Abstract
Purpose: Transforming growth factor-β-activated kinase 1 (TAK1) plays a role in inhibiting apoptosis in response to multiple stresses. In the present study, we investigated the role of TAK1 in cell death induced by heat stress (HS). Materials and methods: TAK1 knockdown HeLa cells and their parental cells were exposed to HS at 44 °C for 15, 30, 45 min followed by colony formation assay. Heat shock proteins (HSPs) induction, NF-κB phosphorylation, and caspase-3 cleavage were estimated by western blotting using specific antibodies. Global gene expression analysis was performed using the GeneChip® system. The anti-apoptotic roles of the identified genes were elucidated using small interfering RNAs targeting those genes. Results: Heat sensitivity estimated by colony formation assay and caspase-3 cleavage increased in TAK1 knockdown cells. This sensitisation was not due to alterations in HSP induction or NF-κB phosphorylation as the expression levels of these proteins did not differ significantly between the TAK1 knockdown and the parent cells after HS exposure. The GeneChip® analysis revealed differences in gene expression between both cell variants after HS exposure and defined the genetic network associated with cell death. TNF-α interacting protein 3 (TNFAIP3) and Interleukin 8 (IL-8) are two of the identified genes. RNA interference against these genes increased the cleavage of caspase-3 and cell death after HS exposure. Conclusion: Our findings reveal the role of TAK1 in thermoresistance and show that the mediation is independent of NF-κB phosphorylation but is dependent on TNFAIP3 and IL-8 induction.
Introduction
Transforming growth factor-β-activated kinase 1 (TAK1) was identified as a MAPK kinase kinase (MAP3K), which can be activated by transforming growth factor-β [Citation1]. TAK1 is also activated by TNF-α, interleukin-1, and ligands of Toll-like receptors, such as lipopolysaccharides [Citation2]. TAK1 is essential for activating the IκB kinase (IKK)/nuclear factor κB (NF-κB) pathways and the stress kinases (i.e. JNK and p38 MAPK) pathways in response to various stimuli [Citation3,Citation4]. TNF receptor-associated factor 6 (TRAF6) facilitates the synthesis of non-degradative lys-63-linked polyubiquitin chains to recruit and activate TAK1 through its really interesting new gene (RING)-dependent ubiquitin ligase activity; this recruitment in turn activates IKK to initiate the NF-κB pathway and activates MKK6 and MKK7 to induce the JNK, p38 MAPK and ERK pathways, respectively [Citation5–8]. Although activation of the JNK pathway is involved in apoptosis [Citation9,Citation10], simultaneous activation of the IKK/NF-κB pathway and MEK/ERK pathway negatively modulates TNF-α-mediated apoptosis [Citation11,Citation12].
Hyperthermia induced by HS inhibits the proliferation of cancer cells and induces their apoptosis [Citation13]. The best characterised heat stress response is induction of heat shock proteins (HSPs). The HSPs are implicated in cell cycle regulation, in resistance to stress-induced apoptosis or necrotic cell death, and in antioxidative defence. A number of studies have shown that HSPs such as Hsp27, 70, 90 might modulate the homeostasis of TAK1 in response to different physiological conditions [Citation14,Citation15].
Previous studies showed that TAK1 is likely involved in cell survival in response to multiple stimuli such as TNF-α, IL-1β, ligands of several TLRs and X-radiation [Citation16–18]. However, it has not been demonstrated whether or not TAK1 is involved in cell survival after heat stress (HS) exposure. The present study was designed to examine the role of TAK1 in HeLa cells exposed to HS. The work provides a better understanding of the molecular mechanism of TAK1 pathway as triggered by HS. The work also suggests TAK1 as a therapeutic target in cancer therapy.
Materials and methods
Cell culture and treatments
The TAK1 knockdown and control HeLa cells were transfected with luciferase as previously described [Citation19]. Regarding the establishment of stable cell lines, HeLa cells were transfected with pSUPERgfp/neo vectors containing short hairpin RNAs (shRNAs) against human TAK1 and firefly luciferase GL2 (Luc) using lipofectamine reagent. The target sequence for TAK1 is 5′-ATGACGATTCATGAGTGTTAG-3′, which is located in the 3′ untranslated region. Stable cell lines were selected by G418 at 1 mg/mL, and green fluorescent protein (GFP) expression was confirmed by flow cytometer (Epics XL, Beckman-coulter, Miami, FL). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)(high glucose) (044-29765, Wako, Osaka, Japan) supplemented with 10% fetal calf serum, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37 °C in 5% CO2. The G418 antibiotic (400 µg/mL) was added to the medium to maintain selection for the transfected plasmids.
