Abstract
Alpha-1-antitrypsin is the most abundant circulating protease inhibitor. It is mainly produced by the liver and secreted into the circulation where it acts to prevent excessive proteolytic damage in the lungs by the enzyme neutrophil elastase. The most common severe deficiency allele is the Z mutation, which causes the protein to self-associate into ordered polymers. These polymers accumulate within hepatocytes to cause liver damage. The resulting lack of circulating α1-antitrypsin predisposes the Z homozygote to proteolytic lung damage and emphysema. Other pathways may also contribute to the development of lung disease. In particular, polymers of Z α1-antitrypsin can form within the lung where they act as a pro-inflammatory stimulus that may exacerbate protease-mediated lung damage. Researchers recognized in the 1980s that plasma α1-antitrypsin levels could be restored by intravenous infusions of purified human protein. Alpha-1-antitrypsin replacement therapy was introduced in 1987 but subsequent clinical trials have produced conflicting results, and to date there remains no widely accepted clinical evidence of the efficacy of α1-antitrypsin replacement therapy. This review addresses our current understanding of disease pathogenesis in α1-antitrypsin deficiency and questions why this treatment in isolation may not be effective. In particular it discusses the possible role of α1-antitrypsin polymers in exacerbating intrapulmonary inflammation and attenuating the efficacy of α1-antitrypsin replacement therapy.
Introduction
Alpha-1-antitrypsin deficiency was first described in 1963 by Laurell and ErikssonCitation1 who noted the absence of the α1 band on serum protein electrophoresis in 5 out of 1500 samples. Three of these individuals had developed early onset emphysema. The association with liver disease was reported 6 years later by Sharp and colleaguesCitation2 and more recently α1-antitrypsin deficiency has been associated with the development of bronchiectasis,Citation3 asthma,Citation4 vasculitis,Citation5 and panniculitis.Citation6
Alpha-1-antitrypsin is a member of the serine protease inhibitor or serpin superfamily of proteins. It is secreted mainly by hepatocytes but also by lung and gut epithelial cells,Citation7–Citation9 neutrophilsCitation10 and alveolar macrophages,Citation11 and is present in the plasma at a concentration of 1.5–3.5 g/L (when measured by an immunodiffusion method). Circulating plasma α1-antitrypsin is a 394-amino-acid, 52 kDa, acute-phase glycoprotein that acts to inhibit the proteolytic enzyme neutrophil elastase. This enzyme is released at sites of inflammation and, if unregulated, causes proteolytic damage to connective tissue. This is particularly important in the lung as it is persistently exposed to inhaled pro-inflammatory stimuli.
Most individuals carry the normal “M” allele of α1-antitrypsin. However more than 100 different alleles have been identified to date, of which over 30 affect either the amount or the function of the molecule in vivo. Most α1-antitrypsin variants are named according to their migration during isoelectric focusing, variants A–L running faster and N–Z slower than the normal “M” protein. The most common severe deficiency mutant is the “Z” (Glu342Lys) allele which is thought to have originated in northern Europe (where the prevalence is 2%–4%)Citation12,Citation13 and is also seen as the predominant severe mutation in North America, Australia, and New Zealand.Citation14 Other important disease-causing alleles include Siiyama (Ser53Phe), prevalent in Japan,Citation15 and Mmalton (Δ52Phe) which is the most common rare deficiency allele seen in SardiniaCitation16 and which has been reported sporadically in the UK and Canada. details the characteristics of recognized deficiency and nullCitation17 alleles.
Table 1 Pathogenic alleles that cause α1-antitrypsin deficiency
Individuals with abnormal alleles have a plasma deficiency due to a lack of secretion of α1-antitrypsin from hepatocytes.Citation18 The severity of the plasma deficiency is predictable with the S allele reducing plasma levels to 60% of normal and the Z allele to 10% of normal; thus an SZ compound heterozygote has plasma levels that are 40% of normal. The resulting lack of elastase inhibition contributes to tissue destruction and panlobular emphysema, particularly in the inflamed lungs of smokers who have a significantly reduced life expectancy when compared with never smokers.Citation19 Indeed α1-antitrypsin deficiency is the only known genetic cause of emphysema and is found in 1%–2% of all cases of chronic obstructive pulmonary disease (COPD).Citation20
Alpha-1-antitrypsin augmentation therapy was developed to replace the deficient circulating protein and so ameliorate the progression of the associated emphysema.Citation21 It is now widely used in many countries for individuals with severe deficiency of circulating α1-antitrypsin. However no randomized controlled studies have convincingly shown it to be an effective strategy in slowing the progression of lung disease or reducing mortality.Citation22 This may be due to a lack of suitably powered studies although the pathogenesis of this condition is complex which may mean that this approach has only limited efficacy. This review considers the factors that may mitigate against the complete or partial effectiveness of α1-antitrypsin augmentation therapy.
Pathogenesis of disease: α1-antitrypsin function, processing and polymerization
Normal “M” α1-antitrypsin is secreted from the liver and acts as a very effective protease inhibitor. It binds neutrophil elastase via a methionine residue at position 358, on the reactive center loop of the protein (). After binding, the enzyme is translocated from one end of the protein to the other in association with insertion of the reactive loop into β-sheet A. This forms a covalently linked complex of enzyme and inhibitor that is cleared from the circulation.
Figure 1 (A) Inhibition of neutrophil elastase by α1-antitrypsin. After docking (left) the neutrophil elastase (grey) is inactivated by movement from the upper to the lower pole of the protein (right). This is associated with insertion of the reactive loop (red) as an extra strand into β-sheet A (green). Reproduced from Lomas et alCitation136 with permission. (B) The structure of α1-antitrypsin is centered on β-sheet A (green) and the mobile reactive center loop (red). Polymer formation results from the Z variant of α1-antitrypsin (Glu342 Lys at P17; arrowed) or mutations in the shutter domain (blue circle) that open β-sheet A to favor partial loop insertion (step 1) and the formation of an unstable intermediate (M*). The patent β-sheet A can either accept the loop of another molecule (step 2) to form a dimer (D), which then extends into polymers (P). A small proportion of the unstable serpin molecules can accept their own loop (step 3) to form an inactive, thermostable, latent conformation (L). The individual molecules of α1-antitrypsin within the polymer are colored red, yellow, and blue. Reproduced from Gooptu et alCitation29 with permission.
