 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
The aim of this study was to develop a generalized in vitro-in vivo relationship (IVIVR) model based on in vitro dissolution profiles together with quantitative and qualitative composition of dosage formulations as covariates. Such a model would be of substantial aid in the early stages of development of a pharmaceutical formulation, when no in vivo results are yet available and it is impossible to create a classical in vitro-in vivo correlation (IVIVC)/IVIVR.
Methods
Chemoinformatics software was used to compute the molecular descriptors of drug substances (ie, active pharmaceutical ingredients) and excipients. The data were collected from the literature. Artificial neural networks were used as the modeling tool. The training process was carried out using the 10-fold cross-validation technique.
Results
The database contained 93 formulations with 307 inputs initially, and was later limited to 28 in a course of sensitivity analysis. The four best models were introduced into the artificial neural network ensemble. Complete in vivo profiles were predicted accurately for 37.6% of the formulations.
Conclusion
It has been shown that artificial neural networks can be an effective predictive tool for constructing IVIVR in an integrated generalized model for various formulations. Because IVIVC/IVIVR is classically conducted for 2–4 formulations and with a single active pharmaceutical ingredient, the approach described here is unique in that it incorporates various active pharmaceutical ingredients and dosage forms into a single model. Thus, preliminary IVIVC/IVIVR can be available without in vivo data, which is impossible using current IVIVC/IVIVR procedures.
Video abstract
Point your SmartPhone at the code above. If you have a QR code reader the video abstract will appear. Or use:
Introduction
An efficient in vitro-in vivo correlation (IVIVC) model is a tool for predicting the in vivo bioavailability of a particular drug based on its in vitro data, and is simple to obtain using reproducible and inexpensive dissolution tests. This correlation enables the bioperformance of various dosage formulations to be evaluated without conducting animal or human studies. Applications of IVIVC include justification of biowaivers, post-approval scale-up changes,Citation1 and establishing dissolution specifications. IVIVC is also used during the early stages of development of a dosage formulation, bringing in some degree of biorelevance to in vitro dissolution tests.Citation2 The classical term, IVIVC, is often broadened to include the in vitro-in vivo relationship (IVIVR), indicating introduction of nonlinear modeling when classical linear IVIVC relationships are not sufficient or are not applicable.Citation3
Modeling based on artificial neural networks (ANNs) is a well established method.Citation4 Although it requires vast computational power, it is very useful because of its self-organizational properties and ability to incorporate many variables and relationships without a predefined model structure, unlike conventional statistical methods. In the case of IVIVR, these features allow inclusion of more variables than just the dissolution profile, such as formulation composition and manufacturing parameters when building a model, and enable direct evaluation of their influence on the in vivo response.
ANNs encompass a vast number of computational techniques which can imitate the human brain morphologically and functionally, and are capable of identifying hidden relationships between many variables. In practice, a neural network is a set of highly interconnected processing elements known as nodes (analogs of biological neurons), which store experimental knowledge by adjusting connection weights and make that knowledge available for use. An ANN is characterized by so-called architecture (arrangement of nodes and their connections), activation function, and training conditions. ANN architecture consists of an input and output layer, and one or more hidden layers. Relevant in this case is so-called supervised training, which is performed by presenting pre-existing task examples (the training data set) with known system responses to the input patterns. This is a crucial step for obtaining a well performing model able to predict output values for unknown patterns.Citation4
Neural modeling is already well established in pharmaceutical science. In pharmaceutical technology, ANNs have been successfully applied for optimization of formulationsCitation5–Citation8 and optimization of preparation technology.Citation9–Citation11 There are also numerous applications of ANNs in pharmacokinetics.Citation12–Citation17 Finally, neural modeling for IVIVC has been studied.Citation18–Citation21 All these applications of ANNs for IVIVC use these networks as a nonlinear mapping tool for in vitro and in vivo profiles, and have been limited to single drugs and/or formulations. So far, to the authors’ knowledge, there has been no attempt to create a universal model encompassing different formulations and different active pharmaceutical ingredients.
