Abstract
Prostate cancer is the most common male cancer and up to one fifth of diagnosed patients will die of their disease. Current prognostic variables including T-category (of the TNM staging), the absolute or kinetics of prostatic specific antigen (PSA) and the pathologic Gleason score (GS) are utilized to place men in low, intermediate and high-risk prostate cancer risk groupings. There is great heterogeneity within the non-indolent intermediate risk group with respect to clinical response. It is therefore imperative that further genetic and other prognostic factors be identified to better individualize treatment. Somatic alterations in prostate cancer. Herein, we review the potential for somatic alterations in tumor-associated genes (based on comparative genomic hybridization (CGH) in prostate cancers to be novel prognostic, and possibly predictive, factors for prostate cancer radiotherapy response. Intermediate risk prostate cancers show alterations in a number of genes thought to be involved in radiosensitivity, DNA repair, cell death and stem cell renewal. These include deletions at 21q (TMPRSS2: ERG), 13q (RB1), 10q (PTEN), 8p (NKX3.1), additions at 8q21 (containing c-Myc)) and haplo-insufficiency for p53, PARP1, ATM and DNA-PKcs. Conclusions. The use of high-resolution CGH for fine-mapping of deletions and amplifications in pre-radiotherapy prostate cancer biopsies is feasible. Genetic alterations may delineate localized prostate cancer from systemic disease and be used as a predictive factor in that patients would be individually triaged to local (surgery versus radiotherapy) and/or adjuvant (adjuvant androgen ablation or post-operative radiotherapy) therapies in a prospective fashion to improve outcome. The knowledge of abnormal DNA repair pathways within in a given patient could allow for the judicious use of targeted agents (PARP/ATM inhibitors) as personalized medicine.
Special considerations for genetic studies in intermediate risk prostate cancer
Prostate cancer is the most common cancer in men. More than 200 000 men are diagnosed in the USA and Canada each year with more than 30 000 to 40 000 men dying of their disease; similar proportions of men are duly affected in Europe. Using the prognostic variables of T-category, the absolute value and doubling time of serum prostate specific antigen (PSA), and the pathologic Gleason score (GS), men with localized prostate cancer are placed in low, intermediate and high-risk groupings based on the relative propensity for local and distant spread. These risk groupings can predict for biochemical failure (based on PSA outcome data), disease-free and cause-specific survival [Citation1,Citation2].
Intermediate risk prostate cancer can be defined as clinical T1-T2 disease, Gleason score <8 and PSA of <20 ng/ml, with at least one of the following adverse factors present: T2b disease, PSA >10, or Gleason score (GS) =7 [Citation3]. Despite close to one third of all prostate cancer diagnosed presenting as intermediate risk disease, this cohort has not been studied closely for novel predictive or prognostic factors. Current management options for men with intermediate prostate cancer include: (1) robotic, open or laparoscopic radical prostatectomy; (2) image-guided and intensity-modulated external beam radiotherapy (IGRT-IMRT) to doses above 74 Gy; (3) IMRT less than 70 Gy but additional concurrent/adjuvant androgen deprivation; or (4), a combination of IMRT plus a brachytherapy boost [Citation2]. Even with the attempted homogenization of patient populations using current prognostic factors, intermediate-risk patients demonstrate significant clinical heterogeneity following modern-era image-guided and intensity modulated external beam radiotherapy (IGRT-IMRT) based on biochemical outcome data. The 5-year PSA-based biochemical failure rates range from 9 to 44% depending on the clinical series. Although some of this heterogeneity relates to variable use of dose-escalation with image-guidance, other prognostic factors may include inherent genetic factors that lead to local radioresistance or occult systemic metastases and/or adverse features associated with the 3-D prostate cancer microenvironment, such as hypoxia. Therefore this risk category has an urgent need for important genomic markers that allow for triaging patients to the best local and adjuvant therapies [Citation2].