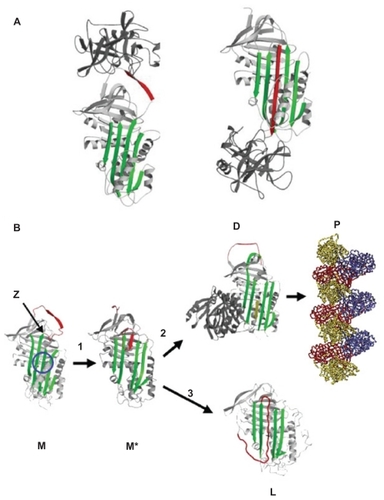
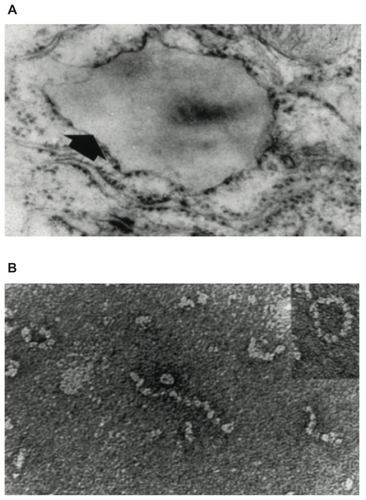
The Z (Glu342 Lys), Siiyama, and probably many other alleles result in a delay in folding in the secretory pathway of hepatocytes.Citation23,Citation24 Much of the protein fails to fold and is “timed out” by the folding sensor manosidase I.Citation25 This material is a substrate for endoplasmic reticulum (ER) associated degradation and is destroyed by the proteasome.Citation26–Citation28 A proportion is folded correctly, trafficked through the endoplasmic reticulum and Golgi apparatus and secreted into the circulation. The remainder is folded to a near-native conformation but does not achieve the native state. The Glu342 Lys mutation results in an expansion of β-sheet A and perturbation of the F helix to form an intermediate that we have termed (M*).Citation29 The reactive center loop of a second α1-antitrypsin molecule can then bind to this intermediate, forming a dimer that extends to form a polymer.Citation18 The resultant polymer has an ordered, repeating structure but no anti-neutrophil elastase activity, because the reactive loop that is central to the polymerization process is buried in β-sheet A of another molecule (). These polymers are sequestered within the ER of hepatocytes where they form diastase resistant, periodic acid-Schiff stain positive inclusions, which are associated with liver disease ().Citation18 The Z α1-antitrypsin that is correctly folded and trafficked through the secretory pathways still has the propensity to form polymers. These have been identified in the lungCitation30 and in biopsies from the skinCitation31 and kidneysCitation32 from Z α1-antitrypsin homozygotes.
Figure 2 (A) Electron microscopy (×20,000) of a hepatocyte from a Z homozygote showing a massive inclusion (arrowed) in the endoplasmic reticulum. Reproduced from Lomas et alCitation18 with permission. (B) The intra-hepatic polymers of mutant Z α1-antitrypsin have the appearance of beads on a string on electron microscopy. Reproduced from Lomas et alCitation137 with permission.
Study of the consequences of abnormally folded protein accumulation in hepatocytes has helped shed light on the mechanism of this gain-of-function toxicity that causes α1-antitrypsin deficiency associated liver disease. The presence of polymers of α1-antitrypsin within the ER causes ER stress. This is defined as a state in which unfolded proteins accumulate and aggregate within the ER, perturbing normal ER function. Misfolded protein accumulation triggers the unfolded protein response (UPR); resident ER chaperones involved in protein folding are upregulated and translation of the abnormal protein is downregulated to restore homeostasis.Citation33 An excess of protein traffic, for example following viral infection of a cell, triggers the ER overload response (EOR), resulting in calcium-dependent NF-κB activation.Citation34
It is striking that polymers of α1-antitrypsin do not activate the UPR in the absence of a second “hit” such as heat or accumulation of other misfolded proteins; this is likely to be due to the ordered nature of the polymers.Citation35 They do however activate the EOR.Citation36,Citation37 This results in inflammatory mediator production,Citation38 relative resistance to cell death,Citation36,Citation39 and an increase in cell proliferation. Though the precise mechanism by which this causes liver disease has not been elucidated, it follows that abnormal cell survival in the setting of a pro-inflammatory environment may lead to both hepatitis and neoplasia.
Clinical features of α1-antitrypsin deficiency related liver disease
Intracellular polymers form inclusions that are associated with neonatal hepatitis, cirrhosis, and hepatocellular carcinoma.Citation40,Citation41 Cholestatic jaundice affects one in ten neonates with α1-antitrypsin deficiency, 15% of whom develop juvenile cirrhosis. Liver disease can also become clinically relevant later in life with almost 50% of patients over the age of 50 having histological features consistent with cirrhosis in an autopsy series.Citation41 Vaccination against hepatitis A and B is recommended as viral hepatitis may predispose to development of chronic liver disease.Citation42 It is recommended that alcohol consumption does not exceed 60 g/day but there is currently no proven association between alcohol excess and the development of liver disease in α1-antitrypsin deficiency.Citation43 Weight control is advisable in view of an association between obesity and cirrhosis in these individuals.Citation44 Screening for cirrhosis and hepatocellular carcinoma (HCC) with liver ultrasound is warranted; some clinicians may choose to use serum alpha-fetoprotein as an additional screening tool for HCC. Treatment of hepatic failure follows that for any other condition; liver transplantation is an option and accounted for 1.1% of adult liver transplants in the US in the period 1995–2004. Liver transplantation cures the circulating deficiency of α1-antitrypsin and 5-year survival rates are excellent at around 83%.Citation45
Alpha-1-antitrypsin deficiency and lung disease
Alpha-1-antitrypsin deficiency is classically associated with early-onset, lower zone emphysema. Smoking individuals with severe circulating deficiency of α1-antitrypsin tend to develop clinical disease in the third or fourth decade with the most common reported symptoms being breathlessness, cough, and wheeze.Citation46 Those with a smoking history develop more severe disease at an earlier age than could be explained by smoking alone. There is often a significant delay from symptom onset to diagnosis;Citation47 a 2003 survey revealed a time lapse of more than 5 years between first symptom and diagnosis in a cohort of 1851 individuals with α1-antitrypsin deficiency.Citation48 A chest radiograph typically shows hyperinflation and a paucity of vascular markings in the lower zone; pulmonary function testing reveals evidence of airflow obstruction, gas trapping, and impaired gas transfer. The natural history of α1-antitrypsin deficiency-associated emphysema is highly variable but disease is often progressive, with faster lung function decline seen in ongoing smokers,Citation49 those with recurrent exacerbations,Citation50 and those with environmental dust exposure.Citation51
It is apparent from the study of the rare null mutations of α1-antitrypsin that the lack of circulating protein (ie, loss-of-function) plays a vital role in the development of emphysema. Indeed, null α1-antitrypsin homozygotes have particularly severe disease.Citation52 There is now increasing evidence that α1-antitrypsin polymers are also important in the development of the panacinar emphysema that is classically seen in this condition. Alpha-1-antitrypsin enters the lung from the circulation by passive diffusionCitation53 and is also produced locally in alveolar macrophages and bronchial and alveolar epithelial cells.Citation7,Citation8,Citation11 In individuals homozygous for the Z allele, all α1-antitrypsin has the propensity to form polymers regardless of its source. Indeed, polymers have been detected in bronchoalveolar lavage fluidCitation54,Citation55 and explanted lung sections from Z homozygotes, where they are seen both around capillaries (consistent with circulating polymers) and epithelial cells (suggesting local synthesis). This local production of polymers is exacerbated by the presence of cigarette smoke.Citation56 The most compelling evidence for pulmonary α1-antitrypsin production of polymers comes from a bronchoalveolar lavage sample from an α1-antitrypsin ZZ homozygote following liver transplantation which contained polymers; because wildtype M antitrypsin is produced from the transplanted liver this confirms that Z α1-antitrypsin is produced locally and then forms polymers.Citation55
In stark contrast to the anti-inflammatory properties of monomeric M α1-antitrypsin, Z α1-antitrypsin polymers are pro-inflammatory, acting as neutrophil chemoattractants. Polymers, largely located in the interstitium, attract neutrophils as they migrate from capillary to alveolus in response to inflammatory mediators induced by cigarette smoke.Citation56 Here they cause neutrophil degranulation and release of elastases and other degradative enzymes.Citation57 Additionally, the lungs of Z α1-antitrypsin homozygotes contain increased levels of chemotactic cytokines including interleukin-8 (IL-8) and leukotriene B4 (LTB4), compared with controls.Citation58 This may be a response of alveolar macrophages to uninhibited neutrophil elastaseCitation59 but may also reflect stress signaling pathways, perhaps including the UPR, induced by intracellular polymers in epithelial cells and alveolar macrophages.