Heat stress exposure
Cells were cultured 12 h before treatment in 6-well plates at a density of 0.5 × 106 cells/mL. The plates were sealed with paraffin film and placed in a hot water bath at 44 °C for 15, 30 or 45 min. Cells were then transferred to an incubator at 37 °C until analysis.
Colony formation assay
Cell survival after HS exposure was measured by colony formation assay. For all experiments, cells were seeded into 60-mm culture dishes on day 0 and allowed to attach for 12 h at 37 °C. The cells were then exposed to HS at the desired dose (day 1), immediately returned to the incubator, and cultured for 12 days. Colonies were stained with Giemsa, and visible colonies containing approximately 50 or more cells were counted. The survival fraction was calculated relative to the number of control cells to determine the plating efficiency in each experiment (number of HS-exposed colonies/number of colonies in control).
Cell counting assay
A cell counting assay was performed using a Cell Counting Kit-8 (CCK-8) according to the protocol provided by the manufacturers (Dojindo, Kumamoto, Japan). Briefly, after transfection with siTNFAIP3 and IL-8, the cells were exposed to HS. After 48 h incubation, cells were trypsinised, then seeded in a 96-well plate and allowed to adhere overnight. Then the cells were incubated in 100 μL DMEM medium (containing 10 μL CCK-8) per well for 2 h at 37 °C in 5% CO2. The absorbance at 450 nm was determined by using Microplate Reader (Bio-Rad, Hercules, CA).
Western blot analysis
Western blotting was performed as described previously [Citation20]. Cells were collected, washed with cold phosphate buffer saline (PBS), and lysed with radio immunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Nonidet P-40 (v/v), 1% sodium deoxycholate, 0.05% SDS, 1 μg/mL each of aprotinin, pepstatin, and leupeptin, and 1 mM phenylmethylsulphonyl fluoride (PMSF)). Lysates were centrifuged at 3,000 rpm for 10 min at 4 °C. Antibodies against caspase-3, phospho-NF-κB Ser536, TNFAIP3 were obtained from Cell Signaling Technology (Danvers, MA). Antibody against TAK1 was obtained from Santa Cruz Biotechnology (CA). Antibodies against Hsp70, 40, and 27 were obtained from MBL (Woburn, MA). Antibody against NF-κB (p65) was from Millipore (Tokyo, Japan). Antibody against IL-8 was obtained from Sigma-Aldrich. With secondary horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse IgGs (Cell Signaling Technology), band signals were visualised using a luminescent image analyser (LAS4000, Fujifilm, Tokyo, Japan) with a chemiluminescence detection system (Nacalai Tesque, Kyoto, Japan). When necessary, band density was quantified and expressed as fold changes over the untreated control using Image J software.
Microarray analysis
Microarray analysis was performed as previously described [Citation20–23]. Cells were exposed to HS and collected after 6 h. Total RNA was extracted using an RNeasy Total RNA Extraction kit (Qiagen, Valencia, CA). Gene expression was analysed using a GeneChip® system with a Human Genome U133-plus 2.0 array (Affymetrix, Santa Clara, CA) spotted with 54,675 probe sets. The scanned chip was analysed using GeneChip analysis suite software (Affymetrix). Data were further analysed using GeneSpring software (Silicon Genetics, Redwood City, CA) to extract the genes with significant expression. To examine molecular functions and genetic networks, the obtained data were analysed using Ingenuity Pathways Analysis tools (Ingenuity Systems, Mountain View, CA), a web-delivered application that enables the identification, visualisation, and exploration of molecular interaction networks in gene expression data.