The aim of this study was to investigate the concept of a generalized IVIVR model, introducing, in addition to in vitro and in vivo profiles, chemoinformatics software providing physicochemical properties and structural data about drug substances and excipients. Use of chemoinformatics ensures the ability of the model to predict the bioavailability of various drugs in different dosage formulations. Such a model might be used earlier than classical IVIVC, given that the latter requires biological data as the dependent variable for the correlation.
Materials and methods
Knowledge database
A knowledge database was acquired from the published literature. Articles concerning bioavailability and dissolution studies of various formulations were scanned, and the relevant data was extracted upon following conditions:
Complete details on qualitative and quantitative composition of immediate-release or modified-release formulations, as well as dissolution tests and bioavailability studies results
Bioavailability studies were carried out in humans, with tablets or capsules administered orally
Release modification was based on the matrix system
The active pharmaceutical ingredient was a small chemical molecule (with peptides and antibodies discarded)
Dissolution tests carried out using USP apparatus I or II.
The data records were characterized by the following variables:
Formulation characteristics, ie, type, and qualitative and quantitative composition
In vitro dissolution profile
In vivo pharmacokinetic profile
In vitro and in vivo assay conditions.
Formulation
The formulation type was encoded as two binary parameters. The first denoted immediate-release versus modified-release formulation, encoded as 0 and 1, respectively, and the second denoted tablet versus capsule, respectively.
Numerical description of the qualitative composition of the formulation was based on chemoinformatics tools. Each ingredient of the formulation was incorporated into the database as a set of molecular descriptors computed with MarvinCitation22 calculator plugins (ChemAxon, Budapest, Hungary) and classified into three groups, ie, active pharmaceutical ingredient, polymeric release-modifying excipients, and nonpolymeric release-modifying excipients.
Computation of molecular descriptors for the active pharmaceutical ingredient and nonpolymeric excipients was straightforward with use of standard software settings. Prior to computation of molecular descriptors, three-dimensional optimization of the molecules was performed using the gradient method with Marvin software.Citation22
For polymeric release-modifying excipients, direct use of chemoinformatics software is difficult or sometimes impossible. This is mainly due to input limitations on compound size in the chemoinformatics software. To overcome this obstacle, polymers were presented as dimers with hydrogen instead of the chain fragment on one side and hydroxyl on the other. For cellulose derivatives, various substitution combinations were taken into account and several sets of descriptors were computed for different substitution variations, which were averaged according to the molar substitution ratio for each group. Calculations for hydroxypropylmethylcellulose were presented schematically in .
Figure 1 Schematic calculation of descriptors set encoding hypromellose.
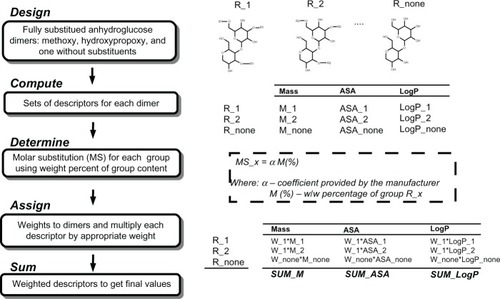
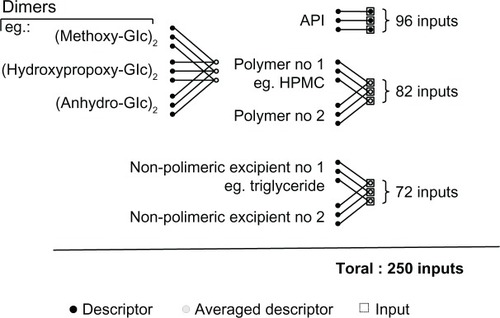
Agents other than polymers still influencing the drug release rate, such as triglycerides, and citric or stearic acid, were classified into a third group of nonpolymeric excipients. In total, 250 descriptors were introduced into the native input vector. An applied schema for merging and weight averaging is shown in . Quantitative composition was simply expressed as the weight percentages for each compound along with the dose of the active pharmaceutical ingredient expressed both as a percentage and in milligrams.
Figure 2 Chemoinformatic description of the excipients included into the native input vector.