The pro-inflammatory environment in the lung is further amplified by a number of other mechanisms: (i) monomeric Z α1-antitrypsin is ten-fold less efficient than wild type protein at inhibiting neutrophil elastase,Citation60 (ii) oxidation of α1-antitrypsin by superoxide radicals may not only reduce its efficacy further,Citation61 it may also stimulate release of IL-8 and monocyte chemoattractant protein-1 from epithelial cells,Citation62 (iii) the reduction in intracellular α1-antitrypsin may lead to loss of inhibition of caspase-3, leading to uncontrolled cellular apoptosis.Citation63 This is in contrast to the relative paucity of apoptosis seen in the liver and perhaps reflects the activation of the UPR in response to the second hit, which is the inflammatory milieu of the lung in Z α1-antitrypsin homozygotes.
Taken together, a model for the pathogenesis of lung disease can be proposed in the Z α1-antitrypsin homozygote involving both loss-of-function and gain-of-function components. The lack of functional α1-antitrypsin (due to reduced secretion, reduced antiprotease activity, and the polymerization of Z α1-antitrypsin) creates a pro-inflammatory and proteolytic environment. This is exacerbated by the presence of interstitial polymers and cytokines, both of which are chemotactic for neutrophils, and can be further driven by oxidation of α1-antitrypsin due to cigarette smoke. Finally, a lack of intracellular α1-antitrypsin prevents inhibition of apoptotic pathways. The resulting inflammation, proteolysis, and cell death lead to the development of panacinar emphysema, the hallmark of α1-antitrypsin deficiency-related lung disease.
Systemic diseases associated with α1-antitrypsin deficiency
Alpha-1-antitrypsin deficiency is associated with other inflammatory conditions including panniculitis and anti-neutrophil cytoplasmic antibody (ANCA) positive vasculitis. The lesions of panniculitis contain neutrophils that co-localize with polymers, though normal skin of the Z α1-antitrypsin homozygote also contains polymers.Citation31 It remains to be seen whether polymers are simply an incidental finding, or whether they play a role in the pathogenesis of the disease. The repeated observation that intravenous α1-antitrypsin replacement therapy can have an effect on flare frequency and severity of panniculitisCitation64 points to loss-of-function being of greater significance in this manifestation of disease.
Case–control studies have confirmed a link between Z α1-antitrypsin deficiency and ANCA-associated vasculitis, and polymers can also be seen in the renal biopsies of affected individuals.Citation32 In addition to the low absolute levels of circulating α1-antitrypsin, it is postulated that polymers may become trapped within glomeruli, promoting neutrophil degranulation and inflammation as they do within the interstitium of the lung.Citation32
Current treatment strategies
The management of patients with α1-antitrypsin deficiency related emphysema closely resembles that of patients with “usual” smoking related COPD. Smoking cessation is crucial to prevent progression of disease with the rate of forced expiratory volume in 1 second (FEV1) decline being significantly less in those who quit successfully.Citation49 Other potentially preventative strategies include minimization of respiratory irritant exposure and pneumococcal and influenza vaccination.Citation43 Inhaled corticosteroids are commonly used and theoretically should reduce neutrophilic airway inflammation; small studies have suggested a possible benefit in terms of lung function in some patientsCitation65,Citation66 and inhaled bronchodilators may result in symptomatic benefit despite little objective evidence of a bronchodilator response. Oxygen used in accordance with national guidelines and pulmonary rehabilitation programs may also be useful.
Lung volume reduction surgery (LVRS) is uncommonly used in individuals with α1-antitrypsin deficiency. Individuals with emphysema of any cause with predominantly basal disease have a worse outcome than those with upper lobe disease.Citation67 Alpha-1-antitrypsin deficient patients undergoing LVRS achieve initial improvements in FEV1 and exercise tolerance comparable with non-deficient patients but the benefits are shorter lived.Citation68 It has been suggested that this is a consequence of disease distribution rather than α1-antitrypsin deficiency per seCitation68 but currently the American Thoracic Society/European Respiratory Society guidelines do not recommend this procedure pending the emergence of further evidence.Citation43 There are case reports of successful use of less invasive procedures including endobronchial valve placementCitation69 though experience remains limited.
Lung transplantation is also an option for patients with end-stage emphysema. Because COPD often develops at an early age in α1-antitrypsin-deficient individuals, they tend to be good candidates. Alpha-1-antitrypsin deficiency accounted for 3.2% of lung transplants (and 10% of transplants for emphysema) in the International Society for Heart and Lung Transplantation Registry in 2009,Citation70 with a 1-year survival rate of 86% and a 3-year survival of 69% for transplants performed between 2006–2010. These figures are comparable with the general COPD population, 84% of whom are alive at 1 year and 67% at 3 years posttransplant.
Alpha-1 antitrypsin replacement therapy: current practice
The concept of using purified human α1-antitrypsin as intravenous replacement therapy was first described in 1981 by Gadek and colleagues who demonstrated normalization of serum α1-antitrypsin levels and establishment of anti-elastase activity within the lower respiratory tract with weekly infusions.Citation21 Alpha-1 antitrypsin replacement therapy was subsequently approved by the United States Food and Drug Administration (FDA) in 1987 based on the demonstration that normal serum levels could be achieved with regular treatment.Citation71 It is also available in Canada and many European countries and replacement therapy is recommended in the joint American Thoracic Society/European Thoracic Society statement on the management of α1-antitrypsin deficiency.Citation43 Many other countries including the UK, New Zealand, and Australia await proof of clinical benefit before licensing. It is not indicated in individuals who have partial α1-antitrypsin deficiency (MZ heterozygotes) and in α1-antitrypsin deficiency-associated liver disease.
Replacement therapy is given as weekly intravenous infusions at a dose of 60 mg/kg. This is based on evidence that with such dosing, trough α1-antitrypsin levels can be kept above the “protective” threshold of 80 mg/dL. This threshold is based on the observation that patients with heterozygous phenotypes whose levels of α1-antitrypsin exceed this level do not usually develop lung disease.Citation71 Four options are available for replacement therapy in the US: Prolastin® (Talecris Biotherapeutics, Research Triangle Park, NC), Aralast® (Baxter Healthcare, Deerfield, IL), Zemaira® (CSL Behring, King of Prussia, PA), and Glassia® (Baxter, Deerfield, IL), with a fifth, Trypsone® (Grifols, Barcelona, Spain), available in Spain. All consist of purified human α1-antitrypsin. The individual characteristics of each have been thoroughly reviewed elsewhereCitation72 but they all appear to be equally effective with subtle differences in storage, preparation, infusion rate, and cost. Glassia®, licensed in October 2009 by the FDA, is the only ready-to-use formulation but requires a slower infusion rate of 0.04 mL/kg/minute, half that of the other preparations. The cost of replacement is dependent on body mass but may be upwards of US$100,000 per annum – and is a lifelong treatment.Citation73
Intravenous α1-antitrypsin is generally considered to be well tolerated and safe. It is contraindicated in patients with IgA deficiency as purified protein may contain small amounts of IgA, prompting a severe hypersensitivity reaction. As it is purified from human plasma there is a theoretical risk of transmission of infectious agents including viruses and prions, but such transmission has never been reported. The commonest reported side effects from the original preparation (Prolastin®) are dizziness, fainting, and dyspnea.Citation74
Alpha-1-antitrypsin replacement therapy: evidence of efficacy
Evidence of a biochemical effect of weekly intravenous antitrypsin was quick to emerge. Serum α1-antitrypsin levels can be maintained above the postulated protective level and anti-elastase activity can concurrently be detected in bronchoalveolar lavage fluid.Citation71 Infused α1-antitrypsin is functionally active and antibodies do not develop following repeated infusion.Citation75 There are inherent difficulties in attempting to establish the clinical efficacy of α1-antitrypsin replacement therapy. Though the gene frequency makes α1-antitrypsin deficiency as common as cystic fibrosis, the heterogeneous nature of the disease means that only around 5%–10% of patients have been diagnosed.Citation76 This means that the pool of patients on whom to undertake trials is relatively small and inevitably results in the majority of evidence coming from larger observational studies rather than the few randomized controlled trials, each of which contains a small number of participants.Citation77 Additionally, the evolution of emphysema is slow, meaning that protracted studies are necessary to show differences in lung function decline and mortality.Citation78 Indeed, Schluchter et al estimated that to detect a 40% reduction in mortality in 5 years, 684 α1-antitrypsin deficient individuals with an FEV1 of 35%–49% predicted would need to be recruited over a 2-year period.Citation79 Furthermore enthusiasm from pharmaceutical companies to fund such trials may be lacking with their products already freely available in many countries.
