Abstract
Background
A formulation of crush-resistant extended-release opioids may deter abuse. The purpose of this study was to evaluate the bioequivalence of oxymorphone extended-release (Oxy-ER) and a crush-resistant formulation of oxymorphone extended-release (Oxy-CRF).
Methods
In three open-label, randomized studies, healthy adults at a clinical research center received two single oral doses of Oxy-ER and two single doses of Oxy-CRF, each separated by a ≥7-day washout. Doses were administered under fasted conditions (study 1, 5 mg doses; study 2, 40 mg doses) or after a high-fat breakfast (study 3, 40 mg doses). Subjects administered 40 mg doses also received naltrexone. The primary endpoint was systemic oxymorphone exposure; the bioequivalence criterion was met if the 90% confidence intervals of the geometric mean ratio (Oxy-CRF/Oxy-ER) for oxymorphone area under the curve from time 0 to the last measured concentration (AUC0–t), AUC from time 0 to infinity (AUC0–inf), and maximum plasma concentration (Cmax) were within 0.8–1.25. Safety was assessed by monitoring adverse events.
Results
In studies 1, 2, and 3, the safety population comprised 30, 37, and 36 subjects and the pharmacokinetics population comprised 27, 30, and 29 subjects, respectively. Oxy-ER and Oxy-CRF produced similar mean ± standard deviation oxymorphone AUC0–t (study 1, 5.05 ± 1.55 versus 5.29 ± 1.52 ng · h/mL; study 2, 31.51 ± 10.95 versus 31.23 ± 10.33 ng · h/mL; study 3, 50.16 ± 14.91 versus 49.01 ± 14.03 ng · h/mL) and Cmax (0.38 ± 0.11 versus 0.37 ± 0.12 ng/mL; 2.37 ± 1.20 versus 2.41 ± 0.94 ng/mL; 5.87 ± 1.99 versus 5.63 ± 2.26 ng/mL) under all conditions. The 90% confidence intervals for plasma oxymorphone AUC0–t, AUC0–inf, and Cmax fulfilled the bioequivalence criterion. Adverse event rates were similar with Oxy-ER and Oxy-CRF (study 1, 25% versus 23%; study 2, 9% versus 16%; study 3, 20% each group).
Conclusion
Oxy-CRF and Oxy-ER (5 mg and 40 mg) are bioequivalent under fasted and fed conditions, suggesting that Oxy-CRF will have clinical efficacy and safety equivalent to Oxy-ER.
Introduction
Between 1997 and 2006, the use of therapeutic opioids (mg/person) in the United States increased by 347%.Citation1 Illicit opioid use, as with therapeutic use, has increased rapidly in the last decade. Between 1999 and 2006, the number of individuals reporting past-month illicit use of pain relievers increased from 2,621,000 to 5,220,000.Citation1
Abuse of extended-release opioids is a particular concern because of the potential for fatal doses to be released if a tablet is crushed or chewed. A survey of prescription drug abusers entering drug rehabilitation found that 80% of abusers crush or chew extended-release opioids in order to abuse them.Citation2 Chewing or crushing can also occur without intention of abuse in patients who have difficulty swallowing intact tablets and do not understand the consequences of misusing their medication in this way. Formulation of extended-release tablets that are resistant to crushing and accidental chewing may deter abuse and prevent adverse events from misuse. Careful patient selection and adherence monitoring are also essential.Citation3,Citation4
Oxymorphone extended-release (Oxy-ER; Opana® ER, Endo Pharmaceuticals Inc, Chadds Ford, PA) is indicated for the relief of moderate to severe pain in patients requiring continuous, around-the-clock opioid treatment for an extended period.Citation5
We report three randomized clinical studies evaluating the bioequivalence of Oxy-ER and a crush-resistant formulation of oxymorphone extended-release (Oxy-CRF; EN3288®, Endo Pharmaceuticals Inc, Chadds Ford, PA) at the highest (40 mg) and lowest (5 mg) supplied dosage strengths in healthy adults. Oxy-ER is formulated using TIMERx® technology that minimizes fluctuation in drug concentrations, providing consistent 12-hour dosing,Citation6 which is advantageous in the setting of chronic pain. Oxy-CRF contains oxymorphone embedded in a hard polymer matrix (distinct from TIMERx®) that is intended to be crush-resistant. In vitro dissolution analyses of Oxy-ER and Oxy-CRF indicated that oxymorphone release from these formulations was not increased in 40% aqueous ethanol compared with 0% ethanol.Citation7 It was hypothesized that Oxy-ER and Oxy-CRF would produce equivalent systemic plasma oxymorphone exposure.
