitor for development of HES and/or an underlying disease.
• Treatment of HES depends on the underlying disease and on the presence of identified therapeutic targets.
• Complete staging of all organ systems, including the skin, lung, heart, bone marrow and blood and GI tract, is essential in the evaluation of all patients with unexplained HE.
• Patients with myeloid or stem cell neoplasms and eosinophila caused by abnormalities in PDGFRA or PDGFRB should be treated with imatinib.
Financial & competing interests disclosure
P Valent has received a Research Grant from Novartis and a Research Grant from BMS, as well as honoraria from Novartis and BMS. AD Klein is supported by the Intramural Program of the NIAID, NIH. KM Leiferman, HU Simon and GJ Gleich have been supported by grants from GSK. M Arock received honorarium from Novartis. P Vandenberghe is a Senior Clinical Investigator of FWO-Vlaanderen. T Haferlach is part owner of the Munich Leukemia Laboratory. A Reiter received Honoraria from Novartis and BMS. BS Bochner is a co-author on existing and pending Siglec-8-related patents. If Siglec-8-related products are developed in the future, BS Bochner may be entitled to a share of royalties received by Johns Hopkins University on the potential sales of such products. The terms of this arrangement are being managed by Johns Hopkins University in accordance with its conflict of interest policies. This study was supported by a research grant of the Medical University of Vienna and by the Division of Intramural Research, NIAID, NIH, Bethesda, MD, USA. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Notes
† In most cases, eosinophilia may be triggered by eosinopoietic cytokines.
‡ In most patients, eosinophils are clonal cells (neoplastic eosinophils).
§ Eosinophilia often develops in patients with ASM, but may also occur in patients with indolent systemic mastocytosis.
AML: Acute myeloid leukemia; ASM: Aggressive systemic mastocytosis; CML: Chronic myeloid leukemia; HE: Hypereosinophilia; HEUS: Hypereosinophilia of undetermined (unknown) significance; IBD: Inflammatory bowel disease; L-HES: Lymphoid variant of hypereosinophilic syndrome; MDS: Myelodysplastic syndrome; MPN: Myeloproliferative neoplasm; NOS: Not otherwise specified.
References
- Gleich GJ, Adolphson CR, Leiferman KM. The biology of the eosinophilic leukocyte. Annu. Rev. Med. 44, 85–101 (1993).
- Weller PF. Eosinophils: structure and functions. Curr. Opin. Immunol. 6(1), 85–90 (1994).
- Fulkerson PC, Rothenberg ME. Origin, regulation and physiological function of intestinal eosinophils. Best. Pract. Res. Clin. Gastroenterol. 22(3), 411–423 (2008).
- Kita H. Eosinophils: multifaceted biological properties and roles in health and disease. Immunol. Rev. 242(1), 161–177 (2011).
- Gleich GJ. Mechanisms of eosinophil-associated inflammation. J. Allergy Clin. Immunol. 105(4), 651–663 (2000).
- Hogan SP, Rosenberg HF, Moqbel R et al. Eosinophils: biological properties and role in health and disease. Clin. Exp. Allergy 38(5), 709–750 (2008).
- Ackerman SJ, Bochner BS. Mechanisms of eosinophilia in the pathogenesis of hypereosinophilic disorders. Immunol. Allergy Clin. North Am. 27(3), 357–375 (2007).
- Ogbogu PU, Rosing DR, Horne MK 3rd. Cardiovascular manifestations of hypereosinophilic syndromes. Immunol. Allergy Clin. North Am. 27(3), 457–475 (2007).
- Kay AB. The eosinophil in infection diseases. J. Infect. Dis. 129(5), 606–613 (1974).
- Capron M. Eosinophils and parasites. Ann. Parasitol. Hum. Comp. 66(S1), 41–45 (1991).
- Walsh SA, Creamer D. Drug reaction with eosinophilia and systemic symptoms (DRESS): a clinical update and review of current thinking. Clin. Exp. Dermatol. 36(1), 6–11 (2011).
- Kargili A, Bavbek N, Kaya A, Koşar A, Karaaslan Y. Eosinophilia in rheumatologic diseases: a prospective study of 1000 cases. Rheumatol. Int. 24(6), 321–324 (2004).
- Tefferi A, Patnaik MM, Pardanani A. Eosinophilia: secondary, clonal and idiopathic. Br. J. Haematol. 133(5), 468–492 (2006).
- Sade K, Mysels A, Levo Y, Kivity S. Eosinophilia: a study of 100 hospitalized patients. Eur. J. Intern. Med. 18(3), 196–201 (2007).
- Lotfi R, Lee JJ, Lotze MT. Eosinophilic granulocytes and damage-associated molecular pattern molecules (DAMPs): role in the inflammatory response within tumors. J. Immunother. 30(1), 16–28 (2007).
- Nutman TB. Evaluation and differential diagnosis of marked, persistent eosinophilia. Immunol. Allergy Clin. North Am. 27(3), 529–549 (2007).
- Simon D, Simon HU. Eosinophilic disorders. J. Allergy Clin. Immunol. 119(6), 1291–1300 (2007).
- Tefferi A. Modern diagnosis and treatment of primary eosinophilia. Acta Haematol. 114(1), 52–60 (2005).