In any systematic approach to study the prostate cancer genome within pre-treatment core biopsies (and relate these findings to radiotherapy outcome), it must be recognized up front that intra- and inter-tumoral (and indeed, intra-biopsy) heterogeneity exists and this can potentially confound the interpretation of the genomic “status” of the entire gland [Citation4]. This must be accounted for when validating any signature for use as a new prognostic or predictive factor. Furthermore, the tumor microenvironment (e.g. hypoxia; epithelial-mesenchymal interactions) may add complexity to a DNA or RNA profile for a given patient. Appropriate sample acquisition using macro- or micro-dissection in close collaboration with prostate cancer pathologists is required for the careful study of genetic factors in intermediate-risk prostate cancer [Citation5]. This is important to validate the genetic factor, despite tumor heterogeneity, as being an additional factor to GS as a primary determinant of radiotherapeutic outcome. This is particularly true for prostate cancer as it is unique in being a multi-focal disease scattered throughout the glandular volume. Each of these foci may be clonal and have differential biology and propensity for aggressive phenotypes, including the capacity for metastasis [Citation4]. These data support the current approach of using image-guided radiotherapy or intra-prostatic brachytherapy to the entire prostatic volume to eradicate all possible tumor clonogens. An increased understanding of biological and genetic heterogeneity within the gland may support the future use of focal therapies to sub-glandular volumes [Citation6]. This approach is currently under active study using MRI-based HDR (high-dose rate) brachytherapy or EBRT (external beam radiotherapy) IMRT boosts.
In one approach to novel molecular prognostic factors, the Radiation Therapy Oncology Group (RTOG) has completed studies on a wide range of immunohistochemical (IHC) markers [Citation7]. These studies used tissues from Phase 3 trials of non-image guided radiotherapy (IGRT) (with and without androgen ablation) for a variety of risk groups. The IHC-based assessment of protein expression for the p53, p16INK4a, Ki-67, MDM2 and BCL2 proteins has been measured by the RTOG and over-expression of these proteins are adverse prognostic markers for outcome post-treatment [Citation7]. However, these IHC protein expression markers have not been sufficiently validated in modern-day IGRT-IMRT cohorts against biochemical outcome or cause-specific survival; but it is hoped that they can be integrated with current prognostic markers to select optimal treatment for individual patients (e.g. dose-escalation versus use of adjuvant androgen deprivation).
Another approach is to study the somatic tumor genetics of patients based on tissues derived from pre-treatment biopsies and utilize genomics to add prognostic power for personalized medicine approaches. We will now discuss the latter approach in intermediate risk prostate cancer in terms of feasibility for the radiation oncologist and patient and high-light some preliminary genetic findings that are provocative for use in outcome prognostication. We will restrict the discussion to tumor genetics although it is appreciated that normal tissue genetics relating to bowel, bladder and penile toxicity also have an important role in determining the overall therapeutic ratio of a treatment protocol.
Somatic alterations in prostate cancer: Focusing on genetic loci that may alter clinical radiocurability
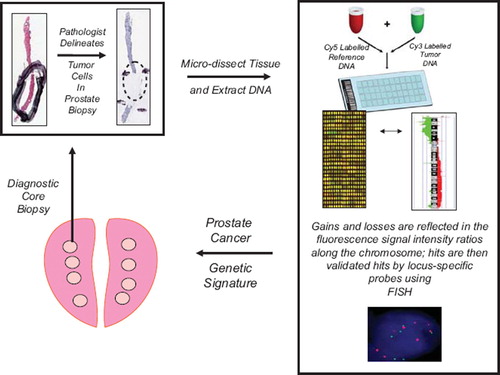
It has been estimated that each tumor contains more than 10 000 mutations where a number of the mutations will be “driver” mutations; yet even more will be “passenger” mutations. Either of these could be helpful to delineate personalized medicine if they involve genetic loci that alter radioresponse in vivo through altered DNA repair, cell cycle control, intracellular signaling, tumor metabolism and cell death. One way to determine somatic mutations in human tumors is to use array comparative genomic hybridization (aCGH). This is a technique to detect DNA copy number variations at the chromosomal and sub-chromosomal levels. DNA from a tumor test sample and a normal human reference sample are labeled using different fluorophores and hybridized to thousands of probes printed on a glass slide. The ratio of the fluorescence intensity of the hybridization of tumor-normal DNA is calculated as a measure of the copy number changes for particular chromosomal loci and software (e.g. SeeGH software) calculates the DNA loss, normal, and or probabilities for a given locus. aCGH can also generate a global measure of genetic instability by calculating the percent of genomic alteration (PGA) per tumor sample, for a given patient. aCGH results are usually confirmed in situ in paraffin sections from the same patient using loci-specific DNA probes during fluorescent-in-situ-hybridization (FISH). See for schematic summary of the aCGH process.