Non-randomized trials
It was not until 1997 that the first observational study with concurrent controls addressing clinical efficacy of α1-antitrypsin replacement therapy was published. Seersholm and colleagues demonstrated a significantly slower rate of FEV1 decline in a cohort of 198 German α1-antitrypsin deficient patients on replacement therapy when compared with 97 Danish controls, particularly in those with an FEV1 of 31%–65% predicted.Citation80 In 1998 the National Heart Lung and Blood Institute α1-antitrypsin deficiency registry study group reported on 927 patients enrolled in their registry. They showed no overall difference in lung function decline between treated and untreated groups. However there was a significant benefit of treatment seen in those with moderate impairment of lung function (FEV1 30%–64% predicted); conversely those with an FEV1 > 80% predicted had a faster annual decline in FEV1 whilst on treatment than untreated individuals (P = 0.09). They also reported overall mortality figures in favor of α1-antitrypsin replacement, though no difference in those with an FEV1 > 50% predicted.Citation81 Wencker et al took a different approach in 2001, comparing decline in lung function in 96 patients before and after initiation of augmentation therapy. These authors observed an overall benefit of treatment, but in contrast to previous observational studies, they demonstrated no significant difference in those with FEV1 in the range 30%–65%. The most significant benefit was seen in the small subgroup with mildly impaired (> 65% predicted at enrolment) but rapidly declining FEV1, with annual FEV1 loss reducing from 255.7 mL/year to 45.8 mL/year on augmentation therapy (P = 0.0016).Citation82 Recently Tonelli et al studied 164 individuals from the Alpha-1 Foundation DNA and Tissue Bank, finding a benefit of treatment in ex-smokers with an FEV1 of less than 50% predicted. It was again noted that those with better lung function (FEV1 > 60% predicted) did worse on augmentation therapy as measured by FEV1 decline.Citation83 This finding may in part be explained by a tendency for those with good but rapidly declining lung function to be started on augmentation therapy. The authors of these observational studies all acknowledge the limitations of their findings. Treatment decisions were made by patients’ physicians and the baseline demographics between groups were unmatched; in particular the baseline FEV1 was lower in the treatment cohort in all studies.
Randomized controlled trials
Alpha-1-antitrypsin replacement therapy had been available in the United States for 12 years before the first of two randomized controlled trials (RCTs) assessing its efficacy was published. In 1999 Dirksen et alCitation84 reported on a cohort of 58 ex-smokers with severe plasma α1-antitrypsin deficiency and an FEV1 of 30%–80% predicted. The active group was treated with 250 mg/kg iv α1-antitrypsin at 4-weekly intervals for at least 3 years. This dose had previously been shown to provide protective serum α1-antitrypsin levels for an average of 25 days of the 28-day interval.Citation85 The study’s primary outcome measure was FEV1; there was no statistically significant difference between groups but a trend toward faster decline of FEV1 was seen in the treated group. In contrast the authors were able to demonstrate a trend towards slower loss of lung density (measured by computed tomography [CT]) in the treated group, albeit with substantial deterioration in both groups. The study was not designed to address mortality.
In 2009 the results of the “exacerbations and computed tomography scan as lung end-points” (EXACTLE trial),Citation86 which used CT-assessed lung density as its primary outcome measure, were published. Participants had severe α1-antitrypsin deficiency, were ex- or never smokers and members of the treatment group were given 60 mg/kg weekly iv α1-antitrypsin replacement (the accepted dosing regimen) for a minimum of 2 years. The authors again reported loss of lung density in both groups with a difference of borderline significance in favor of the treatment group over the entire treatment period in one of four analysis methods. No difference was seen in lung function parameters or exacerbation rate between groups, though a post hoc analysis suggested that exacerbation severity may be milder in the treatment group. A third RCT which plans to address the question of mortality, albeit only as a secondary endpoint, is ongoing.Citation87
A meta-analysis of some of the early trials comprising 1509 patients concluded that FEV1 decline was slower in treated versus untreated individuals.Citation80–Citation82,Citation84,Citation88 This was particularly evident in those with an FEV1 of 30%–65% predicted, who experienced a 26% reduction in the rate of decline in FEV1 as a result of replacement therapy.Citation89 The randomized trials were the subject of two meta-analyses that reached quite different conclusions. A Cochrane review concluded that a lack of evidence regarding mortality combined with conflicting evidence for treatment efficacy from FEV1 and CT data meant that replacement therapy cannot be recommended at present and that further studies should be large enough to detect an effect on mortality.Citation22 A response from the expert community raised concerns over the methodology of this review, particularly regarding differences in drug and dosing regimens, and dropout rates between studies. The authors also questioned whether observational data should have been included in a meta-analysis of this rare condition.Citation77
Stockley and colleagues re-analyzed the 1999 RCT data and, combined with the EXACTLE data, demonstrated a significant reduction in lung density decline in the treated versus untreated group (P = 0.006). They agreed that further studies are warranted, but that there is sufficient evidence of treatment efficacy that in future trials an iv placebo group is not ethically warranted.Citation90
Exacerbations and airway inflammation
One study asking patients to self-report symptoms after initiation of replacement therapy found that 56 of 89 patients reported a definite decrease in exacerbation number from an average of 3–5 per year to 0–1 per year.Citation91 Others have noted ongoing exacerbations despite treatmentCitation92 and the EXACTLE trial did not find a reduction in exacerbation frequency on treatment. Augmentation therapy does however seem to have an effect upon markers of airway inflammation. Stockley et al noted an increase in sputum α1-antitrypsin to the levels seen in nondeficient individuals with intravenous replacement; this was associated with a decrease in sputum LTB4 but there was no significant change in markers of neutrophil number and activation: IL-8 and myeloperoxidase (MPO). They suggest that this may reflect decreased LTB4 secretion by macrophages due to a reduction in uninhibited elastase.Citation93
summarizes the trial data discussed above. The very nature of observational studies along with their conflicting results must limit their role in confirming treatment efficacy. RCTs to date have been small scale. They suggest a lack of treatment benefit as measured by lung function, but raise the possibility of a positive impact on the rate of lung density loss as measured by CT, a parameter thought to be a more accurate measure of disease progression than lung function.Citation94 The effect of augmentation therapy on mortality has not been adequately addressed. As outlined by Schluchter and colleaguesCitation79 the scale of trial required would be likely to require international collaboration. With disparate current clinical practice and a lack of agreement on the ethical design of future trials to assess disease progression, it seems this question will remain unanswered.