- Gotlib J, Cross NC, Gilliland DG. Eosinophilic disorders: molecular pathogenesis, new classification, and modern therapy. Best Pract. Res. Clin. Haematol. 19(3), 535–569 (2006).
- Bain BJ, Fletcher SH. Chronic eosinophilic leukemias and the myeloproliferative variant of the hypereosinophilic syndrome. Immunol. Allergy Clin. North Am. 27(3), 377–388 (2007).
- Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 22(1), 14–22 (2008).
- Haferlach T, Bacher U, Kern W, Schnittger S, Haferlach C. The diagnosis of BCR/ABL-negative chronic myeloproliferative diseases (CMPD): a comprehensive approach based on morphology, cytogenetics, and molecular markers. Ann. Hematol. 87(1), 1–10 (2008).
- Valent P. Pathogenesis, classification, and therapy of eosinophilia and eosinophil disorders. Blood Rev. 23(4), 157–165 (2009).
- Bain BJ. Review: eosinophils and eosinophilic leukemia. Clin. Adv. Hematol. Oncol. 8(12), 901–903 (2010).
- Apperley JF, Gardembas M, Melo JV et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor β. N. Engl. J. Med. 347(7), 481–487 (2002).
- Cools J, DeAngelo DJ, Gotlib J et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N. Engl. J. Med. 348(13), 1201–1214 (2003).
- Pardanani A, Reeder T, Porrata LF et al. Imatinib therapy for hypereosinophilic syndrome and other eosinophilic disorders. Blood 101(9), 3391–3397 (2003).
- Cross NC, Reiter A. Fibroblast growth factor receptor and platelet-derived growth factor receptor abnormalities in eosinophilic myeloproliferative disorders. Acta Haematol. 119(4), 199–206 (2008).
- Simon HU, Rothenberg ME, Bochner BS et al. Refining the definition of hypereosinophilic syndrome. J. Allergy Clin. Immunol. 126(1), 45–49 (1010).
- Bain BJ, Pierre R, Imbert M, Vardiman JW, Brunning RD, Flandrin G. Chronic eosinophilic leukemia and the hypereosinophilic syndrome. In: World Health Organization (WHO) Classification of Tumours. Pathology & Genetics. Tumours of Haematopoietic and Lymphoid Tissues (Volume 1). Jaffe ES, Harris NL, Stein H, Vardiman JW (Eds). IARC Press, Lyon, France, 29–31 (2001).
- Bain BJ, Gilliland DG, Vardiman JW, Horny HP. Chronic eosinophilic leukemia, not otherwise specified. In: World Health Organization (WHO) Classification of Tumours. Pathology & Genetics. Tumours of Haematopoietic and Lymphoid Tissues (Volume 2). Swerdlow, SH, Campo E, Harris NL et al. (Eds). IARC Press, Lyon, France, 51–53 (2008).
- Bain BJ, Gilliland DG, Horny HP, Vardiman JW. Myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB or FGFR1. In: World Health Organization (WHO) Classification of Tumours. Pathology & Genetics. Tumours of Haematopoietic and Lymphoid Tissues (Volume 1). Swerdlow SH, Campo E, Harris NL et al. (Eds). IARC Press, Lyon, France, 68–73 (2008).
- Valent P, Klion A, Horny HP et al. Contemporary consensus on criteria and classification of eosinophil disorders and related syndromes. J. Allergy Clin. Immunol. (2012) (In Press).
- Leary AG, Ogawa M. Identification of pure and mixed basophil colonies in culture of human peripheral blood and marrow cells. Blood 64(1), 78–83 (1984).
- Denburg JA, Telizyn S, Messner H et al. Heterogeneity of human peripheral blood eosinophil-type colonies: evidence for a common basophil–eosinophil progenitor. Blood 66(2), 312–318 (1985).
- Shalit M, Sekhsaria S, Mauhorter S, Mahanti S, Malech HL. Early commitment to the eosinophil lineage by cultured human peripheral blood CD34+ cells: messenger RNA analysis. J. Allergy Clin. Immunol. 98(2), 344–354 (1996).
- Denburg JA. Hemopoietic progenitors and cytokines in allergic inflammation. Allergy 53(Suppl. 45), 22–26 (1998).
- Linden M, Svensson C, Andersson M et al. Circulating eosinophil/basophil progenitors and nasal mucosal cytokines in seasonal allergic rhinitis. Allergy 54(3), 212–219 (1999).
- McNagny K, Graf T. Making eosinophils through subtle shifts in transcription factor expression. J. Exp. Med. 195(11), F43–F47 (2002).
- Clutterbuck E, Shields JG, Gordon J et al. Recombinant human interleukin 5 is an eosinophil differentiation factor but has no activity in standard human B cell growth factor assays. Eur. J. Immunol. 17(12), 1743–1750 (1987).
- Saito H, Hatake K, Dvorak AM et al. Selective differentiation and proliferation of hematopoietic cells induced by recombinant human interleukins. Proc. Natl Acad. Sci. USA 85(7), 2288–2292 (1988).
- Valent P, Schmidt G, Besemer J et al. Interleukin-3 is a differentiation factor for human basophils. Blood 73(7), 1763–1769 (1989).
- Feldmann M, Londei M, Haworth C. T cells and lymphokines. Br. Med. Bull. 45(1), 361–370 (1989).