Real-time quantitative polymerase chain reaction (qPCR) assay
Real-time qPCR assay was performed on a Real-Time PCR system (Mx3000P, Stratagene, Tokyo, Japan) using SYBR PreMix ExTaq (TaKaRa Bio, Otsu Shiga, Japan) or qPCR Master Mix (for the use of TaqMan probes; Eurogentec, Seraing, Belgium) according to the manufacturers’ protocols. Reverse transcriptase reaction (ExScript RT reagent Kit, TaKaRa Bio) was carried out with DNase-treated total RNA using a random primer pd(N)6. Real-time qPCR assay was performed by using the following specific primers; TNFAIP3 Sense 5′-CGGAAGCTTGTGGCGCTGAA-3′, Antisense 5′-AGCGTGCTGAACAGCGCCTT-3′. IL-8 Sense 5′-GAGTGATTGAGAGTGGACC-3′, Antisense 5′-ACTTCTCCACAACCCTCTG-3′. Each mRNA expression level was normalised to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [Citation24].
Transfection of small interfering RNA (siRNA)
siRNA targeting luciferase, TNFAIP3 (5′-AGACACACGCAACUUUAAA-3′), and IL-8 (5′-ACCACCGGAAGGAACCAUC-3′) were obtained from Nippon Gene (Toyama, Japan). siRNA was transfected using RNAimax reagent (Invitrogen, Carlsbad, CA) in accordance with the manufacturer instructions. In brief, siRNA was mixed with 3.2 µL of RNAimax reagent in 200 µL of Opti-MEM (Wako) in a 35-mm culture dish, allowing the formation of complexes for 15 min, then added to resuspended cells (1 × 105 cells/1.8-mL reverse transfection protocol). After 24 h incubation, cells were exposed to HS and incubated for 6 h, then subsequently analysed. Knockdown efficiency was confirmed by real-time PCR analysis.
Statistical analysis
Statistical significance was estimated by two-tailed Student’s t-test using Microsoft Excel software. Statistical significance was set at p < 0.05.
Results
Increased heat sensitivity of TAK1 knockdown HeLa cells
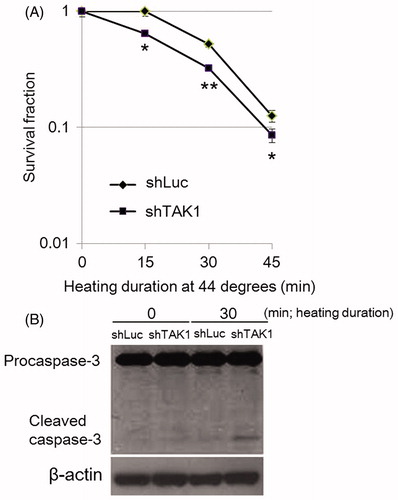
TAK1 depletion has been shown to promote cell death induced by various stimuli [Citation16–18], but the role of TAK1 in HS-induced cell death has not been demonstrated. To determine the effect of the TAK1 knockdown on heat sensitivity, we performed the cell colony formation assay. The data showed that TAK1 knockdown had significant effect in cell survival as compared to parent cells. This corresponded with the enhanced cleavage of caspase-3 which was clearly detected in the TAK1 knockdown cells, conforming that increased heat sensitivity was associated with enhanced apoptosis induced by HS exposure ().
Figure 1. Decreased cell survival in TAK1 knockdown cells (shTAK1) in comparison to parental cells (shLuc) after HS-exposure. (A) Clonogenic survival assay. Cells were exposed to HS for 15, 30, or 45 min. After 12 days incubation, colonies containing approximately 50 or more cells were counted. Data are presented as mean ± SD (n = 3). *indicates statistical significance compared to shLuc (*p < 0.05, **p < 0.01). (B) Western blots for caspase-3. Control and HS-treated cells were incubated for 24 h before analysis.
Increased heat sensitivity independently of HSPs induction and NF-κB phosphorylation in TAK1 knockdown HeLa cells
HS activates responses such as raising signalling pathways and reprogramming gene expression to return to their internal milieu. The most well-characterised heat shock response is the induction of a highly conserved set of polypeptides termed the heat shock proteins (HSPs) [Citation25,Citation26]. The degree of thermotolerance correlates well with the degree of induced HSPs [Citation27,Citation28]. Hsp70 has been shown to play a critical role in the cell survival and thermotolerance in response to stresses, possibly through inhibiting a number of anti-survival pathways such as the SAPK/JNK pathway [Citation29–34] or chaperoning misfolded proteins by interacting with DnaJ/Hsp40 [Citation35]. Hsp27 has also been implicated in acquired thermotolerance [Citation36–38].