Methods
Study design
Three open-label, randomized, single-dose, replicate, cross-over studies were conducted at one site in the United States. In each study, healthy adults received two single doses of Oxy-ER and two single doses of Oxy-CRF during four alternating treatment periods according to one of two randomly assigned sequences. In study 1, subjects were administered 5 mg doses under fasted conditions; in study 2, subjects were administered 40 mg doses under fasted conditions; in study 3, subjects were administered 40 mg doses after a high-fat meal.
The studies were conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation Good Clinical Practice, and US Food and Drug Administration regulations. The protocol and informed consent form were reviewed and approved by the institutional review board (Independent Investigational Review Board Inc, Plantation, FL), and all subjects provided written informed consent before participating.
Subjects
Healthy men and women aged 18–45 years with a body mass index of 18.5–30 kg/m2 and no history of disease or clinically significant findings on physical or laboratory examination were eligible to participate. Women of childbearing potential were required to practice abstinence or use an acceptable method of birth control. Exclusion criteria were smoking, pregnancy, breast-feeding, allergy or hypersensitivity to opioids or naltrexone; a disease or condition that might interfere with drug absorption, distribution, metabolism, or excretion, or otherwise put a subject at risk; positive screen for substances of abuse, recent history of alcohol abuse, drug abuse, or significant mental illness, human immunodeficiency virus or hepatitis; recent (≤14 days) use of other medication, except hormonal contraception, and use of medication known to affect hepatic drug metabolism in the past 30 days.
Treatment
Subjects received two single oral doses of 5 mg or 40 mg Oxy-ER and two single oral doses of Oxy-CRF, each dose separated by a ≥7-day washout. Doses were administered with 240 mL of room temperature water that subjects were instructed to drink in its entirety. Medication was administered according to one of two treatment sequences with alternating treatment periods (ABAB or BABA) based on a computer-generated randomization schedule. During each treatment period, subjects were confined to the study unit from one day before dosing through 48 hours postdose. All medication was administered under supervision by study personnel, and treatment compliance was verified by a mouth and hand check.
Subjects treated under fasted conditions underwent a ≥8-hour (40 mg doses) or ≥10-hour (5 mg doses) overnight fast before drug administration and continued to fast through 4 hours after drug administration. Subjects treated under fed conditions underwent a ≥10-hour overnight fast followed by a high-fat meal (two eggs fried in butter, two strips of bacon, two slices of toast with butter, 4 oz hash brown potatoes, and 8 oz whole milk) initiated 30 minutes before drug administration. Subjects were instructed to eat the entire meal in ≤30 minutes.
To limit the potential for opioid-related adverse events, subjects who were administered 40 mg doses received three single doses of naltrexone 50 mg during each treatment period (12 doses in all). Dosing occurred 12 and 2 hours before and 12 hours after each dose of Oxy-ER and Oxy-CRF. Subjects not tolerating the two initial doses during the first treatment period were not randomized to treatment. Naloxone was readily available to all subjects in the event of respiratory depression.