Table 2 Studies of α1-antitrypsin replacement therapy
Assessing replacement therapy in the context of disease pathogenesis
Practical problems with clinical assessment of intravenous α1-antitrypsin replacement have been outlined above. Putting this treatment in the context of disease pathogenesis may provide more understanding of the questionable efficacy demonstrated to date. Weekly iv α1-antitrypsin provides protective levels of circulating protein as measured by a threshold over which individuals do not develop lung disease. However α1-antitrypsin is an acute phase protein with levels increasing by up to 130% in response to stress and peaking at up to 6 g/L;Citation95 perhaps the ability to mount such a response may be of critical importance rather than the baseline level. Alpha-1-antitrypsin secretion may also be under the control of growth hormone working synergistically with other pituitary hormones, which leads to variable hepatic serum and mRNA levels.Citation96 Replacement therapy cannot address this; it can be envisaged that a patient would be particularly vulnerable shortly before the next infusion is due, especially the significant number of individuals who receive bi-weekly or monthly treatment.Citation81 Based on knowledge of disease pathogenesis it follows that α1-antitrypsin replacement therapy should address some of the drivers of lung disease: (i) it can effectively inhibit neutrophil elastase where the inefficient Z α1-antitrypsin monomers and inactive polymers would otherwise leave uninhibited elastase activity; (ii) the resulting lack of free elastase reduces LTB4 release from alveolar macrophages, thereby reducing the chemotactic signaling to neutrophils.Citation93 Despite this, levels of MPO are unchanged in those on replacement suggesting that equal numbers of neutrophils are present (though they will have their elastase inhibited).
However replacement therapy will not affect (i) the formation of Z α1-antitrypsin polymers that become lodged in the lung interstitium and act as neutrophil chemoattractants; (ii) intracellular Z α1-antitrypsin polymers in epithelial cells and alveolar macrophages that continue to induce stress signaling and release of pro-inflammatory cytokines.
We can postulate that a patient on augmentation therapy may have less tissue destruction as a result of better regulation of neutrophil elastase activity, but will continue to have chronic inflammation in response to intrapulmonary polymers. In the small minority of patients who are α1-antitrypsinnull homozygotes then lung damage entirely reflects the absence of neutrophil elastase inhibition. This group is most likely to benefit from replacement therapy. However in the majority of patients who have polymerogenic mutations it may be that the benefit of replacement therapy is attenuated by the ongoing inflammatory response to polymers.
Clinical and laboratory studies designed specifically to address this question would be of enormous value. A trial comparing treatment efficacy in individuals homozygous for null α1-antitrypsin alleles with those with polymerogenic alleles may shed light on the impact of α1-antitrypsin polymers in attenuating the beneficial effects of augmentation therapy. This would provide crucial information on the relative role of α1-antitrypsin polymers in the pathogenesis of emphysema. Additionally if small molecules that block α1-antitrypsin polymerizationCitation97 can be further developed, these can be tested in vivo to determine whether the absence of intrapulmonary polymers slows the rate of development of emphysema.
Conclusion
The current literature provides good evidence of the safety and biochemical effect of intravenous α1-antitrypsin replacement but there is no widely accepted proof that it affects disease progression or mortality. Further large scale RCTs would help clarify the impact of long-term intravenous α1-antitrypsin replacement on loss of lung density, which is emerging as an important surrogate marker of disease. Combined results from such trials may confirm CT densitometry as the best marker of disease severity and progression in α1-antitrypsin deficiency as well as adding to a body of evidence that could be used to determine any effect on mortality.
The evolving knowledge of the molecular basis of disease requires us to re-evaluate the utility of replacement therapy in individuals with α1-antitrypsin deficiency. A logical approach is to turn away from simply replacing deficient protein and toward tackling the key feature of this disease: the tendency of mutant α1-antitrypsin to polymerize.Citation18 Strategies that result in the secretion of normally folded, functional protein would impact on both intracellular stress signaling and the systemic inflammation caused by polymers and in turn should prevent the development of both liver and lung disease, although whether this will become a reality remains to be seen.
Acknowledgments
JAD is an MRC Clinical Training Fellow. DAL is supported by the Medical Research Council (UK), the Engineering and Physical Sciences Research Council, GlaxoSmithKline, the Alpha-1 Foundation, and Papworth NHS Trust.
Disclosure
The authors report no conflicts of interest in this work.
References
- LaurellC-BErikssonSThe electrophoretic α1-globulin pattern of serum in α1-antitrypsin deficiencyScand J Clin Lab Invest196315132140
- SharpHLBridgesRAKrivitWFreierEFCirrhosis associated with alpha-1-antitrypsin deficiency: a previously unrecognized inherited disorderJ Lab Clin Med19697369349394182334
- KingMAStoneJADiazPTα1-antitrypsin deficiency: evaluation of bronchiectasis with CTRadiology199619911371418633137
- EdenEMitchellDMehlmanBAtopy, asthma, and emphysema in patients with severe alpha-1-antitrypsin deficiencyAm J Respir Crit Care Med1997156168749230728
- GriffithMELovegroveJUGaskinGWhitehouseDBPuseyCDC-anti-neutrophil cytoplasmic antibody positivity in vasculitis patients is associated with the Z allele of alpha-1-antitrypsin, and the P-antineutrophil cytoplasmic antibody positivity with the S alleleNephrol Dial Transplant19961134384438671812
- BleuminkEKlokkeAHRelationship between Weber-Christian panniculitis and the ZZ phenotype of alpha1-antitrypsinArch Dermatol Res198527743283293873915
- CichyJPotempaJTravisJBiosynthesis of α1-proteinase inhibitor by human-derived epithelial cellsJ Biol Chem199727213825082559079644
- VenembrePBouttenASetaNSecretion of alpha 1-antitrypsin by alveolar epithelial cellsFEBS Lett19943462–31711748013628
- GeboesKRayMBRutgeertsPCalleaFDesmetVJVantrappenGMorphological identification of alpha-1-antitrypsin in the human small intestineHistopathology19826155607035326
- PaakkoPKirbyMdu BoisRMGillissenAFerransVJCrystalRGActivated neutrophils secrete stored alpha-1 antitrypsinAm J Respir Crit Care Med19961546 pt 1182918338970377
- CohenABInterrelationships between the human alveolar macrophage and alpha-1-antitrypsinJ Clin Invest19735211279327994201266