In vitro and in vivo profiles
In order to ensure compatibility of the data, the pharmacokinetic and dissolution profiles were preprocessed. For pharmacokinetic profiles, the concentration units were recalculated if necessary and expressed in ng/mL. There was no such problem for the in vitro profiles because their values were always available in percentages of the total drug amount in the formulation. For both the in vitro and in vivo profiles, the average values reported in the published literature were used. Most of the data was extracted from the graphs by g3data software using a simple point-and-click procedure. Because of the manual nature of the above procedure, another member of the team rescanned approximately 5% of the randomly chosen profiles for validation purposes. All time points for the in vitro and in vivo profiles were expressed in hours. The time scale for the pharmacokinetic profiles was 0–120 hours and for dissolution profiles was 0–24 hours. There were up to 18 time points available for the dissolution profiles; for those with a smaller number of points, the remaining points were created artificially with time up to 24 hours and plateau values. The latter was the result of linear extrapolation between the last value present and the maximum possible amount of active pharmaceutical ingredient released (100%) at the time endpoint of 24 hours.
Assay conditions
For the in vitro dissolution profiles, there was information provided about the volume and pH of the medium, together with the paddle or basket rotation speed (rpm). Sodium lauryl sulfate concentration was also introduced as a variable. For pharmacokinetic profiles, in order to ensure eligibility of the maximum number of papers, in vivo characteristics was limited to information about whether the assay was performed in a fasted or fed state.
Neural modeling
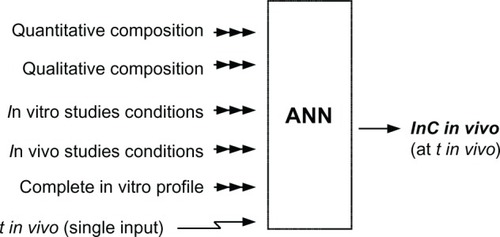
The neural models of IVIVR were designed to predict whole plasma concentration-time profiles based on dissolution curves and the composition of the formulation, as well as data on the conditions of the in vivo and in vitro studies.
All models were multiple-input-single-output type with the natural logarithm of plasma concentration as the output vector. The input vector consisted of the quantitative and qualitative description of the formulation as described above together with the in vitro dissolution profile, in vivo sampling time, and conditions of the in vitro and in vivo assays ().
Figure 3 General structure of artificial neural network models.
Multilayer perceptrons with a back-propagation learning algorithm were applied. The following activation functions were implemented: linear, logistic, hyperbolic tangent, and a combination of logarithmic functions known as “fsr”.Citation23 The ANN architectures varied from one to seven hidden layers. In addition to multilayer perceptrons, neurofuzzy systems of the simplest Mamdani type were employed, with 5–100 nodes in the hidden layer. In order to match nonlinear activation function domains, the data were scaled linearly using output ranges of <0.2, 0.8>, and <−0.8, 0.8>, respectively. A noise addition was performed with amplitude ±5% of the original values and a four times larger number than the original records. The latter procedure is a well known technique which is used to improve the generalizability of neural models.Citation4 Numerous modifications of the classical back-propagation algorithm were used as follows:
Momentum technique with a momentum factor of 0.3
Delta-bar-delta algorithm with an initial learning factor of 0.65
Jog-of-weights technique designed to avoid becoming stuck in the local minima of the cost function; a simple noise addition to the weights where the ANN was not improving its efficiency during 100,000 epochs (the patience criterion).
In total, considering two types of scaling and whether or not noise data were used 336 types of neural networks were trained and tested. Training was conducted up to numerous predefined steps (50,000, 100,000, and up to 10,000,000 iterations), after which training and generalization errors were observed. The error measures were root mean squared error (Equationequation 1(1) ) and normalized root mean squared error (Equationequation 2
(2) )
where obsi is the observed value, predi is the predicted value, and n is the total number of records.
where RMSE is the root mean squared error, MAXobs is the maximum value of the observed results, and MINobs is the minimum value of the observed results.
Two major modes of ANN training were used, ie, sensitivity analysis on the whole dataset and a 10-fold cross-validation technique for assessment of generalizability. These modes were used in their interplay in order to provide the minimum input vector, ie, the set of crucial variables. Optimization of ANN architecture was derived at the same time. A detailed description of this approach has been presented elsewhere.Citation24
An extended 10-fold cross-validation scheme was used, whereby whole formulations were treated as units for construction of pairs of train/test datasets. This was to ensure that the all information about a particular formulation was either in the test or the train dataset, in order to simulate the ractical application of the system, ie, to predict the actual bioperformance of the formulation.