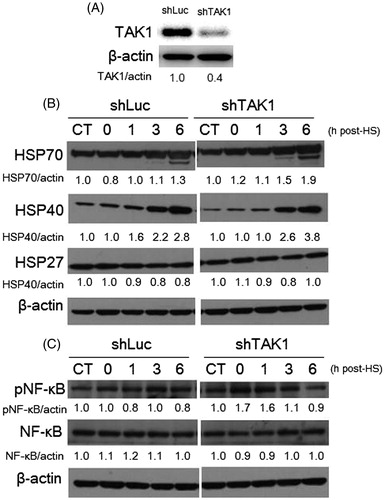
The knockdown of TAK1 was confirmed by western blotting (Figure 2A). To determine whether TAK1 was involved in HSP induction, we detected the expression levels of Hsp70, 40, and 27 in TAK1 knockdown and parental cells after HS treatment (44 °C, 30 min). Consistently with other studies, the HSP induction markedly increased in a time-dependent manner, especially for Hsp70 and 40, whereas the increase of Hsp27 was slight in both cell lines regardless of TAK1 status (). HS is also known to activate NF-κB signaling to promote cell survival [Citation39]. The phosphorylation of NF-κB induced by HS was not significantly suppressed by TAK1 inhibition (). Taken together, these findings suggest that TAK1 is involved in counteracting apoptosis progression in an independent manner from HSPs and NF-κB phosphorylation.
Figure 2. HSP induction and NF-κB phosphorylation in TAK1 knockdown cells in comparison to parental cells exposed to HS. (A) Expression level of TAK1 in control TAK1 knockdown versus its expression in control parent cells. (B) Time-dependent induction of Hsp70, 40, and 27 in cells exposed to HS. (C) Phosphorylation levels of NF-κB induced by HS. Band density was quantified and expressed as fold changes over the untreated control using Image J software.
Global gene expression analysis in control and TAK1 knockdown cells exposed to HS
Our results showed that TAK1 depeletion promoted HS-induced cell death independently of HSP induction and NF-κB-phosphorylation. In order to clearly determine the mechanisms underlying the regulation of HS-induced cell death by TAK1, we analysed the genes involved in driving the apoptotic machinery under the lack of a functional TAK1 compared to the parental cells at 6 h after exposure to HS (44 °C, 30 min). Using cDNA microarrays, we identified 536 and 376 differentially expressed probes in each cell line after treatment. Among the identified genes there were 326 and 468 genes up-regulated in response to HS in TAK1 knockdown cells and parental cells, respectively. Of the up-regulated genes, there were 245 probes common between both cell variants (). Next, to find TAK1 responsive genes we excluded the 81 up-regulated probes of TAK1 knockdown cells and fed the remaining 468 probes into a bioinformatics analytical tool. The analysis using ingenuity pathway analysis (IPA) identified two genetic networks that were associated with molecular function/maintenance or cell death (). Network I consisted of the 245 common up-regulated genes including many HSPs genes, such as the HSPA family genes encoding the Hsp70 family proteins (HSPA1A, 1 L, 5, 6), DNAJ family genes encoding the Hsp40 family proteins (DNAJB1, 4, 9, and DNAJC7), and HSPB family genes encoding the Hsp27 family proteins (HSPB8) (For full gene names, see ). The expression levels of the respective mRNAs were slightly different between the parent and TAK1 knockdown cells exposed to HS, supporting the western blot data (). On the other hand, the network II consisted of 223 genes that were predominantly up-regulated in parent cells. The network contained several genes known to be associated with apoptosis regulation among which TNFAIP3 and IL-8 were markedly different from TAK1 knockdown cells in induction rate and expression level ( and ).
Figure 3. Global gene expression and bioinformatics analyses of TAK1 knockdown cells in comparison to parental cells after HS exposure. (A) The number of differentially expressed genes by a factor of ≥3.0 in HS-treated cells. Venn diagram indicates the commonly and specifically up-regulated genes in each cell line. (B) The identified responsive genetic networks to HS. The up-regulated genes by a factor of ≥3.0 in treated parent cells are included in the networks. Relative gene expression ratios are shown in and II. (C) Differential induction levels of TNFAIP3 and IL-8 proteins in control cells and TAK1 knockdown cells 6 h after HS exposure. Band density was quantified and expressed as fold changes over the untreated control using Image J software.
