
Abstract
The immune system struggles every day between responding to foreign antigens and tolerating self-antigens to delicately maintain tissue homeostasis. If self-tolerance is broken, the development of autoimmunity can be the consequence, as it is in the case of the chronic inflammatory autoimmune disease systemic lupus erythematosus (SLE). SLE is considered to be a multifactorial disease comprising various processes and cell types that act abnormally and in a harmful way. Oxidative stress, infections, or, in general, tissue injury are accompanied by massive cellular demise. Several processes such as apoptosis, necrosis, or NETosis (formation of Neutrophil Extracellular Traps [NETs]) may occur alone or in combination. If clearance of dead cells is insufficient, cellular debris may accumulate and trigger inflammation and leakage of cytoplasmic and nuclear autoantigens like ribonucleoproteins, DNA, or histones. Inadequate removal of cellular remnants in the germinal centers of secondary lymphoid organs may result in the presentation of autoantigens by follicular dendritic cells to autoreactive B cells that had been generated by chance during the process of somatic hypermutation (loss of peripheral tolerance). The improper exposure of nuclear autoantigens in this delicate location is consequently prone to break self-tolerance to nuclear autoantigens. Indeed, the germline variants of autoantibodies often do not show autoreactivity. The subsequent production of autoantibodies plays a critical role in the development of the complex immunological disorder fostering SLE. Immune complexes composed of cell-derived autoantigens and autoantibodies are formed and get deposited in various tissues, such as the kidney, leading to severe organ damage. Alternatively, they may also be formed in situ by binding to planted antigens of circulating autoantibodies. Here, we review current knowledge about the etiopathogenesis of SLE including the involvement of different types of cell death, serving as the potential source of autoantigens, and impaired clearance of cell remnants, causing accumulation of cellular debris.
Introduction
Under physiological conditions, the efficient removal of dead cells and their remnants prevents the accumulation of cellular debris that may serve as a potential source of autoantigens. Abnormal cell demise and impaired or delayed clearance of cellular remnants are considered to play a crucial role in the etiology of autoimmune diseases such as systemic lupus erythematosus (SLE).Citation1,Citation2 It is thought that both environmental factors, such as chemicals, drugs, or pathogens, and genetic predisposition, shown by genome-wide association studies, contribute to the development of this disease.Citation3–Citation5 The progression of the malady is fostered by the formation of immune complexes leading to a plethora of clinical manifestations, such as butterfly rash, nephritis, glomerulonephritis, proteinuria, seizures, arthritis, thrombocytopenia, serositis, and psychosis. SLE affects women (before menopause) nine times more often than men.Citation6 Among the many types of cell death, “suicidal” apoptosis, “passive” necrosis, and neutrophil extracellular trap formation (NETosis) are supposed to be the source of autoantigens that play an important role in the etiology and pathogenesis of SLE.Citation7–Citation9 Insufficient clearance of post-apoptotic cells is an intrinsic and complex defect embracing several entities from defective phagocytosis to erroneous opsonization of autoantigens by autoreactive immunoglobulin G (IgG). This shifts their engulfment to an inflammatory pathway. Among others, poorly functioning phagocytic cells (“lazy macrophages”) and deficiencies of DNase I or C1q mainly contribute to the hampered clearance of dying cells.Citation10,Citation11 In addition, a failure along the pathway from apoptotic cell recognition to TLR9 signaling may be involved in the development of autoimmunity.Citation12 This review discusses how a failure in the above-mentioned mechanisms and their key players contributes to the development and maintenance of SLE.
Apoptosis
Apoptosis is a highly controlled and well-organized process involved in many physiological conditions, such as embryogenesis and normal tissue turnover. The intentional suicide of cells which are no longer required, as is the case with immune cells after resolution of infection or cells whose DNA is damaged, is mandatory for the preservation of tissue homeostasis and prevention of malignancies. During the execution of this form of cell demise, the integrity of the cell membrane is maintained as long as possible, avoiding autoantigen leakage and triggering of immune responses.Citation13–Citation16
Apoptosis can be initiated either intrinsically (mitochondrial) or extrinsically (death receptor-mediated). In both pathways, effector caspases are proteolytically activated. They cleave a plethora of intracellular substrates such as focal adhesion or cytoskeleton proteins, consequently leading to weakening of the plasma membrane scaffold. Moreover, molecules such as DNA fragmentation factorCitation17 and caspase-activated deoxyribonuclease are activated.Citation18 Simultaneous inactivation of poly(ADP-ribose) polymerase consequently results in the degradation of the DNA.Citation19,Citation20 The action of the effector caspases contributes to the characteristic phenotype of an apoptotic cell.