Assessments
Pharmacokinetics
During each treatment period, blood samples for pharmacokinetic analysis were collected predose (≤1 hour before drug administration) and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 16, 24, 36, and 48 hours after drug administration. Samples were kept frozen at −70°C until analysis. Oxymorphone and 6-hydroxy-oxymorphone (6-OH-oxymorphone) concentrations were determined using a simultaneous liquid chromatography-tandem mass spectrometry method validated for the range of 0.025–10.00 ng/mL. Pharmacokinetic parameters (area under the concentration versus time curve from time 0 to infinity [AUC0–inf], AUC from time 0 to the last measured concentration [AUC0–t], maximum plasma concentration [Cmax], and time to Cmax [tmax]) were derived from the plasma concentration data using noncompartmental methods and actual sample times. AUC0–t was calculated using the linear trapezoid rule, and AUC0–inf was calculated as AUC0–t plus last measured plasma concentration/terminal rate constant. The terminal rate constant (λz) was calculated by linear regression of the terminal portion of the linear concentration versus time curve, and the terminal half-life [t1/2] was calculated as ln 2/λz.
Safety
Subjects were monitored for adverse events from one day before treatment through 15 days after the last dose of study medication. Relationship of adverse events to study medication (not, unlikely, possibly, or probably related) and intensity (mild, moderate, or severe) were determined by the investigator. Serious adverse events were defined as an adverse event that was immediately life-threatening, resulted in or prolonged inpatient hospitalization, resulted in death or permanent or substantial disability, was a congenital anomaly/birth defect, or might have jeopardized the subject and required medical intervention to prevent one of these outcomes. Complete physical examination was performed at screening and 48 hours after the last dose of study medication. Vital signs were recorded at screening, one day before each dose of study medication, and at specified times from 2–48 hours after each dose of study medication. Clinical chemistry, hematology, and urinalysis were performed at screening, one day before the first dose of study medication, and 48 hours after the last dose of study medication.
Statistical analysis
Pharmacokinetic parameters were analyzed in all subjects who received ≥1 dose of Oxy-ER and Oxy-CRF and were determined by the pharmacokineticist to have sufficient plasma concentration data to calculate AUC and Cmax. Pharmacokinetic data from subjects who had vomited within the first 12 hours of a treatment period were not analyzed. For calculation of mean concentrations, values below the limit of quantification were set to 0 or, if occurring between two other such concentrations, were indicated as missing. For calculation of pharmacokinetic parameters, plasma concentrations below the limit of quantification were set to 0 if occurring before the first measurable concentration and otherwise were indicated as missing.
Pharmacokinetic parameters were summarized descriptively, using the number of subjects, mean, standard deviation, and coefficient of variation (AUC0–t, AUC0–inf, Cmax, and t1/2) or the median and range (tmax). Repeated measures analysis of variance with treatment as the fixed factor and subject within-treatment sequence as a random factor was performed on the log-transformed values for AUC0–t, AUC0–inf, and Cmax. Geometric mean ratios (Oxy-CRF/Oxy-ER) and 90% confidence intervals (CI) were calculated by the antilog of the least squares mean differences and their CIs. The criterion for bioequivalence was met if the 90% CIs of the geometric mean ratio (Oxy-CRF/Oxy-ER) for oxymorphone AUC0–t, AUC0–inf, and Cmax were within 0.8–1.25.
The safety population comprised all subjects who received ≥1 dose of Oxy-ER, Oxy-CRF, or naltrexone. Safety variables were summarized by treatment and using appropriate descriptive statistics.
SAS® (version 9.2, SAS Institute Inc, Cary, NC) was used for all analyses. A sample size of 26 subjects was planned for each study. Assuming an intrasubject coefficient of variation of 30.2% for oxymorphone, and allowing for a 5% difference between treatment groups, this sample size would provide ≥90% power to demonstrate bioequivalence using this replicated dosing design.