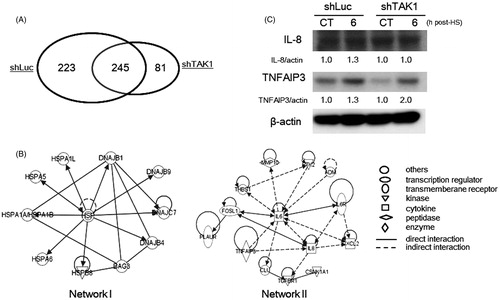
Table I. Relative gene expression levels of differentially expressed genes in identified gene network I.
Table II. Relative gene expression levels of differentially expressed genes in identified gene network II.
To confirm the results of microarray experiments, we examined the expression levels of TNFAIP3 and IL-8 by western blotting. Although the expression levels of these two proteins were not completely comparable to that found by microarray analysis, the expression levels of these two proteins were significantly increased in parent cells after HS exposure ().
Knockdown of TNFAIP3 or IL-8 by siRNA promoted caspase-3 cleavage and cell death after HS exposure
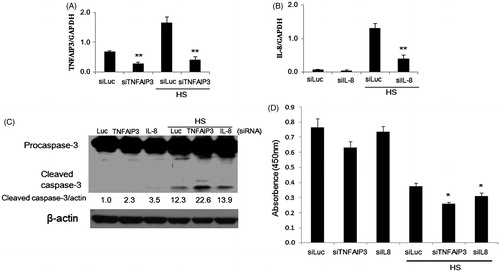
IL-8 is an important member of the ELR+ CXC family of chemokines and is a HS-responsive gene that requires HSF-1 and multiple 5′-flanking HSEs for optimal HS-responsiveness [Citation40]. IL-8 protein was found to decrease the apoptosis rate of human airway smooth muscle cells in vitro [Citation41]. TNFAIP3 (A20), a zinc finger protein originally identified as a tumour necrosis factor (TNF)-responsive gene in endothelial cells (ECs), serves as a dual cytoprotective molecule; providing protection against tumour necrosis factor (TNF)-mediated apoptosis and inhibiting inflammation via blocking the transcription factor NF-κB [Citation42]. To determine whether TNFAIP3 or IL-8 are involved in reversing apoptosis in response to HS, we used siRNA targeting TNFAIP3 or IL-8 in parent HeLa cells. Transfection of siRNA targeting TNFAIP3 (siTNFAIP3) abrogated mRNA levels of TNFAIP3 in both unexposed and HS-exposed cells (). For IL-8, the constitutive expression of the protein was markedly low, so that the effect of the transfected siRNA targeting IL-8 (siIL-8) was marginal; however, siIL-8 decreased the mRNA levels of IL-8 following HS dramatically (). Simultaneous to TNFAIP3 or IL-8 abrogation, the expression level of cleaved caspase-3 following HS-exposure was markedly enhanced in case of siTNFAIP3, while only slight enhancement was observed with siIL-8 (). The result of cell survival analysis by CCK-8 was also consistent with these findings (). These results confirm that both TNFAIP3 and IL-8 function as anti-apoptotic molecules in cells exposed to HS and, therefore, the inability of TAK1 knockdown cells to transcribe these proteins might account for the increased heat sensitivity observed.
Figure 4. Involvement of TNFAIP3 and IL-8 in apoptosis of HeLa cells exposed to HS. (A, B) Densitometric quantification of TNFAIP3 and IL-8 expression analysed by qPCR following knockdown of TNFAIP3 and IL-8 using siRNAs and / or exposure to HS (44 °C, 30 min). Cells were transfected with siRNA 24 h before HS exposure. (C) Promotion of caspase-3 cleavage by knockdown of TNFAIP3 and IL-8 in cells 24 h after HS exposure. Band density was quantified and expressed as fold changes over the untreated control using Image J software. (D) Depression of cell survival by knockdown of TNFAIP3 and IL-8 in cells 48 h after HS-exposure. Cell survival analysis was performed by CCK-8. Data are presented as mean ± SD (n = 3). *Statistical significance from siLuc with or without HS treatment (*p < 0.05, **p < 0.01).
