
Abstract
Aim. The aim of this study was to investigate whether poly(ADP-ribose) polymerase (PARP) inhibition improves endothelin-1 (ET-1)-induced endothelial dysfunction (ED).
Methods. Isolated rat thoracic aorta rings were incubated with ET-1 (10 nmol/L) in the presence or absence of either polyethylene glycol–superoxide dismutase (PEG-SOD; a cell-permeable superoxide radical scavenger, 41 U/mL) plus apocynin (a NADPH oxidase inhibitor, 300 µmol/L) or PJ34 (an inhibitor of polyADP-ribose polymerase, 3 µmol/L) for 18 h. Isometric tension studies were performed in response to acetylcholine (ACh; an endothelium-dependent vasodilator), sodium nitroprusside (SNP; an endothelium-independent vasodilator), and phenylephrine (Phe). PARP-1 and PAR (an end-product of PARP activity) expressions were evaluated by both Western blot and immunohistochemistry.
Results. Incubation of thoracic aorta rings with ET-1 resulted in a significant inhibition of the response to ACh, while SNP-induced relaxation was unaffected. The contractile response to Phe increased in arteries that were incubated with ET-1. PARP-1 and PAR expressions increased after ET-1 incubation. The diminished vasoreactivity as well as changes in expressions of PARP-1 and PAR in ET-1-incubated vessels were improved by both PEG-SOD plus apocynin and PJ34.
Conclusion. Our studies demonstrate that ED induced by ET-1 seems to be effected via oxidative stress in the thoracic aorta endothelium with subsequent activation of the PARP pathway.
Introduction
The healthy endothelium plays an important physiological role in maintaining the cardiovascular homeostasis. Previous studies confirm the importance of endothelial dysfunction (ED), which is associated with the impairment of endothelium-dependent vasodilatation, in the pathophysiology of several cardiovascular diseases such as diabetes, hypertension, and heart failure (Citation1–3). ED refers to the inability of arteries and arterioles to dilate fully in response to vasodilatory stimuli such as acetylcholine or shear stress, which is associated with decreased endothelial nitric oxide (NO) bioavailability (Citation4). Endothelin-1 (ET-1), one of the most potent endogenous vasoconstrictor peptides, primarily generated by endothelial cells, has been reported to contribute to the pathogenesis and maintenance of ED in cardiovascular diseases (Citation5). Several mechanisms can be underlying the harmful effects of ET-1 on the endothelium with one important effect being a decreased NO availability. It has been reported that increased NO inactivation and sequestration by reactive oxygen species (ROS) are responsible for this effect (Citation6). Furthermore, it was also found that NADPH oxidase may explain, at least in part, ET-1-induced vascular ROS production (Citation7), which leads to ED and hypertension. Although the relationship between ROS and ET-1-induced ED has been investigated (Citation7–9), until now, the downstream cellular mechanisms involved after ROS generation have not yet been clearly defined.
Poly(ADP-ribose) polymerase (PARP), also known as poly(ADP-ribose) synthetase (PARS), is an abundant nuclear enzyme that has recently been identified as a major downstream intracellular pathway of oxidative stress (Citation10–13). The PARP pathway is involved in the cellular response to DNA injury (Citation14). Under physiological conditions or limited DNA damage, PARP acts as a survival factor involved in the DNA damage repair process (Citation15). On the other hand, after excessive DNA damage following oxidative stress, PARP can lead to cell death due to depletion of nicotinamide adenine dinucleotide (NAD+) and consequently of adenosine triphosphate (ATP) pools (Citation14). Taking the above into account, it could be hypothesized that activation of the PARP pathway as a consequence of increased oxidative stress, and subsequent DNA strand breaks, may be one of the key mechanisms involved in the development of ED induced by ET-1. To our knowledge, however, there is no study directly evaluating PARP activation in ET-1-induced ED.
In the present study, the contribution of PARP activation in the development of ED has been evaluated in isolated rat thoracic aorta after ET-1 incubation. We also investigated whether pharmacological PARP inhibition may represent a novel therapeutic approach in the treatment of ED caused by ET-1.
Material and methods
Experimental model
This study was registered by the Animal Ethics Committee of Akdeniz University Medical Faculty, Antalya, Turkey. Briefly, male Wistar rats, 6–8 week of age, weighing 250–300 g were used. These rats did not receive any vehicle or chemical. The animals were anaesthetized with an intraperitoneal injection of ketamine hydrochloride (90 mg/kg) and xylazine (10 mg/kg), and decapitated. The full length of thoracic aorta was removed and cleaned of connective tissue. Then, aortas isolated from these rats were cut into 3–4 mm width rings and incubated in Dubelco's Modified Eagle Medium (DMEM) containing an antibiotic mixture (120 U/mL penicillin and 120 μg/mL streptomycin) in the presence of either deionized H2O (control), ET-1 (1 mmol/L), PJ34 (3 μmol/L), a combination of ET-1 (1 mmol/L) and polyethylene glycol–superoxide dismutase (PEG-SOD) (41 U/mL) plus apocynin (300 μmol/L), or a combination of ET-1 (1 mmol/L) and N-(6-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide hydrochloride (PJ34; 3 μmol/L) at 37°C overnight. Then, functional and histological studies were carried out.