Results
Subject disposition and characteristics
Subject disposition is shown in . In study 1, 2, and 3, there were 30, 37, and 36 subjects enrolled and included in the safety population and 28, 30, and 29 subjects who completed the study, respectively. In study 1 (5 mg, fasted), two subjects were excluded from the pharmacokinetics population because they were administered only one dose of Oxy-ER. The pharmacokinetics population comprised 27 subjects who completed the study and one who partially completed the study. In study 2 (40 mg, fasted), 6 subjects were excluded from the pharmacokinetics population because they were not administered any Oxy-ER or Oxy-CRF (n = 3) or because they were administered only one dose of Oxy-ER (n = 3). The pharmacokinetics population comprised all 30 subjects who completed the study and one who partially completed the study. In study 3 (40 mg, fed), six subjects were excluded from the pharmacokinetics population because they were not administered oxymorphone. The pharmacokinetics population comprised 29 subjects who completed the study and one who partially completed the study.
Figure 1 Subject disposition.
Demographics and clinical characteristics were similar in the three studies (). Across the three studies, mean age was 33–35 years, 47%–60% of subjects were women, ≥78% were white, and mean body mass index was 25–26 kg/m2.
Table 1 Subject demographics (safety population)
Plasma oxymorphone pharmacokinetics
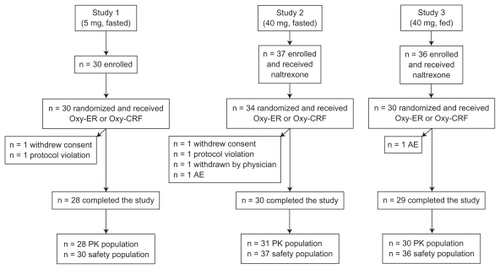
With all doses and administration conditions (fasted or fed), the oxymorphone plasma concentration versus time profile was similar overall for Oxy-ER and Oxy-CRF (). Each profile demonstrated two distinct peaks at approximately 2–3 hours and 5–6 hours after drug administration, followed by a gradual decline in plasma concentration interrupted by a brief plateau at approximately 10–12 hours after drug administration. The second peak was generally larger than the first, but the difference was more notable for Oxy-CRF, which had a smaller initial peak than Oxy-ER. Mean oxymorphone plasma concentrations for Oxy-ER and Oxy-CRF were the same starting 6 (5 mg, fasted), 8 (40 mg, fasted), or 16 hours (40 mg, fed) after administration.
Figure 2 Mean oxymorphone plasma concentrations 0–48 hours after single oral doses of Oxy-ER and Oxy-CRF (A) 5 mg administered under fasted conditions, (B) 40 mg administered under fasted conditions, and (C) 40 mg administered after a high-fat breakfast.
Systemic plasma oxymorphone exposure (AUC and Cmax) was also similar after single doses of Oxy-ER and Oxy-CRF (). Mean ± SD oxymorphone AUC0–t for Oxy-ER and Oxy-CRF, respectively, was 5.05 ± 1.55 and 5.29 ± 1.52 ng · h/mL after a 5 mg dose administered under fasted conditions, 31.51 ± 10.95 and 31.23 ± 10.33 ng · h/mL after a 40 mg dose administered under fasted conditions, and 50.16 ± 14.91 and 49.01 ± 14.03 ng · h/mL after a 40 mg dose administered following a high-fat breakfast. Mean ± SD oxymorphone Cmax for Oxy-ER and Oxy-CRF, respectively, was 0.38 ± 0.11 and 0.37 ± 0.12 ng/mL after a 5 mg dose administered under fasted conditions, 2.37 ± 1.20 and 2.41 ± 0.94 ng/mL after a 40 mg dose administered under fasted conditions, and 5.87 ± 1.99 and 5.63 ± 2.26 ng/mL after a 40 mg dose administered following a high-fat breakfast.