Sensitivity analysis was done using the method described by Żurada et al,Citation25 with some modificationsCitation24 to enable knowledge-based selection of crucial variables and obtain collective results from the set of best trained ANNs.
ANN ensembles (“expert committees”) were prepared in order to enhance the predictive ability of the models obtained. These higher order models were constructed from the best ANNs identified. Their total output was computed as the average of the outputs for the ANNs chosen as members of the ensemble.
Three rankings were prepared for a clear description of the predictability of the model, ie, ability to predict the whole in vivo profile, ability to predict the elimination phase, and ability to predict the absorption phase. The absorption phase and elimination phase were simply chosen as the ranges from t = 0 to tmax and from tmax to the last time point, respectively, thus being the ascending and descending parts of the pharmacokinetic curve. Successful prediction was confirmed if the normalized root mean squared error did not exceed 20%.
Hardware and software environment
The neural analysis was performed using own-written simulator Nets2010 and numerous applications for data preprocessing,Citation24 all working in the Linux environment. Literature in graphic form was digitalized using g3data version 1.5.2.Citation26 Formulation ingredients were encoded by Marvin version 5.3.8 plugins.Citation22 All computations were performed on 29 PC workstations (116 cores). The total number of neural networks trained and tested was approximately 8000.
Results
Our search identified 93 formulations in the literature database. It contained a description of 13 active pharmaceutical ingredients, ie, levosimendan,Citation27,Citation28 ritonavir,Citation29 danazol,Citation30 metoprolol,Citation31–Citation34 griseofulvin,Citation35 diltiazem,Citation36 propranolol,Citation32 alprazolam,Citation37 ketoprofen,Citation38 diclofenac,Citation39 carbamazepine,Citation40 ibuprofen,Citation41,Citation42 and theophylline.Citation43 In total, the database yielded 1067 data records. Initially, there were 307 inputs of the neural model () describing active pharmaceutical ingredients, excipients, and assay conditions.
Table 1 General description of the native input vector
Sensitivity analysis showed the significance of variables describing the type of drug formulation (tablet or capsule, immediate-release/modified-release), bioavailability and dissolution study conditions, and quantitative composition (). The model was less sensitive to inputs covering the dissolution profile. The most important molecular descriptors were active pharmaceutical ingredient, with polymeric excipients lower in the ranking and descriptors for nonpolymeric excipients being discarded.
Figure 4 Results of sensitivity analysis for the most important 28 inputs, with relative importance computed in the context of the native dataset.
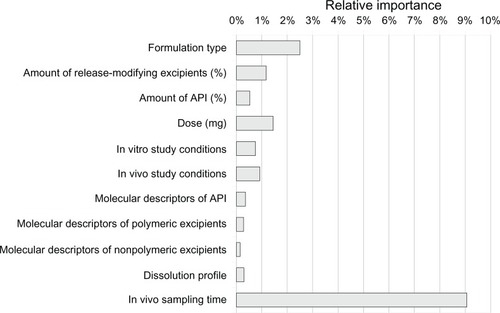
As a result of the input vector reduction procedure, 28 governing variables were chosen (), among which were 13 molecular descriptors of active pharmaceutical ingredients and three descriptors of polymers. Only two sampling times remained for the in vitro profile, resulting in four inputs.
Table 2 Input vector reduced to 28 governing variables
Using the trial and error method, various combinations of both multilayer perceptron nets and fuzzy neural networks were formed into the ANN ensembles and tested for the lowest generalization root mean squared error. Finally, four ANNs were combined (), with a collective root mean squared error of 1.05. The preferred activation functions were fsr and hyperbolic tangent. Fuzzy networks, although capable of predicting profiles for certain formulations, proved to be insufficient for generalization of the different formulations. The number of hidden layers varied from two to seven, within the total number of nodes from 12 to 142. Such diversity shows that different problems (different dosage formulations, model drugs, orders of magnitude of plasma concentrations) were better solved with different ANN architecture, so it is understandable that the collective root mean squared error was lower than for each ANN apart.