Figure 1. Flow diagram of array CGH technology for determining sub-chromosomal losses and gains within the prostate cancer genome (see text for details). Areas of tumor tissue are micro-dissected within pre-treatment biopsies based on a pathologist's markings. DNA is then extracted from the dissected tissue and subjected to aCGH hybridization. The aCGH “hits” are defined by fluorescence-based, image analysis software. Allelic losses and gains can be validated within the same patient's tissues, or amongst groups of patients, using loci-specific fluorescent-in-situ-hybridization (FISH). The final aCGH validated “hits” can be compared between responders and non-responders to radiotherapy to develop novel prognostic and predictive factors.
aCGH and other DNA-based studies have shown that prostate cancer progression is associated with increased gross chromosomal alterations [Citation8–11]. These include: deletions of PTEN and ATBF1, amplification of c-MYC and the prostate cancer fusion gene, TMPRSS2/ERG; androgen receptor gene amplification; KLF5 and KL6 deletion. Deletions have also been reported for the RB1, NKX3-1, E-cadherin, p16INK4A, p27KIP, and SMAD4 genes. However, most of these alterations listed above were identified in samples representing high-risk (T3/T4, Gleason score > 7 or PSA > 20 ng/ml) and metastatic disease and, until recently, it was unknown which of these genes are mutated solely in intermediate risk disease.
Newer aCGH and SNP (single-nucleotide polymorphism) arrays are now available with increased resolution, that can globally survey for DNA copy number alterations using only 100–300 nanograms of DNA and at a resolution of approximately 50–70 kilobases [Citation12]. In a recent study, we showed that it was possible to dissect and extract DNA from individual fresh-frozen prostate biopsies that were taken pre-radiotherapy in patients who received image-guided radiotherapy to a median dose of 76 Gy. Prior to radiotherapy, each patient underwent TRUS-guided insertion of three intra-prostatic fiducial markers for radiotherapy planning. At the same time, they underwent three biopsies (two for formalin-fixation and one flash-frozen in liquid nitrogen) as described [Citation13]. This was an important test of whether gland-derived biopsies containing foci with more than 70% tumor cells could be used for DNA analyses given this is the tissue available to the radiation oncologist upon which potential management decisions are made. Furthermore, there was little intra-biopsy heterogeneity when DNA was extracted from various foci within a biopsy, suggesting that the local prostate gland microenvironment is genetically homogenous. We are currently furthering our studies to see whether there is heterogeneity in alterations between sextant biopsies within a patient.
In our recent study, the number of genetic alterations per DNA sample ranged from 4 to 28, with a mean of 12. Losses were significantly more common than gains, accounting for 81% of all alterations. The majority of DNA alterations were single copy gain or loss. No homozygous deletions were observed [Citation13]. The latter is important as it will be important to test whether the allelic losses observed translate into reduced RNA and protein expression/function within our patient population.
In at least one quarter of the patients, we have observed alterations in loci that were previously reported for high risk prostate cancer, including allelic losses of 21q (TMPRSS2: ERG), 13q (Rb1), 10q (PTEN), 8p (NKX3.1), 17q (p53) and allelic gains at 8q21 (containing c-Myc). We have validated many of these aCGH “hits” using locus-specific FISH within the remaining patient's paraffin based diagnostic biopsy tissues and have shown that aCGH from the frozen biopsy has excellent concordance with FISH in situ. This suggests that at the level of aCGH resolution, many of the microfoci contain the same genetic alteration. Additionally, there were also six novel microdeletions noted in more that 20% of the cohort at 1q, 5q, 20q, and 22q; some of which affect DNA repair gene loci (see below). In a larger group of 115 patients, all these changes have been validated (Ishkanian et al. 2010 in prep.).
In summarizing our published data, the genetic alterations within 25% of intermediate risk patients overlap with the genetic alterations observed for high-risk patients. This suggests that some of these markers may predict not only for local tumor growth and aggression, but also for the propensity for sub-clinical metastases. In an attempt to genetically place patients into local versus systemic risk categories, Lapointe and co-workers [Citation14] used array CGH in tissues from prostate and lymph node metastases to define three sub-types of prostate cancer: one (subtype-1) was linked to clinically favorable behavior while the other two remaining subtypes (subtypes-2 and -3) linked with more aggressive prostate cancer, including metastases. Specifically, sub-type-1 contained prostate cancers with deletions on 6q and 5q and was associated with clinically indolent disease. However, deletions on 8p and 21q (NKX3-1; TMPRSS2-ERG), 10q (PTEN) and gains/amplification of 8q (c-Myc) was associated with locally aggressive and metastatic disease. The authors concluded that at least two states of prostate cancer that could be delineated by genomic alterations: indolent and locally confined versus disease with increased propensity for metastases. We are currently testing as to whether patients with abnormal PTEN-MYC have a decreased radiotherapeutic outcome and preliminary findings in a new cohort of 115 men suggest that this signature is associated with increased biochemical failure at more than five years post-therapy (Zafarana et al. 2010 in prep.).