Table 2 Plasma oxymorphone pharmacokinetic parameters
Median oxymorphone tmax was the only pharmacokinetic parameter to differ between Oxy-ER and Oxy-CRF, being shorter for Oxy-ER versus Oxy-CRF 40 mg (), with differences of 2 hours under fasted conditions (3.0 versus 5.0) and 1.5 hours under fed conditions (3.5 versus 5.0). The difference in median tmax was smaller when 5 mg doses of Oxy-ER and Oxy-CRF were compared (6.0 versus 5.0 hours). The tmax values roughly corresponded to the time at which the two early peaks occurred in the plasma concentration versus time profiles (). The range of tmax was similar for the two formulations in each of the three studies.
Plasma 6-hydroxy-oxymorphone pharmacokinetics
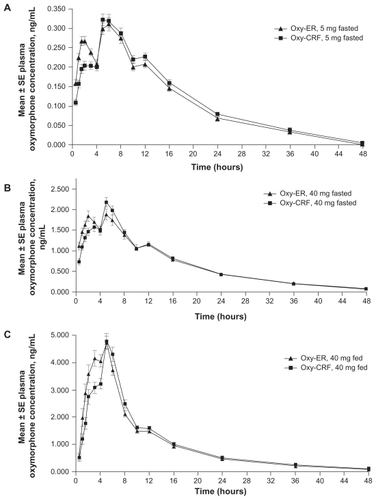
Mean plasma 6-OH-oxymorphone concentration versus time profiles were similar for Oxy-ER and Oxy-CRF, regardless of dose or administration conditions (). Profiles exhibited a single peak at approximately 2–3 hours under fasted conditions and 5 hours under fed conditions. Starting 6 (5 mg, fasted), 12 (40 mg, fasted), or 16 hours (40 mg, fed) after drug administration, mean concentrations were the same for Oxy-ER and Oxy-CRF. Systemic plasma 6-OH-oxymorphone exposure did not differ between the two formulations (), but median tmax was 1.0 (40 mg, fed) to 1.5 hours (5 mg, fasted; 40 mg, fasted) shorter for Oxy-ER versus Oxy-CRF.
Table 3 Plasma 6-OH-oxymorphone pharmacokinetic parameters
Figure 3 Mean 6-OH-oxymorphone plasma concentrations 0–48 hours after single oral doses of Oxy-ER and Oxy-CRF (A) 5 mg administered under fasted conditions, (B) 40 mg administered under fasted conditions, and (C) 40 mg administered after a high-fat breakfast.
Bioequivalence
Within-subject variability in oxymorphone AUC and Cmax ranged from 11% to 24% and was comparable between the two oxymorphone formulations (data not shown). For all doses and under both fasted and fed conditions, the 90% CI for the comparisons of Oxy-ER and Oxy-CRF plasma oxymorphone AUC0–t, AUC0–inf, and Cmax were within 0.8–1.25 (), fulfilling the bioequivalence criterion. Comparisons of Oxy-ER and Oxy-CRF plasma 6-OH-oxymorphone were supportive of the finding of bioequivalence with respect to oxymorphone ().
Table 4 Bioequivalence data: systemic plasma oxymorphone exposure
Table 5 Bioequivalence data: systemic plasma 6-OH-oxymorphone exposure
Safety
The proportion of subjects administered Oxy-ER and Oxy-CRF, respectively, who experienced ≥1 treatment-emergent adverse event was 25% (n = 7/28) and 23% (7/30) for the 5 mg doses administered under fasted conditions, 9% (3/34) and 16% (5/31) for the 40 mg doses administered under fasted conditions, and 20% (6/30, both treatments) for the 40 mg doses administered under fed conditions. The most frequent adverse events, and the only adverse events that occurred in ≥1 subject treated with a particular dose of Oxy-ER or Oxy-CRF, were nausea, headache, vomiting, and dizziness. Treatment-related adverse events were infrequent with both formulations (range for treatment groups, 3%–13% of subjects). There were no severe adverse events, no adverse events, and no deaths. There were no clinically significant changes in physical examination findings, vital signs, or laboratory findings that were deemed to be related to treatment.