- Del Prete G. Human Th1 and Th2 lymphocytes: their role in the pathophysiology of atopy. Allergy 47(5), 450–455 (1992).
- Spencer LA, Szela CT, Perez SA et al. Human eosinophils constitutively express multiple Th1, Th2 and immunoregulatory cytokines that are secreted rapidly and differentially. J. Leukoc. Biol. 85(1), 117–123 (2009).
- Denburg JA. Microenvironmental influences on inflammatory cell differentiation. Allergy 50(S25), 25–28 (1995).
- Shakoory B, Fitzgerald SM, Lee SA, Chi DS, Krishnaswamy G. The role of human mast cell-derived cytokines in eosinophil biology. J. Interferon Cytokine Res. 24(5), 271–281 (2004).
- Lopez AF, Vadas MA, Woodcock JM et al. Interleukin-5, interleukin-3, and granulocyte–macrophage colony-stimulating factor cross-compete for binding to cell surface receptors on human eosinophils. J. Biol. Chem. 266(36), 24741–24747 (1991).
- Lopez AF, Elliott MJ, Woodcock J, Vadas MA. GM-CSF, IL-3 and IL-5: cross-competition on human haemopoietic cells. Immunol. Today 13(12), 495–500 (1992).
- Yoshimura-Uchiyama C, Yamaguchi M, Nagase H et al. Changing expression of IL-3 and IL-5 receptors in cultured human eosinophils. Biochem. Biophys. Res. Commun. 309(1), 26–31 (2003).
- Simon HU, Yousefi S, Schranz C, Schapowal A, Bachert C, Blaser K. Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J. Immunol. 158(8), 3902–3908 (1997).
- Bach MK, Brashler JR, Stout BK et al. Activation of human eosinophils by platelet-derived growth factor. Int. Arch. Allergy Immunol. 97(2), 121–129 (1992).
- Noga O, Englmann C, Hanf G, Grützkau A, Guhl S, Kunkel G. Activation of the specific neurotrophin receptors TrkA, TrkB and TrkC influences the function of eosinophils. Clin. Exp. Allergy 32(9), 1348–1354 (2002).
- Wang JM, Rambaldi A, Biondi A, Chen ZG, Sanderson CJ, Mantovani A. Recombinant human interleukin 5 is a selective eosinophil chemoattractant. Eur. J. Immunol. 19(4), 701–705 (1989).
- Rot A, Krieger M, Brunner T, Bischoff SC, Schall TJ, Dahinden CA. RANTES and macrophage inflammatory protein 1 α induce the migration and activation of normal human eosinophil granulocytes. J. Exp. Med. 176(6), 1489–1495 (1992).
- Dahinden CA, Geiser T, Brunner T et al. Monocyte chemotactic protein 3 is a most effective basophil- and eosinophil-activating chemokine. J. Exp. Med. 179(2), 751–756 (1994).
- Noso N, Proost P, van Damme J, Schröder JM. Human monocyte chemotactic proteins-2 and 3 (MCP-2 and MCP-3) attract human eosinophils and desensitize the chemotactic responses towards RANTES. Biochem. Biophys. Res. Commun. 200(3), 1470–1476 (1994).
- Ponath PD, Qin S, Ringler DJ et al. Cloning of the human eosinophil chemoattractant, eotaxin. Expression, receptor binding, and functional properties suggest a mechanism for the selective recruitment of eosinophils. J. Clin. Invest. 97(3), 604–612 (1996).
- Rothenberg ME, Ownbey R, Mehlhop PD et al. Eotaxin triggers eosinophil-selective chemotaxis and calcium flux via a distinct receptor and induces pulmonary eosinophilia in the presence of interleukin 5 in mice. Mol. Med. 2(3), 334–348 (1996).
- Okada S, Kita H, George TJ, Gleich GJ, Leiferman KM. Transmigration of eosinophils through basement membrane components in vitro: synergistic effects of platelet-activating factor and eosinophil-active cytokines. Am. J. Respir. Cell. Mol. Biol. 16(4), 455–463 (1997).
- Petering H, Götze O, Kimmig D, Smolarski R, Kapp A, Elsner J. The biologic role of interleukin-8: functional analysis and expression of CXCR1 and CXCR2 on human eosinophils. Blood 93(2), 694–702 (1999).
- Bochner BS, Bickel CA, Taylor ML et al. Macrophage-derived chemokine induces human eosinophil chemotaxis in a CC chemokine receptor 3- and CC chemokine receptor 4-independent manner. J. Allergy Clin. Immunol. 103(3), 527–532 (1999).
- White JR, Lee JM, Dede K et al. Identification of potent, selective non-peptide CC chemokine receptor-3 antagonist that inhibits eotaxin-, eotaxin-2-, and monocyte chemotactic protein-4-induced eosinophil migration. J. Biol. Chem. 275(47), 36626–36631 (2000).
- Menzies-Gow A, Ying S, Sabroe I et al. Eotaxin (CCL11) and eotaxin-2 (CCL24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early- and late-phase allergic reactions following cutaneous injection in human atopic and nonatopic volunteers. J. Immunol. 169(5), 2712–2718 (2002).