Table 3 Architecture of ANNs selected for expert committee and their generalization errors
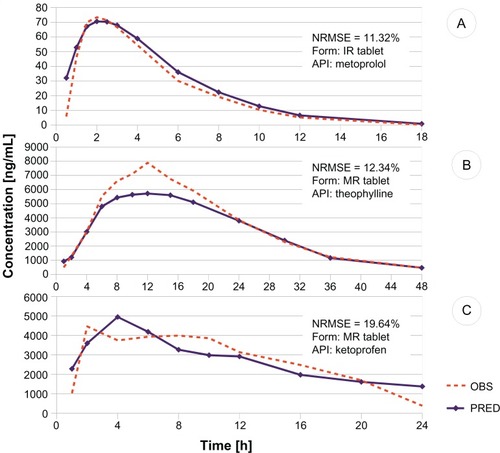
Charts with plasma profiles predicted by an expert committee and profiles observed in the clinical trials were plotted (). According to the aforementioned criterion of normalized root mean squared error < 20%, a ranking was created which showed successful prediction of the whole profile in 37.6% of the formulations. The elimination part of the curve was appropriately predicted for 49.5% of the formulations, whereas absorption was predicted for only 19.4%. Such a tendency for better predictability of the part of the in vivo curve covering the elimination phase over the one describing the absorption process could be explained by the observed higher sensitivity of the model to molecular descriptors of the active pharmaceutical ingredient and lower sensitivity to the inputs responsible for the excipients and the dissolution profile. The process of elimination of a drug involves elimination kinetics, metabolism, and/or redistribution, all of which depend on the physicochemical properties of the active pharmaceutical ingredient rather than the drug formulation. On the other hand, the formulation and its influence on the dissolution rate of the active pharmaceutical ingredient has a great impact on the rate of drug absorption, especially in the case of modified-release formulations.
Figure 5 Examples of in vivo profile predictions.
Discussion
The foundations of the IVIVR are that the in vitro release profile for an active pharmaceutical ingredient should reflect basic release/dissolution processes, which are at least in part responsible for the bioavailability of the active ingredient. The low affinity of ANN models for the dissolution profile suggests that the in vitro data available in the database we collected did not correlate well with the in vivo profiles. A further numerical experiment was performed in order to investigate this problem. The original model based on 28 inputs was enhanced with the whole dissolution profile, despite the previously described sensitivity analysis results. As a result, 58 input-based models were created, trained, and tested according to the methodology described earlier. ANN ensembles for a model based on these 58 inputs were built on the same architecture as that used for the model of 28 inputs. The resulting generalization root mean squared error was found to be 1.51, confirming the relatively low correlation of the in vitro and in vivo profiles in the database analyzed. An increase in error might be associated with the well known “curse of dimensionality”, resulting from the enlarged input vector, but it is noteworthy that all the in vitro assays were carried out using USP apparatus I or II. These methods are still the standard for dissolution testing, so the data sources were available at a level only allowing work such as that presented here. Despite standardization of the methods used, USP apparatus I and II mimic in vivo conditions poorly, sometimes not even achieving sink conditions. Our modeling results presented here are indirect proof of this.
Another issue is the formulation description used, ie, chemical structure only, which was included together with abstract classification as immediate-release or modified-release. This is surely inadequate for thorough description of a pharmaceutical formulation, and some physical parameters should also be included, such as dimensions, hardness, and particle size of the excipients. Unfortunately, no such information was available in the papers selected, and if any such variables were included, they were so scarce that they were not useful for systematic quantitative analysis. A similar dilemma of how to provide a database representative enough to build the model was encountered for the in vivo profiles. Their description was limited only to the fast/fed state because this information was available by default. As a consequence, the model was built on average data, without taking into account any intrasubject and intersubject variability nor mentioning any demographic data. An attempt to introduce the above-mentioned data would be the next stage of research.
The above considerations of model performance are based on the potential for extrapolation, which is not perfect. It is important to remember here that the above-mentioned generalization errors are the result of the 10-fold cross-validation procedure, which is a statistical technique and a derivative of bootstrapping methods. The 10-fold cross-validation procedure tests the predictive ability of the model according to the whole available database, yet with use of the external data, excluded from the training process of the model. Here, external data are a result of the resampling technique applied to the database, ie, 10-fold cross-validation. Therefore, the results presented in the reflect the real situation, where the model is faced with the task of predicting bioperformance of a completely unknown formulation based solely on its in vitro and chemoinformatic characteristics.