Discussion
These three randomized clinical trials evaluated the bioequivalence of the lowest (5 mg) and highest (40 mg) supplied dosage strengths of Oxy-ER and the same doses of Oxy-CRF in healthy adults under fasted (5 mg, 40 mg) and fed (40 mg) conditions. Oxy-ER and Oxy-CRF demonstrated overall similar oxymorphone and 6-OH-oxymorphone plasma concentrations over time. Differences in tmax between Oxy-ER and Oxy-CRF were not clinically relevant. The criterion for bioequivalence of plasma oxymorphone exposure was met for all parameters (AUC0–t, AUC0–inf, Cmax). In these subjects, most of whom also received naltrexone, Oxy-ER and Oxy-CRF were generally well tolerated, with no discernable differences between formulations.
Findings were consistent overall for oxymorphone and its active metabolite, 6-OH-oxymorphone. For 6- OH-oxymorphone, overall systemic plasma exposure, as indicated by the AUC0–t, did not differ between the two formulations, and data were supportive of the finding of bioequivalence with respect to oxymorphone.
In the 40 mg dose studies, naltrexone was administered at the beginning of each treatment period to limit opioid-related adverse events. Although naltrexone is reported to increase oxymorphone peak plasma exposure (data on file, Endo Pharmaceuticals Inc), naltrexone administration in the current study is not likely to have affected the evaluation of bioequivalence because dose and time of administration were standardized across subjects and treatment periods.
The presence of peaks and shoulders in the concentration versus time curves has been observed in previous studies (data on file, Endo Pharmaceuticals Inc). The timing of the later peaks often corresponds with eating and may be related to the known increase in systemic exposure to oxymorphone when administered with food,Citation5 combined with prolonged absorption from these formulations.
The bioequivalence findings indicate a 1:1 correspondence between doses of Oxy-ER and Oxy-CRF. This would facilitate conversion from Oxy-ER to Oxy-CRF. In addition, the ratios for conversion of other oral opioid doses to Oxy-ER can be considered appropriate for Oxy-CRF.Citation5
Study subjects were selected to satisfy US Food and Drug Administration guidelines for bioequivalence studiesCitation8 and not to represent the clinical population that would be using oxymorphone. Moreover, subjects treated with 40 mg doses also received naltrexone. Thus, the safety profile was not the same as in patients or healthy subjects not receiving an opioid antagonist. However, Oxy-ER and Oxy-CRF had a similar safety profile for subjects administered an opioid antagonist (ie, naltrexone) along with an opioid. There were no important differences in the type or frequency of adverse events, regardless of dose or condition of administration (fasted or fed). There was also no indication of new adverse events or an increase in the frequency of adverse events with Oxy-CRF versus Oxy-ER.
The efficacy and safety of Oxy-ER have been demonstrated in randomized controlled trials of up to 12 weeks and extension trials of up to one year in opioid-naive and opioid-experienced patients with moderate to severe chronic low back pain,Citation9,Citation10 cancer pain,Citation11,Citation12 and osteoarthritis.Citation13,Citation14 Other benefits are the low potential for drug–drug interactions (oxymorphone does not inhibit cytochrome P450 enzymes)Citation15 and simplified interpretation of urine testing (oxymorphone does not produce any metabolites that can be mistaken for another prescribed drug).Citation16 The bioequivalence findings of the studies reported here indicate that Oxy-CRF is expected to have clinical efficacy and safety equivalent to Oxy-ER.
Conclusion
Three randomized clinical trials demonstrated bioequivalence of Oxy-ER and Oxy-CRF 5 mg under fasted conditions and 40 mg under fasted and fed conditions. The treatments had similar safety profiles and were generally well tolerated, given that most subjects also received naltrexone.
Acknowledgments
The Crush Resistant Formulation technology was developed by and is property of Gruenenthal, Aachen, Germany. Endo holds an exclusive license for the use of the technology with Opana ER.