- Feistritzer C, Kaneider NC, Sturn DH et al. Expression and function of the vascular endothelial growth factor receptor FLT-1 in human eosinophils. Am. J. Respir. Cell. Mol. Biol. 30(5), 729–735 (2004).
- Ferland C, Flamand N, Davoine F, Chakir J, Laviolette M. IL-16 activates plasminogen-plasmin system and promotes human eosinophil migration into extracellular matrix via CCR3-chemokine-mediated signaling and by modulating CD4 eosinophil expression. J. Immunol. 173(7), 4417–4424 (2004).
- Feistritzer C, Mosheimer BA, Sturn DH, Bijuklic K, Patsch JR, Wiedermann CJ. Expression and function of the angiopoietin receptor Tie-2 in human eosinophils. J. Allergy Clin. Immunol. 114(5), 1077–1084 (2004).
- Kishimoto S, Oka S, Gokoh M, Sugiura T. Chemotaxis of human peripheral blood eosinophils to 2-arachidonoylglycerol: comparison with other eosinophil chemoattractants. Int. Arch. Allergy Immunol. 140(Suppl. 1), 3–7 (2006).
- Jung YJ, Woo SY, Jang MH et al. Human eosinophils show chemotaxis to lymphoid chemokines and exhibit antigen-presenting-cell-like properties upon stimulation with IFN-γ, IL-3 and GM-CSF. Int. Arch. Allergy Immunol. 146(3), 227–234 (2008).
- Bates ME, Sedgwick JB, Zhu Y et al. Human airway eosinophils respond to chemoattractants with greater eosinophil-derived neurotoxin release, adherence to fibronectin, and activation of the Ras-ERK pathway when compared with blood eosinophils. J. Immunol. 184(12), 7125–7133 (2010).
- Bochner BS. Siglec-8 on human eosinophils and mast cells, and Siglec-F on murine eosinophils, are functionally related inhibitory receptors. Clin. Exp. Allergy 39(3), 317–324 (2009).
- Munitz A, Levi-Schaffer F. Inhibitory receptors on eosinophils: a direct hit to a possible Achilles heel? J. Allergy Clin. Immunol. 119(6), 1382–1387 (2007).
- Sillaber C, Geissler K, Scherrer R et al. Type β transforming growth factors promote interleukin-3 (IL-3)-dependent differentiation of human basophils but inhibit IL-3-dependent differentiation of human eosinophils. Blood 80(3), 634–641 (1992).
- Atsuta J, Fujisawa T, Iguchi K, Terada A, Kamiya H, Sakurai M. Inhibitory effect of transforming growth factor β 1 on cytokine-enhanced eosinophil survival and degranulation. Int. Arch. Allergy Immunol. 108(Suppl. 1), 31–35 (1995).
- de Bruin AM, Buitenhuis M, van der Sluijs KF, van Gisbergen KP, Boon L, Nolte MA. Eosinophil differentiation in the bone marrow is inhibited by T cell-derived IFN-γ. Blood 116(14), 2559–2569 (2010).
- Alam R, Forsythe P, Stafford S, Fukuda Y. Transforming growth factor β abrogates the effects of hematopoietins on eosinophils and induces their apoptosis. J. Exp. Med. 179(3), 1041–1045 (1994).
- Park CS, Choi EN, Kim JS et al. Interferon-γ inhibits in vitro mobilization of eosinophils by interleukin-5. Int. Arch. Allergy Immunol. 136(3), 295–302 (2005).
- Peterson AP, Altman LC, Hill JS, Gosney K, Kadin ME. Glucocorticoid receptors in normal human eosinophils: comparison with neutrophils. J. Allergy Clin. Immunol. 68(3), 212–217 (1981).
- Prin L, Lefebvre P, Gruart V et al. Heterogeneity of human eosinophil glucocorticoid receptor expression in hypereosinophilic patients: absence of detectable receptor correlates with resistance to corticotherapy. Clin. Exp. Immunol. 78(3), 383–389 (1989).
- Valent P. The phenotype of human eosinophils, basophils, and mast cells. J. Allergy Clin. Immunol. 94(6), 1177–1183 (1994).
- Bochner BS. Systemic activation of basophils and eosinophils: markers and consequences. J. Allergy Clin. Immunol. 106(Suppl. 5), S292–S302 (2000).
- Tachimoto H, Bochner BS. The surface phenotype of human eosinophils. Chem. Immunol. 76, 45–62 (2000).
- Hartnell A, Moqbel R, Walsh GM, Bradley B, Kay AB. Fc γ and CD11/CD18 receptor expression on normal density and low density human eosinophils. Immunology 69(2), 264–270 (1990).
- Lamas AM, Mulroney CM, Schleimer RP. Studies on the adhesive interaction between purified human eosinophils and cultured vascular endothelial cells. J. Immunol. 140(5), 1500–1505 (1988).
- Bochner BS, Luscinskas FW, Gimbrone MA et al. Adhesion of human basophils, eosinophils, and neutrophils to interleukin 1-activated human vascular endothelial cells: contributions of endothelial cell adhesion molecules. J. Exp. Med. 173(6), 1553–1557 (1991).
- Ebisawa M, Bochner BS, Georas SN, Schleimer RP. Eosinophil transendothelial migration induced by cytokines. I. Role of endothelial and eosinophil adhesion molecules in IL-1 β-induced transendothelial migration. J. Immunol. 149(12), 4021–4028 (1992).