- CoxDWWooSLMansfieldTDNA restriction fragments associated with alpha 1-antitrypsin indicate a single origin for the deficiency allele PI ZNature1985316602379812989709
- BlancoIde SerresFJFernández-BustilloELaraBMiravitllesMEstimated numbers and prevalence of PI*S and PI*Z alleles of α1-antitrypsin deficiency in European countriesEur Respir J2006271778416387939
- de SerresFJWorldwide racial and ethnic distribution of alpha1-antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveysChest200212251818182912426287
- SeyamaKNukiwaTSoumaSShimizuKKiraSAlpha 1-antitrypsin-deficient variant Siiyama (Ser53[TCC] to Phe53[TTC]) is prevalent in Japan. Status of alpha 1-antitrypsin deficiency in JapanAm J Respir Crit Care Med19951526 pt 1211921268520784
- FerrarottiIBaccheschiJZorzettoMTinelliCCordaLBalbiBPrevalence and phenotype of subjects carrying rare variants in the Italian registry for alpha1-antitrypsin deficiencyJ Med Genet200542328228715744045
- LeeJHBrantlyMMolecular mechanisms of alpha1-antitrypsin null allelesRespir Med200094Suppl CS71110954248
- LomasDAEvansDLFinchJTCarrellRWThe mechanism of Z α1-antitrypsin accumulation in the liverNature199235763796056071608473
- LarssonCNatural history and life expectancy in severe alpha1-antitrypsin deficiency, PiZActa Med Scand17982045345351309708
- LiebermanJWinterBSastreAAlpha 1-antitrypsin Pi-types in 965 COPD patientsChest19868933703733485034
- GadekJEKleinHGHollandPVCrystalRGReplacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjectsJ Clin Invest1981685115811657028785
- GøtzschePCJohansenHKIntravenous alpha-1 antitrypsin augmentation therapy for treating patients with alpha-1 antitrypsin deficiency and lung diseaseCochrane Database Syst Rev20107CD00785120614465
- YuM-HLeeKNKimJThe Z type variation of human α1-antitrypsin causes a protein folding defectNat Struct Biol1995253633677664092
- KangHALeeKNYuMHFolding and stability of the Z and Siiyama genetic variants of human α1-antitrypsinJ Biol Chem199727215105168995291
- WuYSwuliusMTMoremenKWSifersRNElucidation of the molecular logic by which misfolded α1-antitrypsin is preferentially selected for degradationProc Natl Acad Sci U S A2003100148229823412815101
- KroegerHMirandaEMacLeodIEndoplasmic reticulum-associated degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpinsJ Biol Chem200928434227932280219549782
- QuDTeckmanJHOmuraSPerlmutterDHDegradation of a mutant secretory protein, α1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activityJ Biol Chem19962713722791227958798455
- LeAFerrellGADishonDSLeQQSiferRNSoluble aggregates of the human PiZ á1-antitrypsin variant are degraded within the endoplasmic reticulum by a mechanism sensitive to inhibitors of protein synthesisJ Biol Chem19922672107210801530934
- GooptuBHazesBChangWSInactive conformation of the serpin alpha(1)-antichymotrypsin indicates two-stage insertion of the reactive loop: implications for inhibitory function and conformational diseaseProc Natl Acad Sci U S A2000971677210618372
- MahadevaRAtkinsonCLiZPolymers of Z α1-antitrypsin co- localize with neutrophils in emphysematous alveoli and are chemotactic in vivoAm J Path2005166237738615681822
- GrossBGrebeMWenckerMStollerJKBjurstenLMJanciauskieneSNew findings in PiZZ alpha1-antitrypsin deficiency-related panniculitis. Demonstration of skin polymers and high dosing requirements of intravenous augmentation therapyDermatology2009218437037519218787
- MorrisHMorganMDWoodsAANCA-associated vasculitis is linked to carriage of the Z allele of α1-antitrypsin and its polymersAnn Rheum Dis2011 In press
- RonDWalterPSignal integration in the endoplasmic reticulum unfolded protein responseNat Rev Mol Cell Biol20078751952917565364
- PahlHLSesterMBurgertHGBaeuerlePAActivation of transcription factor NF-kappaB by the adenvirus E3/19 K protein requires its ER retentionJ Cell Biol199613245115228647884
- GrahamKSLeASifersRNAccumulation of the insoluble PiZ variant of human α1-antitrypsin within the hepatic endoplasmic reticulum does not elevate the steady-state level of grp78/BiPJ Biol Chem19902653320463204682122976
- HidvegiTSchmidtBZHalePPerlmutterDHAccumulation of mutant α1-antitrypsin Z in the endoplasmic reticulum activates caspases -4 and -12, NF-κB, and BAP31 but not the unfolded protein responseJ Biol Chem200528047390023901516183649
- DaviesMJMirandaERousselBDKaufmanRJMarciniakSJLomasDANeuroserpin polymers activate NF-kappaB by a calcium signalling pathway that is independent of the unfolded protein responseJ Biol Chem200928427182021820919423713
- LawlessMWGreeneCMMulgrewATaggartCCO’NeillSJMcElvaneyNGActivation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z α1-antitrypsin deficiencyJ Immunol200417295722572615100318
- RudnickDALiaoYAnJKMugliaLJPerlmutterDHTeckmanJHAnalyses of hepatocellular proliferation in a mouse model of α-1-antitrypsin deficiencyHepatology20043941048105515057909
- SvegerTLiver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infantsN Engl J Med197629424131613211083485
- ErikssonSCarlsonJVelezRRisk of cirrhosis and primary liver cancer in alpha1-antitrypsin deficiencyN Engl J Med1986314127367393485248
- HashemiMAlavianSMGhavamiSHigh prevalence of alpha 1 antitrypsin phenotypes in viral hepatitis B infected patients in IranHepatol Res200533429229716260177
- American Thoracic Society, European Respiratory SocietyAmerican Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiencyAm J Respir Crit Care Med2003168781890014522813
- BowlusCLWillnerIZernMAFactors associated with advanced liver disease in adults with alpha-1-antitrypsin deficiencyClin Gastroenterol Hepatol20053439039615822045
- KemmerNKaiserTZachariasVNeffGWAlpha-1-antitrpysin deficiency: outcomes after liver transplantationTransplant Proc20084051492149418589136
- McElvaneyNGStollerJKBuistASBaseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1-antitrypsin deficiency. Alpha 1-Antitrypsin Deficiency Registry Study GroupChest199711123944039041988
- StollerJKSmithPYangPSprayJPhysical and social impact of alpha 1-antitrypsin deficiency: results of a surveyCleve Clin J Med19946164614677828337
- StollerJKSandhausRATurinoGDicksonRRodgersKStrangeCDelay in diagnosis of alpha1-antitrypsin deficiency: a continuing problemChest200512841989199416236846
- JanusEDPhillipsNTCarrellRWSmoking, lung function, and alpha 1-antitrypsin deficiencyLancet1985184211521542857224
- DowsonLJGuestPJStockleyRALongitudinal changes in physiological, radiological, and health status measurements in alpha(1)-antitrypsin deficiency and factors associated with declineAm J Respir Crit Care Med200116410 pt 11805180911734427
- MayerASStollerJKBucherBBOccupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiencyAm J Respir Crit Care Med20001622 pt 155353810934086
- BamforthFJKalshekerNAAlpha 1 antitrypsin deficiency due to Pi null: clinical presentation and evidence for molecular heterogeneityJ Med Genet198825283872831367
- StockleyRAMeasurement of soluble proteins in lung secretionsThorax19843942412476372152
- ElliottPRBiltonDLomasDALung polymers in Z α-1-antitrypsin deficiency-related emphysemaAm J Respir Cell Mol Biol19981856706749569237