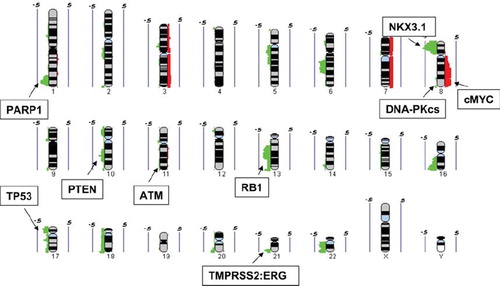
It is of great interest that a number of our aCGH “hits” have involved the allelic loss of genetic loci that normally harbor DNA damage and repair genes including loss of PARP1, ATM, DNA-PKcs, p53, Rb and RAD17. If this leads to functional loss of DNA repair or damage signaling, then these patients may benefit from targeted therapies (e.g. inhibitors of PARP, ATM, DNA-PKcs, MTp53 and CHK1) in addition to the potential tumor cell radiosensitization based on inherently abnormal DNA repair [Citation15,Citation16]. For example, PTEN deficiencies have been linked to deficiencies in RAD51-mediated homologous recombination (HR) during the repair of DNA double stranded breaks. If this leads to an HR deficiency in prostate cancer, it could manifest as an increased sensitivity to agents such as PARP inhibitors which block base excision repair and DNA single strand repair during DNA replication [Citation17]. Indeed, our use of high-resolution CGH identified a novel deletion at 1q42.12-42.3 which contains the PARP-1 gene and these patients may benefit from the use of ATM or DNA-PKcs inhibitors if the therapeutic ratio can be maintained. Similarly, our documented loss of p53 function due to allelic loss or gene mutation in more than 25% of patients ([Citation12] and manuscript in prep. 2010) suggests that some patients could benefit from the use of p53-associated targeted therapies such as the use of CHK1 inhibitors or the use of Nutlins and PRIMA-1 that target p53-defective cells following radiotherapy [Citation16,Citation18,Citation19]. The NKX3.1 protein has been recently implicated the DNA damage response of prostate cancer cells by interacting with the MRE11-ATM signaling axes and a loss of NKX3.1 may lead to increased cellular radiosensitivity [Citation17]. We are currently cataloging all these aCGH alterations and with additional information from RNA analyses, tissue microarray protein expression data and bioinformatics we are placing patients into repair-proficient and repair-deficient groups as an independent prognostic factor in radiotherapy response ().
Figure 2. Summary of DNA copy number gains (red) and losses (green) from 24 intermediate risk prostate samples (adapted from Ishkanian et al. 2009). The values of 0.5 and −0.5 indicates a level of 50% frequency gain or loss, respectively. Regions of copy number alteration containing genes associated with both prostate cancer and tumor cell radiosensitivity are shown.
Finally, the role of the TRMPRSS2:ERG somatic fusion in prostate cancer progression remains controversial and to date, no role has been suggested for this fusion event and altered radiotherapy outcome [Citation20,Citation21]. Deletion at 21q22.2 is observed in more than a third of our patients, corresponding to the TMPRSS2:ERG locus. Future studies will aim to identify frequency of this loss in a larger sample set, and correlate this with clinical outcome in order to better understand its prognostic significance. An important component to this research platform is validation of all aCGH findings in a single frozen biopsy using high-throughput FISH in tissue microarrays derived from similarly treated intermediate- risk patients (e.g. with more than five year median follow-up following dose-escalated treatments using IGRT-IMRT).