The cell executes a series of morphological and biochemical changes comprising chromatin condensation, fragmentation of the nucleus, blebbing, cell shrinkage, and formation of apoptotic bodies. The latter contain modified cell-derived material.Citation21,Citation22 The content of these membranous particles presents a potential source of (often) nuclear autoantigens, which are typically recognized by autoantibodies in patients with SLE.Citation23
Signals during apoptosis
When a cell undergoes apoptosis in vivo, it has to be recognized, ingested, and rapidly degraded in an immunologically silent manner. Thus, an apoptotic cell exposes and secretes a series of signals in order to ensure its quick and efficient engulfment (). Exposing “find me” signals, the apoptotic cell attracts professional and non-professional phagocytes,Citation24 which are able to engulf the dying cell in the presence of “eat me” signals, like phosphatidylserine (PS).Citation25,Citation26 In living cells, PS is mainly restricted to the inner leaflet of the plasma membrane.Citation27 During apoptosis, this asymmetrical distribution is lost.Citation28 In some cells, such as resting B cells.Citation29 or activated CD8+ T cells,Citation30 PS transiently appears on the cells’ surfaces. In these cases, PS exposure does not initiate engulfment by phagocytes. Thus, surface exposure of PS is not sufficient for serving as an “eat me” signal.Citation31 During apoptosis, when the cytoskeleton is fragmented by proteolytic cleavage, the high lateral mobility of its components is discussed to precipitate an adequate recognition and phagocytosis.Citation32,Citation33 On the one hand, recognition of apoptotic cells is directly mediated by receptors on the surfaces of phagocytes, such as Bal-1,Citation34 stabilin-2,Citation35 Tim-1, and Tim-4.Citation36,Citation37 On the other hand, efferocytosis is facilitated indirectly by the soluble bridging molecules, such as CRP, annexin A1, β2GPI, MFG-E8, and Gas-6, and their receptors on phagocytes. The bridging enables a close positioning of the phagocyte and its prey.Citation32–Citation34,Citation37,Citation38 The activation of an intracellular signaling cascade finally results in the remodeling of the actin cytoskeleton and the ingestion of the apoptotic corpse.Citation39 “Stay away” signals, such as lactoferrin, are secreted by the apoptotic cell to prevent granulocyte attraction, inflammation, and the triggering of an immune response. Consequently, the migration of neutrophils to the site of apoptosis is inhibited.Citation40 The uptake of cellular debris causes the secretion of the anti-inflammatory cytokine interleukin 10 (IL-10) by phagocytes, often referred to as a “tolerate me” signal.Citation25 The production of the proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), IL-1, or IL-12, by phagocytes is decreased during ingestion. Finally, the highly regulated process of cell death allows the clearance of dying cells without triggering inflammatory responses and represents a hallmark of apoptosis.Citation33
Table 1 Signals and signaling molecules released during apoptosis and phagocytosis
Failure of dead cell clearance leads to secondary necrosis and SLE
As mentioned earlier, immunological silence during apoptotic cell death is achieved by maintaining the integrity of the cellular membrane and the subsequent secretion of anti-inflammatory cytokines by engulfing phagocytes. The fast and efficient removal of dying cells and their remnants prevents progression of apoptotic cells to the stage of secondary necrosis, accompanied by rupture of the cell membrane and subsequent release of harmful intracellular contents including the damage-associated molecular pattern molecules, HMGB1, ATP, uric acid, and hyaluronic acid. Hampered removal of cells undergoing apoptosis may lead to the induction of inflammation and autoimmune responses.Citation41–Citation44 The production of autoantibodies and the consecutive opsonization of intracellular autoantigens by those antibodies foster the uptake of secondary necrotic cell-derived material by phagocytes. The secretion of the pro-inflammatory cytokines such as IL-8, interferon alpha (IFN-α), TNF-β, IL-18, and IL-1β fuels inflammation and promotes further tissue damage.Citation45 As a consequence, circulating immune complexes are formed and may bind to tissues or deposit, inter alia, in the skin, joints, or kidneys.Citation46 This leads to the activation of the complement system that attracts innate immune cells promoting local inflammation. The activation of plasmacytoid dendritic cells (pDC) by immune complexes results in the secretion of IFN-α, establishing the so-called “IFN-α signature” of leukocytes. As a result of the deposition of the immune complexes and activation of inflammatory processes, severe organ damage occurs, which in turn fuels the vicious cycle of accumulation of secondary necrotic cells and inflammatory clearance driving SLE. The established inflammatory processes encompass both a persistent immunostimulatory status and several rather short-lasting recurrent flares.
In the course of SLE, the canonically immune silent pathway of apoptosis turns into a harmful source of autoantigens and immunostimulatory signals prone to sustain chronic autoimmune responses. Importantly, also in healthy individuals, a few apoptotic cells can undergo secondary necrosis, but fast degradation and removal with the participation of DNase I and C1q ensures the maintenance of immune tolerance.Citation47–Citation49
Primary necrosis in the development of SLE
Necrosis is provoked by exogenous factors that damage the cell, such as toxic events or mechanical injury. As a consequence, swelling of the oncotic cell causes sudden rupture of the membrane. This leads to cellular leakage of modified and deleterious intracellular material into the extracellular space. Neutrophils, lymphocytes, and macrophages migrate to the site of efflux of intracellular content and initiate inflammation.Citation50,Citation51 Thus, tissue damage may contribute to the development of the autoimmune disease SLE. Overwhelming cell death combined with a defective clearance of dying and dead cells is associated with the break of self-tolerance.
The role of NETosis in the development of SLE
Neutrophils are the most abundant granulocytes that play a crucial role in the defense against microbial infections. Attracted by tissue-resident macrophages, resident mast cells, or endothelial cells, they migrate from the blood vessels to the site of injury, where they engulf pathogens, degranulate by releasing antimicrobial molecules, and secrete proinflammatory cytokines.Citation52 Neutrophils are able to attract DC by the secretion of C–C chemokines, like MIP-3α/CCL20 and MIP-3β/CCL19.Citation53 Under healthy conditions, neutrophils undergo apoptosis and are engulfed and cleared by tissue-resident macrophages.Citation54