Isolated organ bath studies
In order to perform functional studies, thoracic aorta rings were carefully suspended by two stainless-steel clips passed through the vessel lumen in 20 mL organ baths filled with PSS (mM: NaCl 118, KCl 5, NaHCO3 25, KH2PO4 1.0, MgSO4 1.2, CaCl2 2.5, and glucose 11.2) maintained at 37°C gassed with 95% O2 and 5% CO2 to obtain a pH of 7.4. Isometric tension was continuously measured with an isometric force transducer (FDT10-A, Commat Ltd, Ankara, Turkey), connected to a computer-based data acquisition system (MP35, Commat Ltd, Ankara, Turkey). Rings were placed at the optimal point of a length–tension relation by gradually stretching them until contraction induced by 20 mM of KCl was maximal at each level of distension. Accordingly, an optimal resting tension of 2 g, which had been obtained in a preliminary study by the construction of a passive length–tension curve, was applied to the rings of the thoracic aorta and allowed to equilibrate for 60 min. Because it was necessary to use endothelium-intact rings in our study, following a 60-min equilibration period, the presence of functional endothelium was confirmed by the ability of acetylcholine (ACh; 1 μmol/L) to produce relaxation of tissues precontracted with phenylephrine (Phe; 1 μmol/L). This concentration was determined from the cumulative contraction response curves to achieve 80% of the maximum contraction. For contraction–relaxation experiments, 10 thoracic aorta rings were used for each group. Relaxation responses were examined with ACh (10 nmol/L–100 μmol/L, an endothelium-specific vasodilator) and sodium nitroprusside (SNP; 0.1 nmol/L–10 μmol/L, an endothelium-independent dilatory agent) in all groups. Briefly, after the tissues had been precontracted with Phe, ACh, and SNP, concentration relaxation responses were obtained by addition of increasing concentrations of agonist to the baths in a cumulative manner. The isometric tension developed by the tissue was then recorded. The tissue response was allowed to reach a stable plateau before each successive addition of the agonist. In the first set of experiments, the effects of ET-1 (10 nmol/L, 18 h) on ACh (endothelium-dependent, 10 nmol/L–10 µmol/L)- and sodium nitroprusside (SNP; endothelium-independent, 0.1 nmol/L-10 µmol/L)-induced relaxation responses of endothelium intact thoracic aorta rings were recorded. In another set of experiments, the effects of ET-1 incubation on Phe-induced (0.1 nmol/L–50 µmol/L) contractions of aorta were investigated. In a second series of experiments, aimed at investigations of the possible contribution of oxidative stress and activation of PARP pathway in the observed inhibitory effect of ET-1, tissues were incubated with either ET-1 alone or in the presence of either PEG-SOD plus apocynin, or the PARP inhibitor PJ34 for 18 h. After incubation, concentration response curves to ACh, SNP, and Phe were recorded. Ten thoracic aortic rings were used for each group (each obtained from one animal).
Tissue processing and immunohistochemistry
In addition to functional studies, histological investigations were carried out on these tissues. Tissues obtained from control, ET-1-incubated, ET-1 and PEG-SOD plus apocynin-incubated, ET-1 and PJ34-incubated, and PJ34-incubated groups were fixed by immersion in 10% formalin and were processed routinely with paraffin embedding. Paraffin-embedded tissue samples were cut into 5-μm-thick sections and mounted on SuperFrost Plus slides (Erie Scientific Company, Portsmouth, NH, USA). For poly(ADP-ribose) (PAR) and PARP-1 immunostainings, fixed sections were deparaffinized in xylene and rehydrated in a graded series of alcohol. For antigen retrieval, slides were placed in 10 mmol/L citrate buffer (pH 6.0) and were microwaved twice for 5 min. Tissue sections were blocked for endogenous peroxidase activity with methanol containing 3% H2O2 for 10 minutes and for non-specific binding with Ultra V Block (Labvision, Freemont, CA, USA) for 7 min at room temperature. Mouse monoclonal PAR (anti-PAR Mab (10H), Alexis, San Diego, CA, USA) and rabbit polyclonal PARP-1 (Ab 6079, Abcam, Cambridge, MA, USA) primary antibodies were applied in a dilution of 2 μg of protein per mL (1:500 dilution) and 6 μg of protein per mL (1:300), respectively, for 2 h at room temperature in a humidified chamber. Sections were washed in phosphate-buffered saline (PBS), incubated with biotinylated horse antimouse IgG and antirabbit IgG (3 mg/mL; Vector Lab., Burlingame, CA, USA) at 1:500 dilution for 45 min at room temperature. After several PBS rinses, the antigen–antibody complex was detected using an avidin-biotin horseradish peroxidase complex with a Universal LSAB Kit (Dako, Glostrup, Denmark). Diaminobenzidine (3,3-diaminobenzidine tetrahydrochloride dihydrate; Sigma Chemical, St. Louis, MO, USA) was used as chromogen, and sections were mounted with Permount (Fisher Chemicals, Springfield, NJ, USA) on glass slides and then evaluated under a light microscope. To test the specificity of the antibodies, sections were incubated with non-immune IgG-3 (MAB007, R&D Systems, Minneapolis, MN, USA) and rabbit serum (X0902, Dako) at the same concentrations as with primary antibodies. Photographs were taken with an Axiophot microscope (Zeiss, Oberkochen, Germany).