- Bochner BS, Schleimer RP. The role of adhesion molecules in human eosinophil and basophil recruitment. J. Allergy Clin. Immunol. 94(3), 427–438 (1994).
- Knol EF, Tackey F, Tedder TF et al. Comparison of human eosinophil and neutrophil adhesion to endothelial cells under nonstatic conditions. Role of L-selectin. J. Immunol. 153(5), 2161–2167 (1994).
- Wein M, Sterbinsky SA, Bickel CA, Schleimer RP, Bochner BS. Comparison of human eosinophil and neutrophil ligands for P-selectin: ligands for P-selectin differ from those for E-selectin. Am. J. Respir. Cell. Mol. Biol. 12(3), 315–319 (1995).
- Seminario MC, Bochner BS. Expression and function of β 1 integrins on human eosinophils. Mem. Inst. Oswaldo Cruz. 92(Suppl. 2), 157–164 (1997).
- Matsumoto K, Sterbinsky SA, Bickel CA, Zhou DF, Kovach NL, Bochner BS. Regulation of α 4 integrin-mediated adhesion of human eosinophils to fibronectin and vascular cell adhesion molecule-1. J. Allergy Clin. Immunol. 99(5), 648–656 (1997).
- Horie S, Okubo Y, Hossain M et al. Intercellular adhesion molecule-1 on eosinophils is involved in eosinophil protein X release induced by cytokines. Immunology 90(2), 301–307 (1997).
- Grayson MH, van der Vieren M, Sterbinsky SA et al. αβ2 integrin is expressed on human eosinophils and functions as an alternative ligand for vascular cell adhesion molecule 1 (VCAM-1). J. Exp. Med. 188(11), 2187–2191 (1998).
- Davenpeck KL, Berens KL, Dixon RA, Dupre B, Bochner BS. Inhibition of adhesion of human neutrophils and eosinophils to P-selectin by the sialyl Lewis antagonist TBC1269: preferential activity against neutrophil adhesion in vitro. J. Allergy Clin. Immunol. 105(4), 769–775 (2000).
- Bochner BS, Schleimer RP. Mast cells, basophils, and eosinophils: distinct but overlapping pathways for recruitment. Immunol. Rev. 179, 5–15 (2001).
- Schleimer RP, Bochner BS. The effects of glucocorticoids on human eosinophils. J. Allergy Clin. Immunol. 94(6), 1202–1213 (1994).
- Kaiser J, Bickel CA, Bochner BS, Schleimer RP. The effects of the potent glucocorticoid budesonide on adhesion of eosinophils to human vascular endothelial cells and on endothelial expression of adhesion molecules. J. Pharmacol. Exp. Ther. 267(1), 245–249 (1993).
- Nagase H, Okugawa S, Ota Y et al. Expression and function of Toll-like receptors in eosinophils: activation by Toll-like receptor 7 ligand. J. Immunol. 171(8), 3977–3982 (2003).
- Aizawa H, Plitt J, Bochner BS. Human eosinophils express two Siglec-8 splice variants. J. Allergy Clin. Immunol. 109(1), 176 (2002).
- Hudson SA, Herrmann H, Du J et al. Developmental, malignancy-related and cross-species analysis of eosinophil, mast cell and basophil Siglec-8 expression. J. Clin. Immunol. 31(6), 1045–1053 (2011).
- Kikly KK, Bochner BS, Freeman S et al. Identification of SAF-2, a novel siglec expressed on eosinophils, mast cells and basophils. J. Allergy Clin. Immunol. 105(6), 1093–1100 (2000).
- Hudson SA, Bovin NV, Schnaar RL, Crocker PR, Bochner BS. Eosinophil-selective binding and proapoptotic effect in vitro of a synthetic Siglec-8 ligand, polymeric 6’-sulfated sialyl Lewis x. J. Pharmacol. Exp. Ther. 330(2), 608–612 (2009).
- Nutku E, Aizawa H, Hudson SA, Bochner BS. Ligation of Siglec-8: a selective mechanism for induction of human eosinophil apoptosis. Blood 101(12), 5014–5020 (2003).
- Wein M, Bochner BS. Adhesion molecule antagonists: future therapies for allergic diseases? Eur. Respir. J. 6(9), 1239–1242 (1993).
- Robinson AJ, Kashanin D, O’Dowd F, Fitzgerald K, Williams V, Walsh GM. Fluvastatin and lovastatin inhibit granulocyte macrophage-colony stimulating factor-stimulated human eosinophil adhesion to inter-cellular adhesion molecule-1 under flow conditions. Clin. Exp. Allergy 39(12), 1866–1874 (2009).
- Bochner BS, Gleich GJ. What targeting eosinophils has taught us about their role in diseases. J. Allergy Clin. Immunol. 126(1), 16–25 (2010).
- Moqbel R, Hamid Q, Sun Y et al. Expression of mRNA and immunoreactivity for the granulocyte-macrophage colony stimulating factor in activated human eosinophils. J. Exp. Med. 174(3), 749–752 (1991).
- Desremaux P, Janin A, Colombel JF et al. Interleukin 5 messenger RNA expression by eosinophils in the intestinal mucosa of patients with coeliac disease. J. Exp. Med. 175(1), 293–296 (1992).