At the site of infection, neutrophils are able to form neutrophil extracellular traps (NETs) decorated with several antimicrobial molecules. This form of suicidal cell death is referred to as NETosis.Citation55 NETs consist of processed chromatin associated with cytoplasmic proteins and granules. The latter are vesicles containing toxic, microbicidal molecules. These granules associate with the NETs during the process of NETosis. Four types of granules can be distinguished: azurophilic (primary) granules, specific (secondary) granules, gelatinase (tertiary) granules, and secretory vesicles. NETosis also occurs in response to invading pathogens and exogenous or endogenous stimuli, like fine dust or monosodium urate crystals. NETosis can also be induced by the proinflammatory cytokines IL-8 and IL-1β or platelet activation factor. Eosinophils and basophils are also able to externalize their chromatin and form extracellular DNA-based traps.Citation56
The formation of NETs involves several steps. First, activated neutrophils get in contact with the stimulus and disintegration of granules occurs. Neutrophil elastase and myeloperoxidase, which are released from azurophilic granules, reach the cellular nucleus orchestrating histone modification and chromatin decondensation.Citation57,Citation58 Another important modification of histones is peptidylarginine deiminase 4 (PAD4)-mediated citrullination. This enzyme is a member of the PAD family, which consists of five isotypes. Only PAD4 possesses a nuclear localization signal.Citation59–Citation61 Citrullination of histone H3 plays an important role in the decondensation of the chromatin during NETosis. As a result, the nuclear envelope is destroyed and the integrity of cell membrane is lost. This leads to the rapid release of DNA-containing net-like structures.Citation62 Neutrophil elastase is able to partially degrade the linker and core histones, thus creating neoantigens. Moreover, histones can undergo other relevant modifications such as acetylation, methylation, hyperubiquitination, or poly(ADP-ribosyl)ation, possibly determining their immunogenic capacities.Citation63 Besides their important function as a scaffold for nucleosome formation, the basic histone proteins can also kill pathogens.Citation64
As mentioned earlier, several patients suffering from SLE are seropositive for antibodies that recognize cytoplasmic autoantigens from neutrophils. These autoantibodies recognize components of NETs like neutrophil elastase, myeloperoxidase, LL-37, or lactoferrin.Citation65 In addition, these autoantibodies promote IFN-α production by pDC and thus contribute to the IFN-α signature. Autoantibodies specific for surface-expressed LL-37 induce the release of NETs by neutrophils. Enhanced LL-37 surface expression in turn fuels inflammation and autoantibody production via IFN-α.Citation66,Citation67 Furthermore, NETosis is accompanied by the release of noxious intracellular constituents such as heat shock proteins, modified histones, or HMGB1. These intracellular proteins are able to activate DC and may thus challenge immune tolerance.Citation68 Since NETs are composed of DNA, they are prone to degradation by DNase I. In the case of patients with SLE, this nuclease may act insufficiently as a result of inhibitors, anti-DNase I antibodies, or genetic variations.Citation69 This may cause persistence of NETs serving as a source for potential autoantigens, thereby challenging self-tolerance (). Importantly, a neutrophil subpopulation enriched in the blood of patients suffering from SLE is more prone to execute NETosis.Citation66,Citation67 It has recently been shown that neutrophils isolated from patients with SLE reveal a robust pattern of demethylation of the IFN-regulated genes, suggesting a pathogenic role for neutrophils in lupus.Citation70
Figure 1 The role of neutrophils in the etiopathogenesis of SLE.
Notes: Activated neutrophils release NETs covered with α-NE, α-LF, α-MPO, or α-LL-37. Chromatin and associated compounds are hallmark antigens of the autoimmune response of patients with SLE. Decreased activity of DNase I combined with a general (anti-inflammatory) clearance deficiency leads to the accumulation of NETs covered with proinflammatory and cytotoxic intracellular constituents. Opsonization of NETs with autoantbodies comes with immune complex formation followed by inflammatory clearance by blood-borne phagocytes. This process causes inflammation and tissue damage, thus stimulating pDC to secrete IFN-α and IL-6, ultimately resulting in the so-called “IFNα signature” typical of SLE. The pro-inflammatory cytokines secreted by pDCs induce long-lived plasma cell formation and massive autoantibody production.
Abbreviations: α-NE, antibodies against neutrophil elastase; α-MPO, antibodies against myeloperoxidase; α-LF, antibodies against lactoferrin; NET, neutrophil extracellular trap; IFN-α, interferon-alpha; IL-6, interleukin-6; pDC, plasmacytoid dendritic cells; SLE, systemic lupus erythematosus; NETosis, neutrophil extracellular trap formation; NETs, neutrophil extracellular traps; MΦ: macrophage.
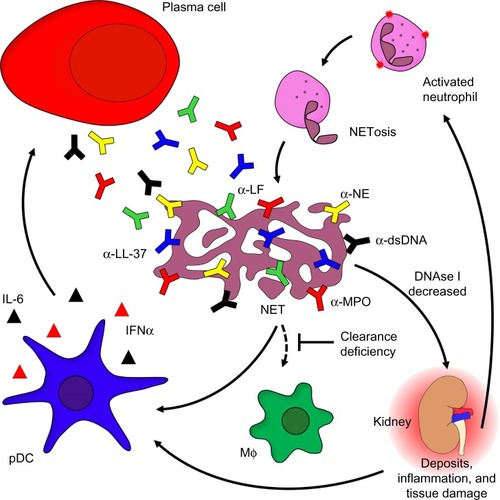
Neutrophils may secrete proinflammatory cytokines and factors able to activate B cells such as a proliferation-inducing ligand, if activated by deposited immune complexes. Nevertheless, aggregated NETs are also important to confine and resolve inflammation. They are able to trap pro-inflammatory cytokines and chemokines, which are consequently proteolytically degraded. This limits the inflammatory response.Citation71
Implications of impaired phagocytosis on autoimmunity
Hampered clearance of cellular debris is discussed to play a substantial role in the development of SLE. It is, among others, the result of failures in the processes of apoptosis and/or phagocytosis. In healthy individuals, cellular debris is cleared in a fast and efficient manner. Signals released by apoptotic cells during their death induce attraction of phagocytes, which engulf and digest cellular remnants before the integrity of the cellular membrane is lost. This prevents the triggering of an inflammatory reaction.
Opsonization facilitates the proper removal of cell remnants. Under healthy conditions, the recognition of dead cells is supported by opsonins like IgM, the complement component C1q, CRP, or MBP. Several patients with SLE reportedly show decreased levels of these components in their sera. In individuals who are genetically predisposed to develop SLE, decreased levels of MBP and C1q have been shown to be associated with the disease.Citation72,Citation73 A cooperation of C1q and DNase I accelerates the degradation of necrotic cells-derived chromatin.Citation49,Citation74 The combination of a failure in the effective and immunologically silent opsonization of dead cell remnants and the abnormal immunogenic opsonization by autoantibodies of secondary necrotic cell-derived materials substantially contributes to the development of autoimmune disease and fuels chronic inflammation. This highlights the important role of opsonization of dead and dying cells for the multifactorial and complex disorder SLE.