Conclusions: A need for better models in pre-clinical prostate cancer radiobiology studies and “next generation” deep sequencing of prostate cancer
As part of our intermediate risk study, we compared our CGH profiles of human intermediate risk prostate cancers with the CGH profiles of normal or malignant prostate epithelial cell lines commonly used in prostate cancer research (i.e. PREC, BPH, 22RV1, PC3, DU145 and LNCAP). As expected, there were no copy number alterations detected in the PREC cell line derived from “normal” prostate epithelium, but huge differences existed amongst these cell lines with alterations that were not consistent with the primary human biopsy data. Only the aCGH data from the 22RV1 cell line/xeonograft (which expresses WTp53, has a karyotype of 50 chromosomes, expresses PSA, and is androgen-sensitive) approximated the changes found in human intermediate risk disease [Citation13]. As such, from a genetics and radiotherapy standpoint, the radiobiology community needs to develop primary prostate cancer xenografts to study radioresponse in vivo as the commonly used prostate cancer models do not recapitulate human intermediate risk prostate cancer.
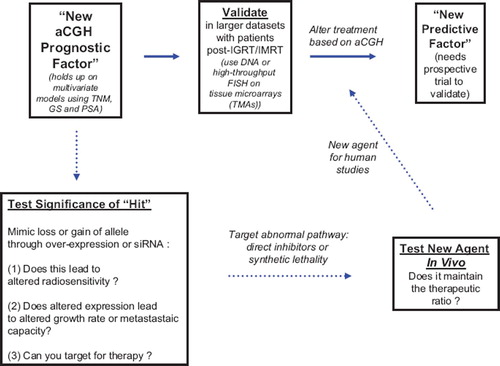
In conclusion, the use of high-resolution CGH for fine-mapping of deletions and amplifications in pre-radiotherapy prostate cancer biopsies is feasible. We have also shown that many regions previously identified in high-risk disease are also present in Gleason 6/7 patients. Genetic alterations may delineate localized prostate cancer from systemic disease and be used as a predictive factor in that patients would be individually triaged to local (surgery versus radiotherapy) and/or adjuvant (adjuvant androgen ablation or post-operative radiotherapy) therapies in a prospective fashion to improve outcome. This iterative approach to pre-clinical and clinical validation of a novel genetic prognostic marker is shown in . Furthermore, if allelic loss is associated with a loss of gene function, this information would be useful to select individualized therapies based on synthetic lethality (e.g. use of PARP inhibitors in patients with loss of ATM, NKX3.1, and DNA-PKcs function) or abnormal intracellular signaling (PTEN). An important concept is that the genetic changes that are currently documented are reflective of the genetics of clonogens within the irradiated volume that are capable of re-growing the tumor if they are resistant to radiotherapy. Finally, although beyond the scope of this review, information regarding loss of DNA repair or signaling in normal tissues based on SNP or functional analyses will also be important to document in order to effectively determine a molecular therapeutic ratio on a patient to patient basis [Citation22–24].
Figure 3. Flow diagram of authors’ approach to iterating “hits” from aCGH studies in prostate cancer into prognostic and predictive factors and using the same information to develop novel targeting strategies in prostate cancer treatment.
As high-throughput sequencing costs decrease, it is anticipated that “next generation” deep sequencing and paired-end analyses will overtake aCGH in the next three to five years for DNA-associated genomic studies. These are powerful technologies in which automated DNA sequencers now have the capacity to sequence millions, and soon billions, of DNA bases per day and could be done for as little at $1 000.00 USD per genome within five years. A Canadian-led project within the International Cancer Genomic Consortium is attempting to utilize these high-throughput technologies to sequence the entire genome of at least 1 500 prostate cancers (www.icgc.org) and relate this to outcome. Although the data are not yet available to be used in clinical practice, this new information will no doubt lead to practical changes pertaining to the treatment of indolent versus non-indolent prostate cancer, radioresistant prostate cancers and prostate cancers that harbor occult metastases at the time of diagnosis.
Acknowledgments
We thank Charles Catton, Michael Milosevic, Brad Wouters, Chad Malloff, Theo van der Kvast, Jeremy Squire, and Wan Lam for helpful discussions. Supported by operating grants from Prostate Cancer Canada, the Terry Fox Research Institute, the Ontario Institute for Cancer Research, the US Army DOD Prostate Program and an infrastructure grant from the Canadian Foundation for Innovation grant to the STTARR Innovation Facility. This research was also funded in part by the Ontario Ministry of Health and Long Term Care. The views expressed do not necessarily reflect those of the OMOHLTC. RGB is a Canadian Cancer Society Research Scientist. AI holds a Canadian Urology Oncology Group Post-Doctoral Award and JT is a CFCRI–OCI Prostate Cancer Fellow.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Mohler J, Bahnson RR, Boston B, Busby JE, D'Amico A, Eastham JA, . NCCN clinical practice guidelines in oncology: Prostate cancer. J Natl Compr Cancer Network 2010;8:162–200.