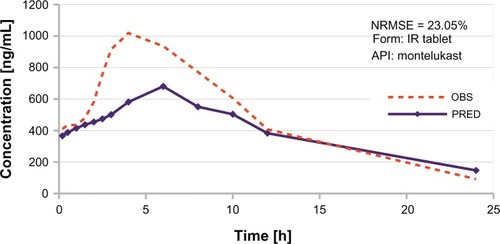
Further, in order to demonstrate the real application of the system, a new dataset was introduced, including data from the recent study published by Zaid et al concerning the in vitro-in vivo characteristics of montelukast,Citation44 which is a completely unknown structure in the system. Prediction of its behavior is presented in . It is not a perfect representation of the pharmacokinetic profile measured in vivo (normalized root mean squared error 23%), but ANNs were able to predict the general characteristics of the pharmacokinetic profile for montelukast from the immediate-release formulation, ie, tmax (overestimated by 1.5 hours) and the last measured concentration. Moreover, ANNs correctly predicted that the concentration of montelukast rises rapidly after administration of the drug, thus the first point measured is around 400 ng/mL. This result is an example of extrapolation beyond the known database, and this can be illustrated by comparison of the values for the Dreiding energy parameter, ie, the maximum value for the database is 137.26, whereas for montelukast it was estimated at 357.48. The numbers quoted above are confirmation of the long-range extrapolation performed by the neural model, and although not entirely precise, are still reasonable and within the range of concentrations measured in vivo ().
Figure 6 Example of in vivo profile prediction for the new test dataset introduced after the model development phase.
Conclusion
While clinical trials have remained time-consuming and expensive despite significant technological improvements over the years, development of information technology has increased rapidly and provides inexpensive hardware with immense computation power as well as superior software. Therefore, it seems reasonable to use its potential in the field of pharmaceutical research. IVIVC/IVIVR is now a standard and necessary tool in the pharmaceutical industry and reduces the cost of clinical trials, but in its classical form has limited applications. Specifically, the classical IVIVC/IVIVR approach involves 2–4 formulations with a single active pharmaceutical ingredient and requires in vivo data along with in vitro release profiles. Therefore, the procedure is limited in its ability to identify a mathematical function describing two sets of data (in vivo versus in vitro), and it is obligatory to have both these datasets.
Any extension of the current capabilities of IVIVC/IVIVR models would be beneficial, providing more scientific justification for decision-making in the early stages of drug development. The model presented here provides such an extension by introducing the complex relationships between active pharmaceutical ingredients and excipients, and mapping the in vitro profile directly to the in vivo profile for a particular drug formulation. The input of the system does not require an in vivo profile, so allows an IVIVR to be created at an earlier stage of drug development than does classical IVIVC/IVIVR.
In this study, neural modeling was demonstrated to be capable of handling complex problems, such as building an IVIVR model using a generalized approach not identified in the published literature. A detailed methodology was established which is suitable for further extension of the database, which is a crucial task in future development of IVIVR for real-life application. It was a first step towards the development of an integrated IVIVC/IVIVR system in silico, which might be used in the early stages of drug development to assess the effects of formulation parameters on the biological properties of a given drug. Such an empirical modeling-based system would be an enhancement, but certainly not a replacement, for current systems based on mechanistic modeling. A major advantage of such neural models is that they can integrate information easily, thus enhancing the models currently used, and provide more detailed information on formulation characteristics and a better description of test subjects if the relevant data were provided in the future.
Acknowledgment
The authors thank Dirk Schmalz from Harke Pharma GmbH for supplying details of formulae for computation of molecular substitution in the hydroxypropylmethylcellulose derivatives.
Disclosure
The authors report no conflicts of interest in this work.