A reduction in the phagocytic capacity and impaired adhesion is a hallmark of monocyte-derived macrophages (MDM) found in patients with SLE. Professional phagocytes and antigen-presenting cells of patients with SLE show a decreased size and capacity to engulf their prey, when compared with cells isolated from the blood of normal healthy donors.Citation75 Additionally, MDMs isolated from patients suffering from SLE exhibit a decreased adhesionCitation76 and an impaired production of the adhesion molecule CD44, which also contributes to the clearance of apoptotic neutrophils.Citation77 Consequently, the removal of their apoptotic remnants by MDMs is impaired. There is also evidence that MDMs isolated from patients with SLE die faster than those isolated from normal healthy donors.Citation78
Under healthy conditions, cellular remnants of cells undergoing apoptosis in the germinal centers (GCs) of secondary lymphoid organs are taken up and digested by tingible body macrophages without their activation as antigen-presenting cells. Since the number and size of tingible body macrophages are decreased in patients with SLE, the persistence of cellular debris might favor the binding of apoptotic cell-derived autoantigens by follicular dendritic cells (fDC) and the subsequent presentation of nuclear autoantigens to autoreactive B cells, resulting in their survival, activation, and expansion.Citation79
Moreover, it has been shown that the levels of CRP are atypically low in patients with SLE. CRP serves as bridging molecule between phagocytes and their prey and reduces the inflammatory potential of apoptotic corpses.Citation80 This may result from its recognition by autoreactive antibodies. The fact that several components of the complex and diverse interplay in apoptotic cell clearance are reportedly defective in SLE emphasizes the multifactorial genesis of this disease. Finally, failures in opsonization and consequently in the clearance of apoptotic and necrotic cells result in the accumulation of autoantigens challenging immunological tolerance and causing the fueling of inflammation.
Autoantibodies as disease-driving factors
In healthy individuals, central and peripheral tolerance checkpoints ensure the elimination of autoreactive T and B cells, protecting the organism against the development of autoimmunity. However, in the case of SLE, B- and T-cell tolerance is challenged and subsequently lost. This may be a consequence of a failure of clonal deletion of autoreactive T or rescue of autoreactive B cells.Citation79,Citation81–Citation85
In healthy GCs, fDC present immune complexes to maturing and mutating B cells, and provide survival signals in the case of cognate interaction of the B cell with the antigen of the immune complexes. In the lymph nodes of several SLE patients, uncleared nuclear remnants accumulate, which bind to fDC and provide survival signals for autoreactive B cells that had been generated by chance during the process of somatic hypermutation. The improper exposure of nuclear autoantigens in this delicate location is consequently prone to break self-tolerance to nuclear autoantigens. Indeed, the germline variants of autoantibodies often do not display auto-reactivity.Citation86 It is important to note that fDC are not derived from bone marrow, do not ingest particulate antigens, and are neither related to myeloid nor to plasmacytoid DC.
B cells that are positively selected in the GC by either immune complexes (immune B cells) or by nuclear debris (autoimmune B cells) can receive long-term survival signals from autoreactive CD4+ T cells in the mantle zone of the lymphatic tissue. B cells subsequently differentiate into memory or plasma cells which produce autoreactive antibodies. As already mentioned, autoantibodies are able to recognize cytoplasmic and nuclear autoantigens derived from cellular remnants, thereby opsonizing circulating self-antigens and facilitating the formation of inflammatory immune complexes. Thus, the production of autoantibodies is an important and characteristic feature in the course of SLE that greatly contributes to its etiopathogenesis. In patients with SLE, autoantibodies undergo isotype switch from IgM to IgG and affinity maturation.Citation87 Opsonization of self-antigens by autoantibodies of the IgG isotype leads to Fcγ receptor-mediated inflammatory phagocytosis. However, autoantibodies of the IgM isotype may serve as an ameliorative factor in SLE, since they are discussed to opsonize autoantigens for an anti-inflammatory engulfment, thus avoiding the challenge of self-tolerance.Citation51,Citation88,Citation89
Importantly, the status of glycosylation of complexed IgG seems to have an impact on disease activity, since lectin binding to native complexed IgG is increased in patients with SLE. Immune complexes with highly fucosylated autoantibodies may be engulfed more efficiently by phagocytes and, thereby, foster inflammation.Citation90 In contrast, the loss of terminal galactose in the Fc part of IgG results in increased complement activationCitation91 favoring the clearance of dead cell remnants, and the absence of terminal sialic acid reportedly suppresses antibody-dependent cell-mediated cytotoxicity.Citation92
Dendritic cells and SLE
DC are a heterogeneous group of cells with two major populations: myeloid DC (mDC) and pDC. DC are scattered over the entire body, where they serve as sentinels (mDC and pDC) and scavengers (mDC) for pathogens. mDC finally process and present phagocytosed antigens to naïve T cells in an immune-stimulatory or tolerance inducing fashion. Note that fDC are a completely different cell type that is not human leukocyte antigen-restricted and not derived from bone marrow precursors. fDC present surface-bound antigens in immune complexes to follicular B cells.