- MulgrewATTaggartCCLawlessMWZ α-1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractantChest200412551952195715136414
- AlamSLiZJanciauskieneSMahadevaROxidation of Z{alpha}1-antitrypsin by cigarette smoke induces polymerisation: a novel mechanism of early-onset emphysemaAm J Respir Cell Mol Biol2010 [Epub ahead of print]
- ParmarJSMahadevaRReedBJPolymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysemaAm J Respir Cell Mol Biol200226672373012034572
- WoolhouseISBayleyDLStockleyRASputum chemotactic activity in chronic obstructive pulmonary disease: effect of alpha(1)-antitrypsin deficiency and the role of leukotriene B(4) and interleukin 8Thorax200257870971412149532
- HubbardRCFellsGGadekJPacholokSHumesJCrystalRGNeutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophagesJ Clin Invest19918838918971653278
- OgushiFFellsGAHubbardRCStrausSDCrystalRGZ-type alpha 1-antitrypsin is less competent than M1-type alpha-1 antitrypsin as an inhibitor of neutrophil elastaseJ Clin Invest1987805136613743500183
- JohnsonDTravisJThe oxidative inactivation of human alpha-1-proteinase inhibitor. Further evidence for methionine at the reactive centerJ Biol Chem19792541040224026312289
- LiZAlamSWangJSandstromCSJanciauskieneSMahadevaROxidized alpha1-antitrypsin stimulates the release of monocyte chemotactic protein-1 from lung epithelial cells: potential role in emphysemaAm J Physiol Lung Cell Mol Physiol20092972L38840019525388
- PetracheIFijalkowskaIMedlerTRAlpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosisAm J Pathol200616941155116617003475
- BlancoILaraBde SerresFEfficacy of alpha1-antitrypsin augmentation therapy in conditions other than pulmonary emphysemaOrphanet J Rare Dis201161421486454
- WilckeJTDirksenAThe effect of inhaled glucocorticosteroids in emphysema due to alpha1-antitrypsin deficiencyRespir Med19979152752799176645
- CordaLBertellaELa PianaGEBoniERedolfiSTantucciCInhaled corticosteroids as additional treatment in alpha-1-antitrypsin-deficieny-related COPDRespiration2008761616818319586
- FishmanAMartinezFNaunheimA randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysemaN Engl J Med2003348212059207312759479
- DauriatGMalHJebrakGFunctional results of unilateral lung volume reduction surgery in alpha1-antitrypsin deficient patientsInt J Chron Obstruct Pulmon Dis20061220120618046897
- RemundKFBartosikWWoodAEEganJJEndobronchial valve treatment in patients with advanced emphysema and alpha-1-antitrypsin deficiencyThorax200863Suppl VIIA12
- The International Society for Heart and Lung Transplantation Available at: http://www.ishlt.org/registriesAccessed March 2, 2011
- WewersMDCasolaroMASellersSEReplacement therapy for alpha 1-antitrypsin deficiency associated with emphysemaN Engl J Med198731617105510623494198
- KueppersFThe role of augmentation therapy in alpha-1 antitrypsin deficiencyCurr Med Res Opin201127357958821226542
- SilvermanEKSandhausRAClinical Practice: alpha1-antitrypsin deficiencyN Engl J Med2009360262749275719553648
- StollerJKFallatRSchluchterMDAugmentation therapy with alpha1-antitrypsin: patterns of use and adverse eventsChest200312351425143412740257
- SchmidtEWRascheBUlmerWTReplacement therapy for alpha-1-protease inhibitor deficiency in PiZ subjects with chronic obstructive diseaseAm J Med1988846A63693289388
- CamposMAWannerAZhangGSandhausRATrends in the diagnosis of symptomatic patients with α1-antitrypsin deficiency between 1968 and 2003Chest200512831179118616162704
- Feedback on Cochrane review; see ref 20 Available at: http://onlinelibrary.wiley.com/o/cochrane/clsysrev/articles/CD007851/frame.htmlAccessed August 4, 2011
- HutchinsonDCHughesMDAlpha-1-antitrypsin replacement therapy: will its efficacy ever be proved?Eur Respir J19971010219121939387938
- SchluchterMDStollerJKBarkerAFFeasibility of a clinical trial of augmentation therapy for α1-antitrypsin deficiencyAm J Respir Crit Care Med20001613 pt 179680110712324
- SeersholmNWenckerMBanikNDoes α1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary α1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study groupEur Respir J19971010226022639387950
- The Alpha-1-Antitrypsin Deficiency Registry Study GroupSurvival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsinAm J Respir Crit Care Med1998158149599655706
- WenckerMFuhrmannBBanikNKonietzkoNLongitudinal follow-up of patients with α1-protease inhibitor deficiency before and during therapy with IV α1-protease inhibitorChest2001119373774411243951
- TonelliARRouhaniFLiNSchreckPBrantlyMLAlpha-1-antitrypsin augmentation therapy in deficient individuals enrolled in the Alpha-1 Foundation DNA and Tissue BankInt J Chron Obstruct Pulmon Dis2009444345220054436
- DirksenADijkmanJHMadsenFA randomized clinical trial of α1-antitrypsin augmentation therapyAm J Respir Crit Care Med19991605 pt 11468147210556107
- HubbardRCSellersSCzerskiDStephensLCrystalRGBiochemical efficacy and safety of monthly augmentation therapy for alpha 1-antitrypsin deficiencyJAMA19882609125912643261353
- DirksenAPiitulainenEParrDGExploring the role of CT densitometry: a randomised study of augmentation therapy in α1-antitrypsin deficiencyEur Respir J20093361345135319196813
- Zemaira in Subjects With Emphysema Due to Alpha1-Proteinase Inhibitor (API) DeficiencyClinicalTrials.gov Identifier: NCT00261833
- ChapmanKRBradiACPatersonDSlower lung function decline during augmentation therapy in patients with alpha 1-antitrypsin efficiency (A1ATD): results from the Canadian AIR registryProc Am Thorac Soc2005A808
- ChapmanKRStockleyRADawkinsCAugmentation therapy for alpha1-antitrypsin deficiency: a meta-analysisCOPD20096317718419811373
- StockleyRAParrDGPiitulainenETherapeutic efficacy of alpha-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometryRespir Res20101113620920370
- LiebermanJAugmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting dataChest200011851480148511083705
- CamposMAAlazemiSZhangGExacerbations in subjects with alpha-1 antitrypsin deficiency receiving augmentation therapyRespir Med2009103101532153919464158
- StockleyRABayleyDLUnsalIDowsonLJThe effect of augmentation therapy on bronchial inflammation in α1-antitrypsin deficiencyAm J Respir Crit Care Med2002165111494149812045122
- DirksenAFriisMOlesenKPSkovgaardLTSorensenKProgressof emphysema in severe α1-antitrypsin deficiency as assessed by annual CTActa Radiol1997385826832
- VoulgariFCumminsPGardeckiTIBeechingNJStonePCStuartJSerum levels of acute phase and cardiac proteins after myocardial infarction, surgery and infectionBr Heart J19824843523566181800
- SchwarzenburgSJSharpHLBerrySAMantheiRDSeeligSHormonal regulation of serum alpha 1-antitrypsin and hepatic alpha 1-antitrypsin mRNA in ratsBiochem Biophys Res Commun198714739369413499151
- MallyaMPhillipsRLSaldanhaSASmall molecules block the polymerization of Z α1-antitrypsin and increase the clearance of intracellular aggregatesJ Med Chem200750225357536317918823