- Weller PF. Eicosanoids, cytokines and other mediators elaborated by eosinophils. In: Eosinophils: Biological and Clinical Aspects. Makino S, Fukuda T (Eds). CRC Press, FL, USA, 125–154 (1993).
- Moqbel R, Levi-Schaffer F, Kay AB. Cytokine generation by eosinophils. J. Allergy Clin. Immunol. 94(6), 1183–1188 (1994).
- Moqbel R, Ying S, Barkans, J et al. Identification of messenger RNA for IL-4 in human eosinophils with granule localisation and release of the translated product. J. Immunol. 155(10), 4939–4947 (1995).
- Kay AB, Barata L, Meng Q, Durham SR, Ying S. Eosinophils and eosinophil-associated cytokines in allergic inflammation. Int. Arch. Allergy Immunol. 113(1–3), 196–199 (1997).
- Walsh GM. Advances in the immunobiology of eosinophils and their role in disease. Crit. Rev. Clin. Lab. Sci. 36(5), 453–496 (1999).
- Lehrer RI, Szklarek D, Barton A, Ganz T, Hamann KJ, Gleich GJ. Antibacterial properties of eosinophil major basic protein and eosinophil cationic protein. J. Immunol. 142(12), 4428–4434 (1989).
- Hamann KJ, Barker RL, Ten RM, Gleich GJ. The molecular biology of eosinophil granule proteins. Int. Arch. Allergy Appl. Immunol. 94(1–4), 202–209 (1991).
- Plager DA, Stuart S, Gleich GJ. Human eosinophil granule major basic protein and its novel homolog. Allergy 53(Suppl. 45), 33–40 (1998).
- Yousefi S, Gold JA, Andina N et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 14(9), 949–953 (2008).
- Dworski R, Simon HU, Hoskins A, Yousefi S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J. Allergy Clin. Immunol. 127(5), 1260–1266 (2011).
- Simon D, Hoesli S, Roth N, Staedler S, Yousefi S, Simon HU. Eosinophil extracellular DNA traps in skin diseases. J. Allergy Clin. Immunol. 127(1), 194–199 (2011).
- Schmid-Grendelmeier P, Altznauer F, Fischer B et al. Eosinophils express functional IL-13 in eosinophilic inflammatory diseases. J. Immunol. 169(2), 1021–1027 (2002).
- Swartz JM, Byström J, Dyer KD, Nitto T, Wynn TA, Rosenberg HF. Plasminogen activator inhibitor-2 (PAI-2) in eosinophilic leukocytes. J. Leukoc. Biol. 76(4), 812–819 (2004).
- Kato M, Kephart GM, Talley NJ et al. Eosinophil infiltration and degranulation in normal human tissue. Anat. Record. 252(3), 418–425 (1998).
- Noguchi H, Kephart GM, Colby TV, Gleich GJ. Tissue eosinophilia and eosinophil degranulation in syndromes associated with fibrosis. Am. J. Pathol. 140(2), 521–528 (1992).
- Neves JS, Weller PF. Functional extracellular eosinophil granules: novel implications in eosinophil immunobiology. Curr. Opin. Immunol. 21(6), 694–699 (2009).
- Hardy WR, Anderson RE. The hypereosinophilic syndromes. Ann. Intern. Med. 68(6), 1220–1229 (1968).
- Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine 54(1), 1–27 (1975).
- Wilkins HJ, Crane MM, Copeland K, Williams WV. Hypereosinophilic syndrome: an update. Am. J. Hematol. 80(2), 148–157 (2005).
- Weller PF, Bubley GJ. The idiopathic hypereosinophilic syndrome. Blood 83(10), 2759–2779 (1994).
- Vandenberghe P, Wlodarska I, Michaux L et al. Clinical and molecular features of FIP1L1-PDFGRA (+) chronic eosinophilic leukemias. Leukemia 18(4), r734–742 (2004).
- Liapis H, Ho AK, Brown D, Mindel G, Gleich G. Thrombotic microangiopathy associated with the hypereosinophilic syndrome. Kidney Int. 67(5), 1806–1811 (2005).
- Hii MW, Firkin FC, MacIsaac AI, Yii M. Obstructive prosthetic mitral valve thrombosis in idiopathic hypereosinophilic syndrome: a case report and review of the literature. J. Heart Valve Dis. 15(5), 721–725 (2006).
- Numagami Y, Tomita T, Murakami K, Masaki I, Kubo K, Michiharu N. Sinus thrombosis in idiopathic hypereosinophilic syndrome causing fatal cerebral haemorrhage. J. Clin. Neurosci. 15(5), 585–587 (2008).
- Take M, Sekiguchi M, Hiroe M et al. Clinical spectrum and endomyocardial biopsy findings in eosinophilic heart disease. Heart Vessels Suppl. 1, 243–249 (1985).
- Corradi D, Vaglio A, Maestri R et al. Eosinophilic myocarditis in a patient with idiopathic hypereosinophilic syndrome: insights into mechanisms of myocardial cell death. Hum. Pathol. 35(9), 1160–1163 (2004).
- Tai PC, Ackerman SJ, Spry CJ, Dunnette S, Olsen EG, Gleich GJ. Deposits of eosinophil granule proteins in cardiac tissues of patients with eosinophilic endomyocardial disease. Lancet 1(8534), 643–647 (1987).