DC need to mature from antigen-capturing toward antigen-presenting cells. During this process, they increase the surface expression of major histocompatibility complex as well as costimulatory molecules (CD80, CD86, and CD40). Maturation is also accompanied by the secretion of high levels of proinflammatory cytokines such as IL-12, IL-6, and TNF-α, which enable DC to drive the type of T-cell polarization.Citation93 Mature DC may present self-antigens to T cells leading to their differentiation into two subsets, T follicular helper and Th17 cells, and inhibit the development of regulatory T cells with tolerogenic capacity.Citation94 Immature DC are crucial for the maintenance of epitope-specific tolerance, when they present antigen in the absence of costimulation to T cells leading to anergy or deletion of autoreactive T lymphocytes and development of regulatory T cells.Citation95 In the context of SLE, mDC present modified macromolecules in an immunogenic manner and challenge the tolerance against nuclear autoantigens. DC employ pattern recognition receptors such as toll-like receptors (TLR) to recognize their targets.Citation96 mDC express most of the known TLR, whereas pDC only express TLR7 and TLR9 that recognize ssRNA and CpG DNA, respectively.Citation97–Citation99 Opsonization of autoantigens by autoantibodies leads to the activation of pDC, the natural IFN-producing cells. This results in elevated expression of IFN-α-regulated genes in patients with SLE, referred to as “type-1 interferon signature”. As a consequence, CD80 and CD86 are upregulated and serve as signals for survival and expansion of autoreactive CD4+ helper T cells. The latter may support autoreactive B cells and CD8+ cytotoxic T cells. Upregulation of TLR7 by IFN-α leads to an enhanced response against immune complexes containing nucleic acids and in an elevated production of IFN-α, thus fueling an inflammatory loop.Citation100
Therapeutic options
In order to treat SLE, nonsteroidal anti-inflammatory drugs (NSAIDs) as well as immunosuppressive corticosteroids are commonly used. In the combination with these drugs, plasma exchange is used in patients with life-threatening manifestations to remove autoreactive antibodies and harmful immune complexes.Citation101 These conventional therapies have limitations and are frequently accompanied by adverse side effects. As mentioned earlier, autoreactive IgG contributes to the immune pathology of SLE. IgG antibodies are required and sufficient to protect the organism against certain microbial infections. This dual role is referred to as intravenous IgG paradox. Thus, substitution of IgG levels by intravenous immunoglobulin preparations is an important treatment option for patients with SLE after plasma exchange. Intravenous immunoglobulin preparations consist of IgG from pooled serum of thousands of donors.Citation102,Citation103 Another therapeutic option for patients with refractory SLE is B-cell depletion by rituximab, a chimeric anti-CD20 antibody that temporarily depletes B cells as well as short-lived plasma cells.Citation104 Belimumab, a monoclonal antibody that specifically targets the B lymphocyte stimulator and therefore disturbs B-cell activation, proliferation, and survival, has been approved to treat SLE.Citation105 In few patients with refractory SLE with high uncontrolled disease activity, autologous bone marrow transplantation and inhibition of the proteasome with bortezomib may be employed as last option.Citation106,Citation107
In recent years, targeted therapies emerged as new potential therapeutic options, due to a better understanding of the etiopathogenesis of SLE. As discussed earlier, changes in the glycosylation pattern of IgG may either foster or ameliorate inflammatory diseases. Since it is known that deglycosylated IgG is unable to elicit antibody-dependent cell-mediated cytotoxicity and complement activation, one therapeutic option might be modulation of antibody glycosylation.Citation108 A potent glycosylation modulator is endoglycosidase EndoS, which specifically cleaves the sugar moiety of the Fc portion of IgGCitation102,Citation109 making it a potential tool for SLE immunotherapy. Another promising target is the pathogenic loop of DC activation and IFN-α production, which precipitates the development of SLE. Monoclonal antibodies targeting type-I IFNCitation110–Citation112 or molecules inhibiting TLR signaling are promising agents interfering with the above-mentioned pathogenic cycle. The antimalarial drugs chloroquine and hydroxychloroquine display antithrombotic and anti-lipidemic effects, prevent lupus flares, and increase the long-term survival of patients with SLE.Citation113,Citation114 Another putative target for therapeutic intervention is IL-6 which, inter alia, contributes to the maturation of B cells into plasma cells. Increased levels of this proinflammatory cytokine correlate with disease activity of patients with SLE.Citation115 Tocilizumab, a monoclonal antibody targeting IL-6 receptor, is a promising agent that can effectively block IL-6 signaling. Moreover, autoreactive IgM came into focus, since it is considered to be an ameliorative factor in the course of SLE and may be used in antibody therapy.Citation51,Citation88,Citation89 Nevertheless, additional studies are necessary to explore and exploit this therapeutic repertoire.
Conclusion and future direction of research
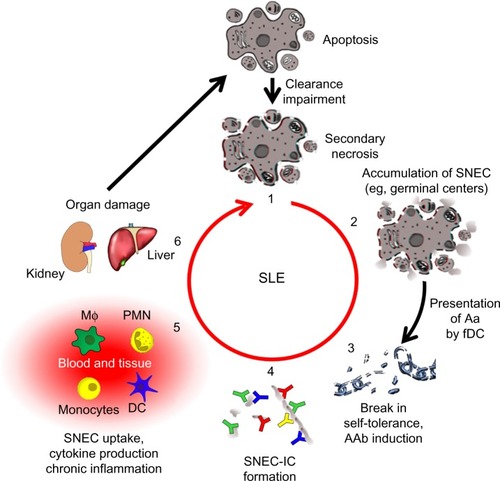
Despite the fact that the knowledge about the etiopathogenesis of SLE has increased in the past few years, there are still unanswered questions. It is well established that a deficiency of the fast and efficient removal of dead cell corpses and debris plays a central role in the course of the disease (). Therefore, therapeutic intervention in the vicious cycle of SLE by rescuing the failure of dead cell clearance is a promising approach. Recent studies have given new insights into the course of SLE, providing new possibilities for the development of targeted therapeutic strategies. So far, secondary necrosis was thought to be the major source of autoantigens triggering autoimmunity. Recently, NETosis has come into focus as another form of cell death with similar effects. This raises the question of whether therapeutic targeting of neutrophils is a new treatment option.
Figure 2 The vicious cycle of SLE.
Notes: A deficiency in the clearance of apoptotic cells leads to autoimmunity and chronic inflammation (1). when apoptotic cells fail to be cleared in time, they get secondary necrosis, leading to the accumulation of SNEC (2). Self-tolerance is broken when SNEC-derived autoantigens (Aag) are presented to autoreactive B cells by fDC. with help from autoreactive helper T cells, these B cells undergo affinity maturation and differentiate into memory B cells, thus establishing autoimmunity (3). IC are formed when autoantibodies (AAb) encounter SNEC in circulation or tissue (4). Newly formed SNEC-IC are then processed by blood-borne phagocytes and dendritic cells (DC) accompanied by the secretion of pro-inflammatory cytokines (5). This in turn leads to severe organ damage and cell death fueling the vicious cycle that maintains chronic inflammation (6).
Abbreviations: SNEC, secondary necrotic cell-derived material; fDC, follicular dendritic cells; IC, immune complex; SLE, systemic lupus erythematosus; DC, dendritic cells; MΦ: macrophage, PMN: polymorphonuclear leukocytes.
The current treatment of SLE with NSAIDs or immuno-suppressive drugs generally ameliorates disease manifestations. Nevertheless, the patient’s life quality is affected. An important aspect is reaching a balance between inhibition of disease-inducing inflammation and maintenance of the beneficiary host immune response. The powerful tools, which are provided by research, may be exploited in combination with commonly used drugs in future therapeutic interventions. However, the possible side effects of these therapeutic agents, such as deglycosylated IgG or anti-inflammatory IgM antibodies as well as agents targeting DC and type-I IFN, have to be examined carefully. The complexity of the disease emphasizes the need of combined therapy specifically targeting the inflammatory loop driving the pathology of SLE.