References
- US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research Guidance for Industry SUPAC-MR: modified release solid oral dosage forms scale-up and postapproval changes; chemistry, manufacturing, and controls; in vitro dissolution testing and in vivo bioequivalence documentation Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070640.pdf Accessed February 17, 2013
- Emami J In vitro-in vivo correlation: from theory to applications J Pharm Sci 2006 9 169 189
- Polli JE IVIVR versus IVIVC Available from: http://www.dissolution-tech.com/DTresour/800Articles/800_art1.html Accessed February 17, 2013
- Żurada JM Introduction to Artificial Neural Systems St Paul, MN West Publishing Company 1992
- Chen Y McCall TW Baichwal AR Meyer MC The application of an artificial neural network and pharmacokinetic simulations in the design of controlled-release dosage forms J Control Release 1999 59 33 41 10210720
- Bourquin J Shmidli H van Hoogevest P Leuenberger H Advantages of artificial neural networks (ANNs) as alternative modeling technique for data sets showing non-linear relationship using data from a galenical study on a solid dosage form Eur J Pharm Sci 1998 7 5 16 9845773
- Mendyk A Jachowicz R Neural network as a decision support system in the development of pharmaceutical formulation – focus on solid dispersions Expert Syst Appl 2005 28 285 294
- Mitra A Wu Y Use of in vitro-in vivo correlation (IVIVC) to facilitate the development of polymer-based controlled release injectable formulations Recent Pat Drug Deliv Formul 2010 4 94 104 20214657
- Türkoğlu M Özarslan R Sakr A Artificial neural network analysis of a direct compression tabletting study Eur J Pharm Biopharm 1995 41 315 322
- Rocksloh K Rapp FR Abed Abu S Optimization of crushing strength and disintegration time of a high-dose plant extract tablet by neural networks Drug Dev Ind Pharm 1999 25 1015 1025 10518241
- Belic A Skrjanc I Bozic DZ Karba R Vrecer F Minimisation of the capping tendency by tableting process optimisation with the application of artificial neural networks and fuzzy models Eur J Pharm Biopharm 2009 73 172 178 19465122
- Brier ME Żurada JM Neural network predicted peak and trough gentamicin concentrations Pharm Res 1995 12 406 412 7617529
- Chow HH Tolle KM Roe DJ Elsberry V Chen H Application of neural networks to population pharmacokinetic data analysis J Pharm Sci 1997 86 840 845 9232526
- Opara J Primozic S Cvelbar P Prediction of pharmacokinetic parameters and the assessment of their variability in bioequivalence studies by artificial neural networks Pharm Res 1999 16 944 948 10397618
- Yamamura S Kawada K Takehira R Artificial neural network modeling to predict the plasma concentration of aminoglycosides in burn patients Biomed Pharmacother 2004 58 239 244 15183849
- Agatonovic-Kustrin S Turner JV Glass BD Quantitative structure-retention-pharmacokinetic relationship studies Drug Metab Lett 2008 2 130 137 19356082
- Poynton MR Choi BM Kim YM Machine learning methods applied to pharmacokinetic modelling of remifentanil in healthy volunteers: a multi-method comparison J Int Med Res 2009 37 1680 1691 20146865
- De Matas M Shao Q Silkstone VL Chrystyn H Evaluation of an in vitro in vivo correlation for nebulizer delivery using artificial neural networks J Pharm Sci 2007 96 3293 3303 17630647
- Dowell J Hussain A Devane J Young D Artificial neural networks applied to the in vitro-in vivo correlation of an extended-release formulation: initial trials and experience J Pharm Sci 1999 88 154 160 9874718
- Parojčić J Ibrić S Djurić Z Jovanović M Corrigan OI An investigation into the usefulness of generalized regression neural network analysis in the development of level A in vitro-in vivo correlation Eur J Pharm Sci 2007 30 264 272 17188851
- Fatouros DG Nielsen FS Douroumis D Hadjileontiadis LJ Mullertz A In vitro-in vivo correlations of self-emulsifying drug delivery systems combining the dynamic lipolysis model and neuro-fuzzy networks Eur J Pharm Biopharm 2008 69 887 898 18367386
- ChemAxon. [homepage on the Internet] ChemAxon company Available from: http://www.chemaxon.com Accessed December 6, 2012