Axel Juan and Audrey Martinez, both of SeaView Research Inc (Miami, FL) were principal investigators for these studies. Clinical monitoring of the studies was performed by Paula Allen. Bioanalytical analyses were performed at Cetero Research (Houston, TX).
Editorial Support (literature search, document retrieval, medical writing, and copy editing) for the development of this manuscript was provided by Nicole Strangman, PhD, and Robert Gatley, MD, of Complete Healthcare Communications, Inc (Chadds Ford, PA) with funding from Endo Pharmaceuticals Inc (Chadds Ford, PA).
Disclosure
All authors were employees of Endo Pharmaceuticals Inc during the course of these studies. Endo Pharmaceuticals Inc (Chadds Ford, PA) provided funding for the study and manuscript development.
References
- ManchikantiLSinghATherapeutic opioids: a ten-year perspective on the complexities and complications of the escalating use, abuse, and nonmedical use of opioidsPain Physician2008112 SupplS638818443641
- PassikSDHaysLEisnerNKirshKLPsychiatric and pain characteristics of prescription drug abusers entering drug rehabilitationJ Pain Palliat Care Pharmacother200620251316702131
- ChouRFanciulloGJFinePGClinical guidelines for the use of chronic opioid therapy in chronic noncancer painJ Pain200910211313019187889
- TrescotAMHelmSHansenHOpioids in the management of chronic non-cancer pain: an update of American Society of the Interventional Pain Physicians’ (ASIPP) GuidelinesPain Physician2008112 SupplS56218443640
- OPANA® ER (oxymorphone hydrochloride extended-release tablets)Full Prescribing InformationChadds Ford, PAEndo Pharmaceuticals Inc2008
- AdamsMPAhdiehHPharmacokinetics and dose-proportionality of oxymorphone extended release and its metabolites: results of a randomized crossover studyPharmacotherapy200424446847615098800
- FiskeWJobesJXiangQChangSBenedekIThe effects of ethanol on the bioavailability of oxymorphone extended-release tablets and oxymorphone crush-resistant extended-release tabletsJ Pain2011 In press
- US Department of Health and Human ServicesGuidance for industry: bioavailability and bioequivalence studies for orally administered drug products-general considerations Available at: http://www.fda.gov/downloads/Durgs/Guidancecomplianceregulatoryinformation/guidances/ucm070124.pdfAccessed October 29, 2010
- HaleMEAhdiehHMaTRauckREfficacy and safety of Opana ER (oxymorphone extended release) for relief of moderate to severe chronic low back pain in opioid-experienced patients: a 12-week, randomized, double-blind, placebo-controlled studyJ Pain20078217518417145204
- KatzNRauckRAhdiehHA 12-week randomized placebo-controlled trial assessing the safety and efficacy of oxymorphone extended release for opioid-naive patients with chronic low back painCurr Med Res Opin200723111712817257473
- GabrailNYDvergstenCAhdiehHEstablishing the dosage equivalency of oxymorphone extended release and oxycodone controlled release in patients with cancer pain: a randomized controlled studyCurr Med Res Opin200420691191815200750
- SlatkinNERhinerMIGouldEMMaTAhdiehHLong-term tolerability and effectiveness of oxymorphone extended release in patients with cancerJ Opioid Manag20106318119120642247
- KivitzAMaCAhdiehHGalerBSA 2-week, multicenter, randomized, double-blind, placebo-controlled, dose-ranging, phase III trial comparing the efficacy of oxymorphone extended release and placebo in adults with pain associated with osteoarthritis of the hip or kneeClin Ther200628335236416750450
- McIlwainHAhdiehHSafety, tolerability, and effectiveness of oxymorphone extended release for moderate to severe osteoarthritis pain: a one-year studyAm J Ther200512210611215767827
- SmithHSOpioid metabolismMayo Clin Proc200984761362419567715
- ConeEJCaplanYHUrine toxicology testing in chronic pain managementPostgrad Med200912149110219641275