Disclosure
The authors report no conflicts of interest in this work.
References
- MunozLEJankoCSchulzeCAutoimmunity and chronic inflammation – two clearance-related steps in the etiopathogenesis of SLEAutoimmun Rev2010101384220817127
- PerniokAWedekindFHerrmannMSpeckerCSchneiderMHigh levels of circulating early apoptic peripheral blood mononuclear cells in systemic lupus erythematosusLupus1998721131189541096
- RhodesBVyseTJThe genetics of SLE: an update in the light of genome-wide association studiesRheumatology (Oxford)200847111603161118611920
- CooperGSMillerFWPandeyJPThe role of genetic factors in autoimmune disease: implications for environmental researchEnviron Health Perspect1999107Suppl 569370010502533
- EdwardsCJEnvironmental factors and lupus: are we looking too late?Lupus200514642342516038104
- Pons-EstelGJAlarconGSScofieldLReinlibLCooperGSUnderstanding the epidemiology and progression of systemic lupus erythematosusSemin Arthritis Rheum201039425726819136143
- IppolitoAWallaceDJGladmanDAutoantibodies in systemic lupus erythematosus: comparison of historical and current assessment of seropositivityLupus201120325025521362750
- PradhanVDBadakereSSBichileLSAlmeidaAFAnti-neutrophil cytoplasmic antibodies (ANCA) in systemic lupus erythematosus: prevalence, clinical associations and correlation with other autoantibodiesJ Assoc Physicians India20045253353715645975
- BoutsYMWolthuisDFDirkxMFApoptosis and NET formation in the pathogenesis of SLEAutoimmunity201245859760122913420
- MunozLEGaiplUSFranzSSLE – a disease of clearance deficiency?Rheumatology (Oxford)20054491101110715928001
- LuJHTehBKWangLThe classical and regulatory functions of C1q in immunity and autoimmunityCell Mol Immunol20085192118318990
- MilesKHeaneyJSibinskaZA tolerogenic role for Toll-like receptor 9 is revealed by B-cell interaction with DNA complexes expressed on apoptotic cellsProc Natl Acad Sci U S A2012109388789222207622
- RenehanAGBachSPPottenCSThe relevance of apoptosis for cellular homeostasis and tumorigenesis in the intestineCan J Gastroenterol200115316617611264570
- HensonPMHumeDAApoptotic cell removal in development and tissue homeostasisTrendsImmunol2006275244250
- RavichandranKSLorenzUEngulfment of apoptotic cells: signals for a good mealNat Rev Immunol200771296497418037898
- SavillJApoptosis in resolution of inflammationJ Leukoc Biol19976143753809103222
- LiuXZouHSlaughterCWangXDFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosisCell19978921751849108473
- SakahiraHEnariMNagataSCleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosisNature1998391666296999422513
- LazebnikYAKaufmannSHDesnoyersSPoirierGGEarnshawWCCleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICENature199437164953463478090205
- GuYSarneckiCAldapeRALivingstonDJSuMSCleavage of poly(ADP-ribose) polymerase by interleukin-1 beta converting enzyme and its homologs TX and Nedd-2J Biol Chem19952703218715187187642516
- ChaurioRAMunozLEMaueroderCThe progression of cell death affects the rejection of allogeneic tumors in immune-competent mice – implications for cancer therapyFront Immunol2014556025426116
- ParthasarathyGPhilippMTReview: apoptotic mechanisms in bacterial infections of the central nervous systemFront Immunol2012330623060884
- BilyyROShkandinaTTominAMacrophages discriminate glycosylation patterns of apoptotic cell-derived microparticlesJ Biol Chem2012287149650322074924
- PeterCWesselborgSHerrmannMLauberKDangerous attraction: phagocyte recruitment and danger signals of apoptotic and necrotic cellsApoptosis20101591007102820157780
- VollREHerrmannMRothEAStachCKaldenJRGirkontaiteIImmunosuppressive effects of apoptotic cellsNature199739066583503519389474
- FadokVAde CathelineauADalekeDLHensonPMBrattonDLLoss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblastsJ Biol Chem200127621071107710986279
- ChaurioRAJankoCMunozLEFreyBHerrmannMGaiplUSPhospholipids: key players in apoptosis and immune regulationMolecules200914124892491420032867
- WilliamsonPSchlegelRATransbilayer phospholipid movement and the clearance of apoptotic cellsBiochim Biophys Acta200215852–3536312531537
- DillonSRManciniMRosenASchlisselMSAnnexin V binds to viable B cells and colocalizes with a marker of lipid rafts upon B cell receptor activationJ Immunol200016431322133210640746
- FischerKVoelklSBergerJAndreesenRPomorskiTMackensenAAntigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cellsBlood2006108134094410116912227
- SegawaKSuzukiJNagataSConstitutive exposure of phosphatidylserine on viable cellsProc Natl Acad Sci U S A201110848192461925122084121
- JankoCJeremicIBiermannMCooperative binding of Annexin A5 to phosphatidylserine on apoptotic cell membranesPhys Biol201310606500624304966
- BiermannMMaueroderCBraunerJMSurface code – biophysical signals for apoptotic cell clearancePhys Biol201310606500724305041
- GregoryCDPoundJDCell death in the neighbourhood: direct microenvironmental effects of apoptosis in normal and neoplastic tissuesJ Pathol2011223217719421125674
- KimSParkSYKimSYCross talk between engulfment receptors stabilin-2 and integrin alphavbeta5 orchestrates engulfment of phosphatidylserine-exposed erythrocytesMol Cell Biol201232142698270822566688
- KobayashiNKarisolaPPena-CruzVTIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cellsImmunity200727692794018082433
- IshimotoYOhashiKMizunoKNakanoTPromotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6J Biochem2000127341141710731712
- KruseKJankoCUrbonaviciuteVInefficient clearance of dying cells in patients with SLE: anti-dsDNA autoantibodies, MFG-E8, HMGB-1 and other playersApoptosis20101591098111320198437
- KinchenJMRavichandranKSJourney to the grave: signaling events regulating removal of apoptotic cellsJ Cell Sci2007120Pt 132143214917591687