- Bilski J The back-propagation learning with logarithmic transfer function Abstract presented at the Fifth Conference Neural Networks and Soft Computing June 6–10, 2000 Zakopane, Poland
- Mendyk A Jachowicz R Unified methodology of neural analysis in decision support systems built for pharmaceutical technology Expert Syst Appl 2007 32 1124 1131
- Żurada JM Malinowski A Usui S Perturbation method for deleting redundant inputs of perceptron networks Neurocomputing 1997 14 177 193
- g3data. [homepage on the Internet] g3data software for extracting data from graphs Available from: http://www.frantz.f/software/g3data.php Accessed December 6, 2012
- Kortejärvi H Malkki J Marvola M Level A in vitro-in vivo correlation (IVIVC) model with Bayesian approach to formulation series J Pharm Sci 2006 95 1595 1605 16732564
- Kortejärvi H Mikkola J Bäckman M Antila S Marvola M Development of level A, B and C in vitro-in vivo correlations for modified-release levosimendan capsules Int J Pharm 2002 241 87 95 12086724
- Rossi RC Dias CL Donato EM Development and validation of dissolution test for ritonavir soft gelatin capsules based on in vivo data Int J Pharm 2007 338 119 124 17343999
- Sunesen VH Vedelsdal R Kristensen HG Christrup L Müllertz A Effect of liquid volume and food intake on the absolute bioavailability of danazol, a poorly soluble drug Eur J Pharm Sci 2005 24 297 303 15734296
- Eddington N Marroum P Uppoor R Hussain A Augsburger L Development and internal validation of an in vitro-in vivo correlation for a hydrophilic metoprolol tartrate extended release tablet formulation Pharm Res 1998 15 466 473 9563079
- Eddington N Rekhi G Lesko L Augsburger L Scale-up effects on dissolution and bioavailability of propranolol hydrochloride and metoprolol tartrate tablet formulations AAPS Pharm Sci Tech 2000 1 a14
- Rekhi GS Eddington ND Fossler MJ Evaluation of in vitro release rate and in vivo absorption characteristics of four metoprolol tartrate immediate-release tablet formulations Pharm Dev Tech Technol 1997 2 11 24
- Sirisuth N Eddington N The influence of first pass metabolism on the development and validation of an IVIVC for metoprolol extended release tablets Eur J Pharm Biopharm 2002 53 301 309 11976018
- Ahmed IS Aboul-Einien MH In vitro and in vivo evaluation of a fast-disintegrating lyophilized dry emulsion tablet containing griseofulvin Eur J Pharm Sci 2007 32 58 68 17628451
- Korhonen O Kanerva H Vidgren M Urtti A Ketolainen J Evaluation of novel starch acetate-diltiazem controlled release tablets in healthy human volunteers J Control Release 2004 95 515 520 15023462
- Mahaguna V Talbert R Peters J Influence of hydroxypropyl methylcellulose polymer on in vitro and in vivo performance of controlled release tablets containing alprazolam Eur J Pharm Biopharm 2003 56 461 468 14602191
- Roda A Sabatini L Mirasoli M Baraldini M Roda E Bioavailability of a new ketoprofen formulation for once-daily oral administration Int J Pharm 2002 241 165 172 12086732
- Rani M Mishra B Comparative in vitro and in vivo evaluation of matrix, osmotic matrix, and osmotic pump tablets for controlled delivery of diclofenac sodium AAPS Pharm Sci Tech 2004 5 153 159
- Jung H Milán R Girard M León F Montoya M Bioequivalence study of carbamazepine tablets, in vitro/in vivo correlation Int J Pharm 1997 152 37 44
- Honkanen O Pia L Janne M Bioavailability and in vitro oesophageal sticking tendency of hydroxypropyl methylcellulose capsule formulations and corresponding gelatine capsule formulations Eur J Pharm Sci 2002 15 479 488 12036724
- Nykänen P Lempää S Aaltonen M Citric acid as excipient in multiple-unit enteric-coated tablets for targeting drugs on the colon Int J Pharm 2001 229 155 162 11604268
- Hayashi T Kanbe H Okada M In vitro and in vivo sustained-release characteristics of theophylline matrix tablets and novel cluster tablets Int J Pharm 2007 341 105 113 17512147
- Zaid AN Natour S Qaddomi A Abu Ghoush A Formulation and in vitro and in vivo evaluation of film coated montelukast sodium tablets using Opadry yellow 20A82938 on an industrial scale Drug Des Devel Ther 2013 7 25 31