- MahadevaRChangWSDaffornTHeteropolymerization of S, I, and Z α1-antitrypsin and liver cirrhosisJ Clin Invest19991037999100610194472
- MirandaEPerezJHadzicNA novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with α1-antitrypsin deficiencyHepatology20105231078108820583215
- PollerWMerkleinFSchneider-RaspSMolecular characterization of the defective alpha 1-antitrypsin alleles PI Mwurzburg (Pro369Ser), Mheerlen (Pro369 Leu), and Q0lisbon (Thr68Ile)Eur J Hum Genet19997332133110234508
- FraizerGCHarroldTRHofkerMHCoxDWIn-frame single codon deletion in the Mmalton deficiency allele of α1-antitrypsinAm J Hum Genet19894468949022786335
- CurielDTVogelmeierCHubbardRCStierLECrystalRGMolecular basis of alpha 1-antitrypsin deficiency and emphysema associated with the alpha 1-antitrypsin Mmineral springs alleleMol Cell Biol199010147561967187
- MatsunagaEShiokawaSNakamuraHMaruyamaTTsudaKFukumakiYMolecular analysis of the gene of the alpha 1-antitrypsin deficiency variant, MnichinanAm J Hum Gene1990463602612
- FaberJ-PPollerWWeidingerSIdentification and DNA sequence analysis of 15 new alpha-1 antitrypsin variants, including 2 PiQO alleles and one deficient PiM alleleAm J Hum Genet1994556111311217977369
- TakahashiHNukiwaTSatohKCharacterization of the gene and protein of the alpha 1-antitrypsin “deficiency” allele MprocidaJ Biol Chem19882633015528155343262617
- JardiRRodriguez-FriasFLopez-TalaveraJCCharacterization of the new alpha-1-antitrypsin-deficient PI M-type allele, PI M(vall d’hebron) (Pro(369)-->Ser)Hum Hered200050532032110878477
- ConiPPiliEConvertinoGMVarallo: a new M(Like) alpha 1-antitrypsin- deficient alleleDiagn Mol Pathol200312423723914639110
- OwenMCBrennanSOLewisJHCarrellRWMutation of antitrypsin to antithrombin. Alpha 1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorderN Engl J Med1983309126946986604220
- FaberJPWeidingerSGoeddeHWOleKThe deficient alpha-I-antitrypsin phenotype PI P is associated with an A-to-T transversion in exon III of the geneAm J Hum Genet19894511611632787118
- GrahamAKalshekerNANewtonCRBamforthFJPowellSJMarkhamAFMolecular characterisation of three alpha-1-antitrypsin deficiency variants: proteinase inhibitor (Pi) nullcardiff (Asp25614018 Val); PiMmalton (Phe5114018deletion) and PiI (Arg3914018Cys)Hum Genet198984155582606478
- HildesheimJKinsleyGBissellMPierceJBrantlyMGenetic diversity from a limited repertoire of mutations on different common allelic backgrounds: alpha 1-antitrypsin deficiency variant PduarteHum Mutat1993232212288364590
- ColiABigottiGAlpha-1-antitrypsin protease inhibitor SZ phenotype and liver cirrhosisAppl Pathol19897154602784975
- HolmeJStockleyRACT scan appearance, densitometry, and health status in protease inhibitor SZ alpha1-antitrypsin deficiencyChest200913651284129019892672
- BlancoIBustilloEFRodriguezMCDistribution of α1-antitrypsin PI S and PI Z frequencies in countries outside Europe: a meta-analysisClin Genet200160643144111846735
- YuasaISugimotoYIchinoseMPI*S(iiyama), a deficiency gene of alpha 1-antitrypsin: evidence for the occurrence in western JapanJpn J Hum Genet19933821851918358043
- HolmesMDBrantlyMLFellsGACrystalRGAlpha 1-antitrypsin Wbethesda: molecular basis of an unusual alpha 1-antitrypsin deficiency variantBiochem Biophys Res Commun19901703101310202390072
- JardiRRodriguezFMiravitllesMIdentification and molecular characterization of the new alpha-1-antitrypsin deficient allele PI Y barcelona (Asp256-->Val and Pro391-->His). Mutations in brief no.174. OnlineHum Mutat199812321310651487
- SørheimICBakkePGulsvikAα1-antitrypsin protease inhibitor MZ heterozygosity is associated with airflow obstruction in two large cohortsChest201013851125113220595457
- GraziadeiIWJosephJJWiesnerRHTherneauTMBattsKPPoraykoMKIncreased risk of chronic liver failure in adults with heterozygous alpha1-antitrypsin deficiencyHepatology1998284105810639755243
- WeidingerSJahnWCujnikFSchwarzfischerFAlpha-1-antitrypsin: evidence for a fifth PI M subtype and a new deficiency allele PI*Z augsburgHum Genet198571127293875547
- WhitehouseDBAbbottCMLovegroveJUGenetic studies on a new deficiency gene (PI*Ztun) at the PI locusJ Med Genet198926127447492575668
- GrahamAKalshekerNABamforthFJNewtonCRMarkhamAFMolecular characterisation of two alpha-1-antitrypsin deficiency variants: proteinase inhibitor (Pi) Null(Newport) (Gly11514018Ser) and (Pi) Z Wrexham (Ser-1914018Leu)Hum Genet19908555375402227940
- SatohKNukiwaTBrantlyMEmphysema associated with complete absence of alpha 1- antitrypsin in serum and the homozygous inheritance [corrected] of a stop codon in an alpha 1-antitrypsin-coding exonAm J Hum Genet198842177833257351
- FraizerGCHarroldTRHofkerMHCoxDWIn-frame single codon deletion in the Mmalton deficiency allele of alpha 1-antitrypsinAm J Hum Genet19894468949022786335
- ZorzettoMFerrarottiICampoIIdentification of a novel alpha1-antitrypsin null variant (Q0Cairo)Diagn Mol Pathol200514212112415905697
- BrantlyMLeeJHHildesheimJAlpha1-antitrypsin gene mutation hot spot associated with the formation of a retained and degraded null variantAm J Respir Cell Mol Biol19971632252319070606
- NukiwaTTakahashiHBrantlyMCourtneyMCrystalRGalpha 1-Antitrypsin nullGranite Falls, a nonexpressing alpha 1-antitrypsin gene associated with a frameshift to stop mutation in a coding exonJ Biol Chem19872622511999120043040726
- SifersRNBrashears-MacateeSKiddVJMuenschHWooSLA frameshift mutation results in a truncated alpha 1-antitrypsin that is retained within the rough endoplasmic reticulumJ Biol Chem198826315733073353259232
- TakahashiHCrystalRGAlpha 1-antitrypsin Null (isola di procida): an alpha 1-antitrypsin deficiency allele caused by deletion of all alpha 1-antitrypsin coding exonsAm J Hum Genet19904734034131975477
- FrazierGCSiewertsenMAHofkerMHBrubacherMGCoxDWA null deficiency allele of alpha 1-antitrypsin, QOludwigshafen, with altered tertiary structureJ Clin Invest1990866187818842254451
- CurielDBrantlyMCurielEStierLCrystalRGAlpha-1-antitrypsin deficiency caused by the alpha-1-antitrypsin nullmattawa geneJ Clin Invest1989834114411522539391
- Vaz RodriguesLCostaFMarquesPMendonçaCRochaJSeixasSSevere α-1 antitrypsin deficiency caused by Q0(Ourém) allele: clinical features, haplotype characterization and historyClin Genet2011 [Epub ahead of print]
- PollerWFaberJPWeidingerSOlekKDNA polymorphisms associated with a new alpha 1-antitrypsin PIQ0 variant (PIQ0riedenburg)Hum Genet19918655225241673114
- LeeJNovoradovskayaNRundquistBRewineJSaltiniCBrantlyMAlpha-1-antitrypsin nonsense mutation associated with retained truncated protein and reduced mRNAMol Genet Metab19986342702809635295
- LaubachVERyanWJBrantlyMCharacterization of a human alpha 1-antitrypsin null allele involving aberrant mRNA splicingHum Mol Genet199327100110058364536
- LomasDABelorgeyDMallyaMMolecular mousetraps and the serpinopathiesBiochem Soc Trans200533pt 232133015787598
- LomasDAFinchJTSeyamaKNukiwaTCarrellRWAlpha1-antitrypsin Siiyama (Ser53Phe). Further evidence for intracellular loop-sheet polymerizationJ Biol Chem19932682115333153358340361