- BournazouIPoundJDDuffinRApoptotic human cells inhibit migration of granulocytes via release of lactoferrinJ Clin Invest20091191203219033648
- VermesIHaanenCRichelDJSchaafsmaMRKalsbeek-BatenburgEReutelingspergerCPApoptosis and secondary necrosis of lymphocytes in cultureActa Haematol19979818139210907
- WuXMolinaroCJohnsonNCasianoCASecondary necrosis is a source of proteolytically modified forms of specific intracellular autoantigens: implications for systemic autoimmunityArthritis Rheum200144112642265211710720
- KonoHRockKLHow dying cells alert the immune system to dangerNat Rev Immunol20088427928918340345
- UrbonaviciuteVFurnrohrBGMeisterSInduction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLEJ Exp Med2008205133007301819064698
- MunozLEJankoCGrossmayerGERemnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosusArthritis Rheum20096061733174219479824
- MunozLELauberKSchillerMManfrediAAHerrmannMThe role of defective clearance of apoptotic cells in systemic autoimmunityNat Rev Rheumatol20106528028920431553
- GaiplUSBeyerTDBaumannIExposure of anionic phospholipids serves as anti-inflammatory and immunosuppressive signal – implications for antiphospholipid syndrome and systemic lupus erythematosusImmunobiology20032071738112638907
- RekvigOPMortensenESImmunity and autoimmunity to dsDNA and chromatin – the role of immunogenic DNA-binding proteins and nuclease deficienciesAutoimmunity201245858859223013317
- GaiplUSBeyerTDHeyderPCooperation between C1q and DNase I in the clearance of necrotic cell-derived chromatinArthritis Rheum200450264064914872509
- KerrJFWyllieAHCurrieARApoptosis: a basic biological phenomenon with wide-ranging implications in tissue kineticsBr J Cancer19722642392574561027
- BiermannMHVeissiSMaueroderCThe role of dead cell clearance in the etiology and pathogenesis of systemic lupus erythematosus: dendritic cells as potential targetsExpert Rev Clin Immunol20141091151116425081199
- Witko-SarsatVRieuPDescamps-LatschaBLesavrePHalbwachs-MecarelliLNeutrophils: molecules, functions and pathophysiological aspectsLab Invest200080561765310830774
- ScapiniPLaudannaCPinardiCNeutrophils produce biologically active macrophage inflammatory protein-3alpha (MIP- 3alpha)/CCL20 and MIP-3beta/CCL19Eur J Immunol20013171981198811449350
- IbaTHashiguchiNNagaokaITabeYMuraiMNeutrophil cell death in response to infection and its relation to coagulationJ Intens Care20131113
- BrinkmannVReichardUGoosmannCNeutrophil extracellular traps kill bacteriaScience200430356631532153515001782
- SchornCJankoCLatzkoMChaurioRSchettGHerrmannMMonosodium urate crystals induce extracellular DNA traps in neutrophils, eosinophils, and basophils but not in mononuclear cellsFront Immunol2012327722969769
- PapayannopoulosVMetzlerKDHakkimAZychlinskyANeutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular trapsJ Cell Biol2010191367769120974816
- MetzlerKDFuchsTANauseefWMMyeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunityBlood2011117395395920974672
- NakashimaKHagiwaraTIshigamiAMolecular characterization of peptidylarginine deiminase in HL-60 cells induced by retinoic acid and 1alpha,25-dihydroxyvitamin D(3)J Biol Chem199927439277862779210488123
- NakashimaKHagiwaraTYamadaMNuclear localization of peptidylarginine deiminase V and histone deimination in granulocytesJ Biol Chem200227751495624956812393868
- VossenaarERZendmanAJvan VenrooijWJPruijnGJPAD, a growing family of citrullinating enzymes: genes, features and involvement in diseaseBioessays200325111106111814579251
- FuchsTAAbedUGoosmannCNovel cell death program leads to neutrophil extracellular trapsJ Cell Biol2007176223124117210947
- PieterseEvan der VlagJBreaking immunological tolerance in systemic lupus erythematosusFront Immunol2014516424782867
- MillerBFAbramsRDorfmanAKleinMAntibacterial properties of protamine and histoneScience194296249742843017729719
- HoffmannMHBrunsHBackdahlLThe cathelicidins LL-37 and rCRAMP are associated with pathogenic events of arthritis in humans and ratsAnn Rheum Dis20137271239124823172753
- LandeRGangulyDFacchinettiVNeutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosusSci Transl Med201137373ra19
- Garcia-RomoGSCaielliSVegaBNetting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosusSci Transl Med201137373ra20
- UrbonaviciuteVVollREHigh-mobility group box 1 represents a potential marker of disease activity and novel therapeutic target in systemic lupus erythematosusJ Int Med20112704309318
- HakkimAFurnrohrBGAmannKImpairment of neutrophil extracellular trap degradation is associated with lupus nephritisProc Natl Acad Sci U S A2010107219813981820439745
- CoitPYalavarthiSOgnenovskiMEpigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophilsJ Autoimmun201558596625638528
- SchauerCJankoCMunozLEAggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokinesNat Med201420551151724784231
- IpWKChanSYLauCSLauYLAssociation of systemic lupus erythematosus with promoter polymorphisms of the mannose-binding lectin geneArthritis Rheum1998419166316689751100
- RicklinDHajishengallisGYangKLambrisJDComplement: a key system for immune surveillance and homeostasisNat Immunol201011978579720720586
- JogNRFrisoniLShiQCaspase-activated DNase is required for maintenance of tolerance to lupus nuclear autoantigensArthritis Rheum20126441247125622127758
- HerrmannMVollREZollerOMHagenhoferMPonnerBBKaldenJRImpaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosusArthritis Rheum1998417124112509663482
- MunozLEChaurioRAGaiplUSSchettGKernPMoMa from patients with systemic lupus erythematosus show altered adhesive activityAutoimmunity200942426927119811273