Discussion
It has been reported that NF-κB can promote cell cycling and inhibit apoptosis induced by a range of stress stimuli [Citation43,Citation44]. Activated TAK1 can phosphorylate inhibitors of NF-κB kinase (IKK) to activate the NF-κB pathway [Citation7]. Also, the induction of heat shock proteins (HSPs) is the most well-characterised and typical heat shock response. In the present study it was demonstrated that the knock down of TAK1 could not affect the association of NF-κB phosphorylation and HSP induction with heat stress response. The independence of NF-κB on TAK1 functionality has been also observed in response to X-radiation [Citation18]. Consequently, it seems that the reported dependence of NF-κB phosphorylation on TAK1 may be specific to some cell type and degree of stimuli. The question that poses itself here would be: what are the novel targets of TAK1 that mediate cell survival other than NF-κB phosphorylation and HSPs? In pursuing an answer for this question we carried out a genetic microarray analysis of parent and TAK1 knockdown cells exposed to similar HS. We have been able to identify several genes which were differentially expressed in both cell variants such as IL-8, TNFAIP3, CXCL2, MMP10, for example. All these genes were members of the genetic network II associated with molecular function/maintainance or cell death and were up-regulated in the parent cells. The knockdown of IL-8 and TNFAIP3 with siRNA resulted in sensitisation of the parent HeLa cells to HS, confirming their anti-apoptotic role. TNFAIP3, a zinc finger protein, can negatively regulate IKK and NF-κB signalling pathway which is activated by TAK1 [Citation45,Citation46]. And its deficiency in mice results in excessive NF-κB activity and multi-organ inflammation [Citation46,Citation47]. In contrast to its so far universal inhibitory effect on NF-κB activation, the anti-apoptotic activity of TNFAIP3 remains controversial and appears to be specific to cell type and stimulus [Citation42]. It has been reported that overexpression of TNFAIP3 can protect from TNF-mediated apoptosis by blocking the activation of the apical caspase-8 and 2, preventing subsequent activation of downstream effectors (caspase-3, 6 and Bid) and maintaining the mitochondrial integrity [Citation42]. In this study it was found that knockdown of TNFAIP3 could increase HS-induced cleavage of caspase-3 and sensitise cells to HS.
IL-8 (CXCL-8), a member of the human CXC family of ELR+ chemokines, was identified as a new HS-responsive gene to delineate a novel pattern of HS responsiveness [Citation40]. Heat shock factor-1 (HSF-1) could act to regulate IL-8 gene transcription. However, unlike the classical HSP genes, HS is not sufficient to activate IL-8, but requires co-activation with a proinflammatory agonist. IL-8 protein increases the expression of mouse CXCL-2 (MIP-2) protein that is also increased by thioglycolate in peritoneum [Citation48]. Combined IL-6 and sIL-6R can induce the expression of IL-8 in endothelial cells [Citation49]. The type I TGF-beta receptor (TGFβR 1) engages TRAF6 to activate TAK1 in a receptor kinase-independent manner [Citation50]. Using an in vitro model, TGFβ1 and TGFβ receptors expression was up-regulated by IL-8, IL-1, and TGFβ1 itself. However, IL-8 receptor expression was decreased by IL-8 and TGFβ1. This suggests that in a cascade IL-8 would feed forward to promote the TGFβ system, whereas TGFβ1 feeds backward to inhibit responsiveness to IL-8 [Citation51].
The detailed mechanisms by which TAK1 regulates TNFAIP3 and IL-8 expression independently of NF-κB phosphorylation remains unclear; however, for the first time, we demonstrated that the defect in TAK1-TNFAIP3/IL-8 pathway was involved, at least in part, in the increased heat sensitivity of TAK1 knockdown HeLa cells. Moreover, our findings support the idea that TAK1 might be a promising target for cancer therapy in not only chemo- or radiotherapy, but also in hyperthermia.
Declaration of interest
This work was supported in part by a Grant-in-Aid for Scientific Research (B) (22390229), Japan Society for the Promotion of Science. The authors alone are responsible for the content and writing of the paper.