- Pardanani A, Ketterling RP, Brockman SR et al. CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in systemic mastocytosis associated with eosinophilia and predicts response to imatinib mesylate therapy. Blood 102(9), 3093–3096 (2003).
- Metzgeroth G, Walz C, Erben P et al. Safety and efficacy of imatinib in chronic eosinophilic leukaemia and hypereosinophilic syndrome: a Phase-II study. Br. J. Haematol. 143(5), 707–715 (2008).
- Krauth MT, Binder T, Ohler L, Jäger U, Valent P. Improvement of cardiac function, mitral regurgitation and pulmonary hypertension in a patient with chronic eosinophilic leukemia (CEL) after low dose imatinib therapy. Leuk. Res. 32(11), 1779–1783 (2008).
- Klion A. Hypereosinophilic syndrome: current approach to diagnosis and treatment. Annu. Rev. Med. 60, 293–306 (2009).
- Roufosse F, Weller PF. Practical approach to the patient with marked hypereosinophilia. J. Allergy Clin. Immunol. 126(1), 39–44 (2010).
- Peters MS, Gleich GJ, Dunnette SL, Fukuda T. Ultrastructural study of eosinophils from patients with the hypereosinophilic syndrome: a morphological basis of hypodense eosinophils. Blood 71(3), 780–785 (1988).
- Horny HP, Sotlar K, Valent P. Eosinophil, basophil, and mast infiltrates in the bone marrow: crossing the boundaries of diagnosis. J. Hematopathol. 4, 101–111 (2011).
- Simon HU, Plötz SG, Dummer R, Blaser K. Abnormal clones of T cells producing interleukin-5 in idiopathic eosinophilia. N. Engl. J. Med. 341(15), 1112–1120 (1999).
- Roufosse F, Schandené L, Sibille C et al. Clonal Th2 lymphocytes in patients with the idiopathic hypereosinophilic syndrome. Br. J. Haematol. 109(3), 540–548 (2000).
- Gleich GJ, Schroeter AL, Marcoux JP, Sachs MI, O’Connell EJ, Kohler PF. Episodic angioedema associated with eosinophilia. N. Engl. J. Med. 310(25), 1621–1626 (1984).
- Butterfield JH, Leiferman KM, Abrams J et al. Elevated serum levels of interleukin-5 in patients with the syndrome of episodic angioedema and eosinophilia. Blood 79(3), 688–692 (1992).
- Roufosse F, Cogan E, Goldman M. The hypereosinophilic syndrome revisited. Annu. Rev. Med. 54, 169–184 (2003).
- Gross WL. Churg–Strauss syndrome: update on recent developments. Curr. Opin. Rheumatol. 14(1), 11–14 (2002).
- Keogh KA, Specks U. Churg–Strauss syndrome. Semin. Respir. Crit. Care Med. 27(2), 148–157 (2006).
- Pagnoux C, Guillevin L. Churg–Strauss syndrome: evidence for disease subtypes? Curr. Opin. Rheumatol. 22(1), 21–28 (2010).
- Silver RM. Eosinophilia-myalgia syndrome, toxic-oil syndrome, and diffuse fasciitis with eosinophilia. Curr. Opin. Rheumatol. 4(6), 851–856 (1992).
- Kaufman LD, Krupp LB. Eosinophilia-myalgia syndrome, toxic-oil syndrome, and diffuse fasciitis with eosinophilia. Curr. Opin. Rheumatol. 7(6), 560–567 (1995).
- Belongia EA, Gleich GJ. The eosinophilia-myalgia syndrome revisited. J. Rheumatol. 23(10), 1682–1685 (1996).
- Klion AD, Bochner BS, Gleich GJ et al. Approaches to the treatment of hypereosinophilic syndromes: a workshop summary report. J. Allergy Clin. Immunol. 117(6), 1292–1302 (2006).
- Ogbogu PU, Bochner BS, Butterfield JH et al. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J. Allergy Clin. Immunol. 124(6), 1319–1325 (2009).
- Simon HU, Cools J. Novel approaches to therapy of hypereosinophilic syndromes. Immunol. Allergy Clin. North. Am. 27(3), 519–527 (2007).
- Klion AD. How I treat hypereosinophilic syndromes. Blood 114(18), 3736–3741 (2009).
- Plötz SG, Simon HU, Darsow U et al. Use of an anti-interleukin-5 antibody in the hypereosinophilic syndrome with eosinophilic dermatitis. N. Engl. J. Med. 349(24), 2334–2339 (2003).
- Garrett JK, Jameson SC, Thomson B et al. Anti-interleukin-5 (mepolizumab) therapy for hypereosinophilic syndromes. J. Allergy Clin. Immunol. 113(1), 115–119 (2004).
- Rothenberg ME, Klion AD, Roufosse FE et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N. Engl. J. Med. 358(12), 1215–1228 (2008).
- Roufosse F, de Lavareille A, Schandené L et al. Mepolizumab as a corticosteroid-sparing agent in lymphocytic variant hypereosinophilic syndrome. J. Allergy Clin. Immunol. 126(4), 828–835 (2010).