- Nichol AM, Warde P, Bristow RG. Optimal treatment of intermediate-risk prostate carcinoma with radiotherapy: Clinical and translational issues. Cancer 2005;104:891–905.
- D'Amico AV, Whittington R, Malkowicz SB, Schultz D, Blank K, Broderick GA, . Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 1998;280:969–74.
- Onik G, Miessau M, Bostwick DG. Three-dimensional prostate mapping biopsy has a potentially significant impact on prostate cancer management. J Clin Oncol 2009;27: 4321–6.
- Van der Kwast TH, Evans A, Lockwood G, Tkachuk D, Bostwick DG, Epstein JI, . Variability in diagnostic opinion among pathologists for single small atypical foci in prostate biopsies. Am J Surg Pathol 2010;34:169–77.
- Ahmed HU, Pendse D, Illing R, Allen C, van der Meulen JH, Emberton M. Will focal therapy become a standard of care for men with localized prostate cancer? Natl Clin Pract Oncol 2007;4:632–42.
- Roach M, 3rd, Waldman F, Pollack A. Predictive models in external beam radiotherapy for clinically localized prostate cancer. Cancer 2009;115(13 Suppl):3112–20.
- Jenkins RB, Qian J, Lieber MM, Bostwick DG. Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res 1997;57:524–31.
- Kim JH, Dhanasekaran SM, Mehra R, Tomlins SA, Gu W, Yu J, . Integrative analysis of genomic aberrations associated with prostate cancer progression. Cancer Res 2007;67: 8229–39.
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, . PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997; 275(5308):1943–7.
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, . Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310(5748):644–8.
- Ishkanian AS, Malloff CA, Watson SK, DeLeeuw RJ, Chi B, Coe BP, . A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet 2004;36: 299–303.
- Ishkanian AS, Mallof CA, Ho J, Meng A, Albert M, Syed A, . High-resolution array CGH identifies novel regions of genomic alteration in intermediate-risk prostate cancer. Prostate 2009;69:1091–100.
- Lapointe J, Li C, Giacomini CP, Salari K, Huang S, Wang P, . Genomic profiling reveals alternative genetic pathways of prostate tumorigenesis. Cancer Res 2007;67:8504–10.
- Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer 2008;8: 180–92.
- Bristow RG, Ozcelik H, Jalali F, Chan N, Vesprini D. Homologous recombination and prostate cancer: A model for novel DNA repair targets and therapies. Radiother Oncol 2007;83: 220–30.
- Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, . Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med 2009;1: 315–22.
- Supiot S, Hill RP, Bristow RG. Nutlin-3 radiosensitizes hypoxic prostate cancer cells independent of p53. Mol Cancer Ther 2008;7:993–9.
- Supiot S, Zhao H, Wiman K, Hill RP, Bristow RG. PRIMA-1(met) radiosensitizes prostate cancer cells independent of their MTp53-status. Radiother Oncol 2008;86:407–11.
- Barwick BG, Abramovitz M, Kodani M, Moreno CS, Nam R, Tang W, . Prostate cancer genes associated with TMPRSS2-ERG gene fusion and prognostic of biochemical recurrence in multiple cohorts. Br J Cancer 2010;102:570–6.
- Prensner JR, Chinnaiyan AM. Oncogenic gene fusions in epithelial carcinomas. Curr Opin Genet Dev 2009;19: 82–91.
- Burri J, Haldimann M, Dudler V. Selenium status of the Swiss population: Assessment and change over a decade. J Trace Elem Med Biol 2008;22:112–9.
- Damaraju S, Murray D, Dufour J, Carandang D, Myrehaug S, Fallone G, . Association of DNA repair and steroid metabolism gene polymorphisms with clinical late toxicity in patients treated with conformal radiotherapy for prostate cancer. Clin Cancer Res 2006;12:2545–54.
- Pugh TJ, Keyes M, Barclay L, Delaney A, Krzywinski M, Thomas D, . Sequence variant discovery in DNA repair genes from radiosensitive and radiotolerant prostate brachytherapy patients. Clin Cancer Res 2009;15:5008–16.