References
- Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 1995;270:2008–11
- Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem 2005;280:7359–68
- Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 2005;6:1087–95
- Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev 2005;19:2668–81
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000;103:351–61
- Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptorassociated Factor 6 auto-ubiquitination is a critical determinant of IkappaB kinase activation. J Biol Chem. 2007;282:4102–12
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001;412:346–51
- Lamothe B, Lai Y, Xie M, Schneider MD, Darnay BG. TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol Cell Biol 2013;33:582–95
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000;288:870–74
- Meriin AB, Sherman MY. Role of molecular chaperones in neurodegenerative disorders. Int J Hyperthermia 2005;21:403–19
- Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, et al. Inhibition of JNK activation through NF-kappaB target genes. Nature 2001;414:313–17
- Bhattacharya S, Ray RM, Johnson LR. Prevention of TNF-alpha-induced apoptosis in polyamine-depleted IEC-6 cells is mediated through the activation of ERK1/2. Am J Physiol-Gastr L 2004;286:G479–90
- Hsu SF, Chao CM, Huang WT, Lin MT, Cheng BC. Attenuating heat-induced cellular autophagy, apoptosis and damage in H9c2 cardiomyocytes by pre-inducing Hsp70 with heat shock preconditioning. Int J Hyperthermia 2013;29:239–47
- Cao X, Yue L, Song J, Wu Q, Li N, Luo L, et al. Inducible Hsp70 antagonizes IL-1β cytocidal effects through inhibiting NF-κB activation via destabilizing TAK1 in HeLa cells. PLoS One 2012;7:e50059
- Alford KA, Glennie S, Turrell BR, Rawlinson L, Saklatvala J, Dean JL. Heat shock protein 27 functions in inflammatory gene expression and transforming growth factor-beta-activated kinase-1 (TAK1)-mediated signaling. J Biol Chem 2007;282:6232–41
- Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 2005;6:1087–95
- Hirata Y, Sugie A, Matsuda A, Matsuda S, Koyasu S. TAK1-JNK axis mediates survival signal through Mcl1 stabilization in activated T cells. J Immunol 2013;190:4621–6
- Furusawa Y, Wei ZL, Sakurai H, Tabuchi Y, Li P, Zhao QL, et al. TGF-β-activated kinase 1 promotes cell cycle arrest and cell survival of X-ray irradiated HeLa cells dependent on p21 induction but independent of NF-κB, p38 MAPK and ERK phosphorylations. Radiat Res 2012;77:766–74
- Nishimura M, Shin MS, Singhirunnusorn P, Suzuki S, Kawanishi M, Koizumi K, et al. TAK1-mediated serine/threonine phosphorylation of epidermal growth factor receptor via p38/extracellular signal-regulated kinase: NF-{kappa}B–independent survival pathways in tumor necrosis factor alpha signaling. Mol Cell Biol 2009;29:5529–39
- Furusawa Y, Tabuchi Y, Takahashi A, Wada S, Ohtsuka K, Kondo T. Gene networks involved in apoptosis induced by hyperthermia in human lymphoma U937 cells. Cell Biol Int 2009;33:1253–62
- Furusawa Y, Tabuchi Y, Wada S, Takasaki I, Ohtsuka K, Kondo T. Identification of biological functions and gene networks regulated by heat stress in U937 human lymphoma cells. Int J Mol Med 2011;28:143–51
- Furusawa Y, Zhao QL, Hassan MA, Tabuchi Y, Takasaki I, Wada S, et al. Ultrasound-induced apoptosis in the presence of Sonazoid. Cancer Lett 2010;288: 107–15
- Ahmed K, Furusawa Y, Tabuchi Y, Emam HF, Piao JL, Hassan MA, et al. Chemical inducers of heat shock proteins derived from medicinal plants and cytoprotective genes response. Int J Hyperthermia 2012;28:1–8
- Wada S, Tabuchi Y, Kondo T, Cui ZG, Zhao QL, Takasaki I, et al. Gene expression in enhanced apoptosis of human lymphoma U937 cells treated with the combination of different free radical generators and hyperthermia. Free Radic Res 2007;41:73–81
- Morimoto RI, Tissieres A, Georgopoulos C. The stress response, function of the proteins, and perspectives. In: Morimoto RI, Tissières A, Georgopoulos C, editors. Stress Proteins in Biology and Medicine. New York: Cold Spring Harbor Laboratory Press; 1990. pp. 1–36