- Reiter A, Grimwade D, Cross NC. Diagnostic and therapeutic management of eosinophilia-associated chronic myeloproliferative disorders. Haematologica 92(2), 1153–1158 (2007).
- Gotlib J, Cools J. Five years since the discovery of FIP1L1-PDGFRA: what we have learned about the fusion and other molecularly defined eosinophilias. Leukemia 22(11), 1999–2010 (2008).
- Tefferi A. Molecular drug targets in myeloproliferative neoplasms: mutant ABL1, JAK2, MPL, KIT, PDGFRA, PDGFRB and FGFR1. J. Cell. Mol. Med. 13(2), 215–237 (2009).
- Elling C, Erben P, Walz C et al. Novel imatinib-sensitive PDGFRA-activating point mutations in hypereosinophilic syndrome induce growth factor independence and leukemia-like disease. Blood 117(10), 2935–2943 (2011).
- Stover EH, Chen J, Lee BH et al. The small molecule tyrosine kinase inhibitor AMN107 inhibits TEL-PDGFRβ and FIP1L1-PDGFRα in vitro and in vivo. Blood 106(9), 3206–3213 (2005).
- Baumgartner C, Gleixner KV, Peter B et al. Dasatinib inhibits the growth and survival of neoplastic human eosinophils (EOL-1) through targeting of FIP1L1-PDGFR α. Exp. Hematol. 36(10), 1244–1253 (2008).
- Tabouret E, Charbonnier A Mozziconacci MJ, Ivanov V. Low-dose Nilotinib can maintain complete molecular remissions in FIP1L1/PDGFRA-positive hyper-eosinophilic syndrome. Leuk. Res. 35(1), 136 (2011).
- Jovanovic JV, Score J, Waghorn K et al. Low-dose imatinib mesylate leads to rapid induction of major molecular responses and achievement of complete molecular remission in FIP1L1-PDGFRA-positive chronic eosinophilic leukemia. Blood 109(11), 4635–4640 (2007).
- Wang LN, Pan Q, Fu JF et al. FIP1L1-PDGFRα alone or with other genetic abnormalities reveals disease progression in chronic eosinophilic leukemia but good response to imatinib. Chin. Med. J. 121(10), 867–873 (2008).
- Salemi S, Yousefi S, Simon D et al. A novel FIP1L1-PDGFRA mutant destabilizing the inactive conformation of the kinase domain in chronic eosinophilic leukemia/hypereosinophilic syndrome. Allergy 64(6), 913–918 (2009).
- Lierman E, Michaux L, Beullens E et al. FIP1L1-PDGFRα D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFRα T674I eosinophilic leukemia with single agent sorafenib. Leukemia 23(5), 845–851 (2009).
- Cools J, Stover EH, Boulton CL et al. PKC412 overcomes resistance to imatinib in a murine model of FIP1L1-PDGFRα-induced myeloproliferative disease. Cancer Cell 3(5), 459–469 (2003).
- Lierman E, Folens C, Stover EH et al. Sorafenib is a potent inhibitor of FIP1L1-PDGFRα and the imatinib-resistant FIP1L1-PDGFRα T674I mutant. Blood 108(4), 1374–1376 (2006).
- von Bubnoff N, Gorantla SP, Thöne S, Peschel C, Duyster J. The FIP1L1-PDGFRA T674I mutation can be inhibited by the tyrosine kinase inhibitor AMN107 (nilotinib). Blood 107(12), 4970–4971 (2006).
- Metzgeroth G, Erben P, Martin H et al. Limited clinical activity of nilotinib and sorafenib in FIP1L1-PDGFRA positive chronic eosinophilic leukemia with imatinib-resistant T674I mutation. Leukemia 26(1), 162–164 (2012).
- Kalac M, Quintás-Cardama A, Vrhovac R, Kantarjian H, Verstovsek S. A critical appraisal of conventional and investigational drug therapy in patients with hypereosinophilic syndrome and clonal eosinophilia. Cancer 110(5), 955–964 (2007).
- Verstovsek S, Tefferi A, Kantarjian H et al. Alemtuzumab therapy for hypereosinophilic syndrome and chronic eosinophilic leukemia. Clin. Cancer Res. 15(1), 368–373 (2009).
- Burgstaller S, Kreil S, Waghorn K et al. The severity of FIP1L1-PDGFRA-positive chronic eosinophilic leukaemia is associated with polymorphic variation at the IL5RA locus. Leukemia 21(12), 2428–2432 (2007).
- Chen J, DeAngelo DJ, Kutok JL et al. PKC412 inhibits the zinc finger 198-fibroblast growth factor receptor 1 fusion tyrosine kinase and is active in treatment of stem cell myeloproliferative disorder. Proc. Natl Acad. Sci. USA 101(40), 14479–14484 (2004).
- Chase A, Grand FH, Cross NC. Activity of TKI258 against primary cells and cell lines with FGFR1 fusion genes associated with the 8p11 myeloproliferative syndrome. Blood 110(10), 3729–3734 (2007).
- Wasag B, Lierman E, Meeus P, Cools J, Vandenberghe P. The kinase inhibitor TKI258 is active against the novel CUX1-FGFR1 fusion detected in a patient with T-lymphoblastic leukemia/lymphoma and t(7;8)(q22;p11). Haematologica 96(6), 922–926 (2011).