- CairnsAPCrockardADMcConnellJRCourtneyPABellALReduced expression of CD44 on monocytes and neutrophils in systemic lupus erythematosus: relations with apoptotic neutrophils and disease activityAnn Rheum Dis2001601095095511557652
- GaiplUSMunozLEGrossmayerGClearance deficiency and systemic lupus erythematosus (SLE)J Autoimmun2007282–311412117368845
- BaumannIKolowosWVollREImpaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosusArthritis Rheum200246119120111817590
- KravitzMSShoenfeldYAutoimmunity to protective molecules: is it the perpetuum mobile (vicious cycle) of autoimmune rheumatic diseases?Nat Clin Prac Rheumatol200629481490
- VollRERothEAGirkontaiteIHistone-specific Th0 and Th1 clones derived from systemic lupus erythematosus patients induce double-stranded DNA antibody productionArthritis Rheum19974012216221719416853
- TheocharisSSfikakisPPLipnickRNKlippleGLSteinbergADTsokosGCCharacterization of in vivo mutated T cell clones from patients with systemic lupus erythematosusClin Immunol Immunopathol19957421351427828367
- ShivakumarSTsokosGCDattaSKT cell receptor alpha/beta expressing double-negative (CD4−/CD8−) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritisJ Immunol198914311031122525144
- CamachoSAKosco-VilboisMHBerekCThe dynamic structure of the germinal centerImmunol Today199819115115149818545
- MevorachDMascarenhasJOGershovDElkonKBComplement-dependent clearance of apoptotic cells by human macrophagesJ Exp Med199818812231323209858517
- SchroederKHerrmannMWinklerTHThe role of somatic hypermutation in the generation of pathogenic antibodies in SLEAutoimmunity201346212112723181829
- WinklerTHFehrHKaldenJRAnalysis of immunoglobulin variable region genes from human IgG anti-DNA hybridomasEur J Immunol1992227171917281623920
- SaikiOSaekiYTanakaTDevelopment of selective IgM deficiency in systemic lupus erythematosus patients with disease of long durationArthritis Rheum19873011128912923689463
- WitteTIgM antibodies against dsDNA in SLEClin Rev Allergy Immunol200834334534718097774
- SjowallCZapfJvon LohneysenSAltered glycosylation of complexed native IgG molecules is associated with disease activity of systemic lupus erythematosusLupus201424656958125389233
- MalhotraRWormaldMRRuddPMFischerPBDwekRASimRBGlycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding proteinNat Med1995132372437585040
- KanekoYNimmerjahnFRavetchJVAnti-inflammatory activity of immunoglobulin G resulting from Fc sialylationScience2006313578767067316888140
- BlancoPPaluckaAKPascualVBanchereauJDendritic cells and cytokines in human inflammatory and autoimmune diseasesCytokine Growth Factor Rev2008191415218258476
- LiaoJChangCWuHLuQCell-based therapies for systemic lupus erythematosusAutoimmun Rev2015141434825308529
- BanchereauJSteinmanRMDendritic cells and the control of immunityNature199839266732452529521319
- FransenJHvan der VlagJRubenJAdemaGJBerdenJHHilbrandsLBThe role of dendritic cells in the pathogenesis of systemic lupus erythematosusArthritis Res Ther201012220720423534
- SeitzHMMatsushimaGKDendritic cells in systemic lupus erythematosusInt Rev Immunol201029218420920367140
- DieboldSSKaishoTHemmiHAkiraSReis e SousaCInnate antiviral responses by means of TLR7-mediated recognition of single-stranded RNAScience200430356631529153114976261
- Ahmad-NejadPHackerHRutzMBauerSVabulasRMWagnerHBacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartmentsEur J Immunol20023271958196812115616
- GangulyDChamilosGLandeRSelf-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8J Exp Med200920691983199419703986
- PagnouxCPlasma exchange for systemic lupus erythematosusTransfus Aphere Sci2007362187193
- AllhornMOlinAINimmerjahnFCollinMHuman IgG/Fc gamma R interactions are modulated by streptococcal IgG glycan hydrolysisPloS One200831e141318183294
- NimmerjahnFRavetchJVThe antiinflammatory activity of IgG: the intravenous IgG paradoxJ Exp Medicine200720411115
- NgKPLeandroMJEdwardsJCEhrensteinMRCambridgeGIsenbergDARepeated B cell depletion in treatment of refractory systemic lupus erythematosusAnn Rheum Dis200665794294516269424
- LutaloPMD’CruzDPUpdate on belimumab for the management of systemic lupus erythematosusExpert Opin Biol Ther201414111701170825303323
- NeubertKMeisterSMoserKThe proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritisNat Med200814774875518542049
- IlleiGGCerveraRBurtRKCurrent state and future directions of autologous hematopoietic stem cell transplantation in systemic lupus erythematosusAnn Rheum Dis201170122071207421873334
- NoseMWigzellHBiological significance of carbohydrate chains on monoclonal antibodiesProc Natl Acad Sci U S A19838021663266366579549
- CollinMOlsenAEndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgGEMBO J200120123046305511406581
- MerrillJTWallaceDJPetriMSafety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised studyAnn Rheum Dis201170111905191321798883
- McBrideJMJiangJAbbasARSafety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebo-controlled, double-blind, dose-escalation studyArthritis Rheum201264113666367622833362
- LauwerysBRHachullaESpertiniFDown-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon alpha-kinoidArthritis Rheum201365244745623203821
- WallaceDJGudsoorkarVSWeismanMHVenuturupalliSRNew insights into mechanisms of therapeutic effects of antimalarial agents in SLENat Rev Rheumatol20128952253322801982
- Ruiz-IrastorzaGRamos-CasalsMBrito-ZeronPKhamashtaMAClinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic reviewAnn Rheum Dis2010691202819103632
- Linker-IsraeliMDeansRJWallaceDJPrehnJOzeri-ChenTKlinenbergJRElevated levels of endogenous IL-6 in systemic lupus erythematosus. A putative role in pathogenesisJ Immunol199114711171232051017