- Welch WJ, Kang HS, Beckmann RP, Mizzen LA. Response of mammalian cells to metabolic stress: Changes in cell physiology and structure/function of stress proteins. Curr Top Microbiol Immunol 1991;167:31–55
- Landry J, Bernier D, Chretien P, Nicole LM, Tanguay RM, Marceau N. Synthesis and degradation of heat shock proteins during development and decay of thermotolerance. Cancer Res 1982;42:2457–61
- Li GC, Werb Z. Correlation between synthesis of heat shock proteins and development of thermotolerance in Chinese hamster fibroblasts. Proc Natl Acad Sci USA 1982;79:3218–22
- Kim D, Ouyang H, Li GC. Heat shock protein hsp70 accelerates the recovery of heat-shocked mammalian cells through its modulation of heat shock transcription factor HSF1. Proc Natl Acad Sci 1995;92:2126–30
- Gabai VL, Meriin AB, Mosser DD, Caron AW, Rits S, Shifrin VI, et al. Hsp70 prevents activation of stress kinases: A novel pathway of cellular thermotolerance. J Biol Chem 1997;272:18033–7
- Huang L, Mivechi NF, Moskophidis D. Insights into regulation and function of the major stress-induced hsp70 molecular chaperone in vivo: Analysis of mice with targeted gene disruption of the hsp70.1 or hsp70.3 gene. Mol Cell Biol 2001;21:8575–91
- Calderwood SK, Asea A. Targeting Hsp70-induced thermotolerance for design of thermal sensitizers. Int J Hyperthermia 2002;18:597–608
- Shim EH, Kim JI, Bang ES, Heo JS, Lee JS, Kim EY, et al. Targeted disruption of hsp70.1 sensitizes to osmotic stress. EMBO Rep 2002;3:857–61
- Zhao M, Tang D, Lechpammer S, Hoffman A, Asea A, Stevenson MA, et al. Double-stranded RNA-dependent protein kinase (PKR) is essential for thermotolerance, accumulation of Hsp70, and stabilization of ARE-containing Hsp70 mRNA during stress. J Biol Chem 2002;277:44539–47
- Takayama S, Reed JC, Homma S. Heat-shock proteins as regulators of apoptosis. Oncogene 2003;22:9041–7
- Landry J, Chretien P, Laszlo A, Lambert H. Phosphorylation of Hsp27 during development and decay of thermotolerance in Chinese hamster cells. J Cell Physiol 1991;147:93–101
- Ciocca DR, Oesterreich S, Chamness GC, McGuire WL, Fuqua SA. Biological and clinical implications of heat shock protein 27,000 (Hsp27): a review. J Natl Cancer Inst 1993;85:1558–70
- James M, Crabbe C, Hepburne-Scott HW. Small heat shock proteins (sHSPs) as potential drug targets. Curr Pharm Biotechnol 2001;2:77–111
- Zheng SQ, Su XW, Qiu PX, Chen LJ, Wan X, Yan GM. Relationship between heat stress suppression of neuroapoptosis and activation of nuclear kappa B in primary cultured rat cerebellar granule cells. Sheng Li Xue Bao 2001;53:193–7
- Singh IS, Gupta A, Nagarsekar A, Cooper Z, Manka C, Hester L, et al. Heat shock co-activates interleukin-8 transcription. Am J Respir Cell Mol Biol 2008;39:235–42
- Halwani R, Al-Abri J, Beland M, Al-Jahdali H, Halayko AJ, Lee TH, et al. CC and CXC chemokines induce airway smooth muscle proliferation and survival. J Immunol 2011;186:4156–63
- Daniel S, Arvelo MB, Patel VI, Longo CR, Shrikhande G, Shukri T, et al. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood 2004;104:2376–84
- Nesic D, Grumont R, Gerondakis S. The nuclear factor-κB and p53 pathways function independently in primary cells and transformed fibroblasts responding to genotoxic damage. Mol Cancer Res 2008;6:1193–203
- Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. Current insights into the regulation of programmed cell death by NF-κB. Oncogene 2006;25:6800–16
- Adhikari A, Xu M, Chen ZJ. Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 2007;26:3214–26
- Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 2004;5:1052–60
- Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000;289:2350–54
- Remick DG, Green LB, Newcomb DE, Garg SJ, Bolgos GL, Call DR. CXC chemokine redundancy ensures local neutrophil recruitment during acute inflammation. Am J Pathol 2001;159:1149–57
- Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 1997;6:315–25
- Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol 2008;10:1199–207
- Hatthachote P, Gillespie JI. Complex interactions between sex steroids and cytokines in the human pregnant myometrium: evidence for an autocrine signaling system at term. Endocrinology 1999;140:2533–40