SDS polyacrylamide gel electrophoresis and Western blotting
Thoracic aorta tissues were weighed and placed in homogenation buffer (10 mM Tris–HCl, 1 mM EDTA, 2.5% SDS, 1 mM phenylmethylsulfonylfluoride and 1 mg/mL leupeptin) supplemented with CompleteR protease inhibitor cocktail (Boehringer, Mannheim, Germany). After homogenization and sonication, samples were centrifuged at 10,000 g for 10 min. Supernatants were collected, and protein concentrations were determined using a Standard BCA assay, and 100 µg protein were applied per lane. Prior to electrophoresis, samples were heated for 5 min at 95°C. Samples were then subject to SDS polyacrylamide gel electrophoresis under Standard conditions and then transferred onto PVDF membranes in a buffer containing 0.2 mol/L glycine, 25 mM Tris, and 20% methanol, overnight. The membranes were blocked for 1 h with 5% non-fat dry milk and were then incubated for 1 h at room temperature with the same rabbit antisera against PARP-1 (0.5 µg/mL) and mouse antisera against PAR (3 µg/mL) used for immunohistochemistry. After washing, the membranes were incubated with antirabbit and antimouse IgG horseradish peroxidase conjugate diluted 1:5,000 for 1 h at room temperature. Immunolabelling was visualized using the chemiluminescence-based SuperSignal CL HRP Substrate System (Pierce Chemical, Rockford, IL, USA) and the membranes were exposed to Hyperfilm. As an internal standard to confirm the equal loading of the proteins, β-actin was loaded to the gels.
Drugs
Acetylcholine chloride, L-phenylephrine hydrochloride, sodium nitroprusside, ET-1, PEG-SOD, apocynin, and PJ34 were used. All drugs and the salts for the physiological salt solution were purchased from Sigma Chemical (St. Louis, MO, USA). All drugs were prepared fresh daily.
Statistical analysis
All values are expressed as mean ± SEM. Responses to ACh and SNP are expressed as percentages of the reversal of the tension developed in response to Phe. Statistical analyses of the results were performed by one-way analysis of variance (ANOVA). Post hoc comparisons were carried out using Tukey's multiple comparison post-test. A p Value lower than 0.05 was considered significant.
Results
Effects of ET-1 administration on vascular reactivity of rat thoracic aorta
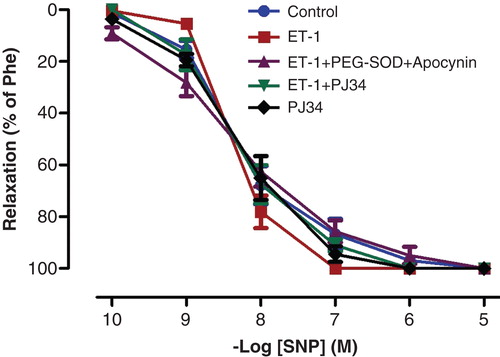
ACh and SNP evoked endothelium-dependent and -independent relaxation of isolated thoracic aorta rings precontracted with Phe, respectively. ET-1 administration attenuated the relaxation response to ACh, which is an endothelium-dependent vasodilator agent (; p < 0.05). The maximal relaxation response induced by ACh decreased from 96.29% ± 1.93% to 44.50% ± 6.22% after ET-1 incubation. Moreover, Phe-induced contraction responses were augmented in vessels incubated with ET-1 (; p < 0.05). The vasodilator effects of SNP, an endothelium-independent vasodilator agent, were not changed by ET-1 incubation (; p > 0.05).
Figure 1. Endothelium-dependent relaxation responses to ACh in controls, ET-1-incubated, ET-1 and PEG-SOD plus apocynin-incubated, ET-1 and PJ34-incubated, and PJ34-incubated thoracic aorta rings. All values are expressed as mean ± standard error of the mean; *p < 0.05 as compared with controls; **p < 0.05 as compared with ET-1-incubated rings; n = 10 for all groups.
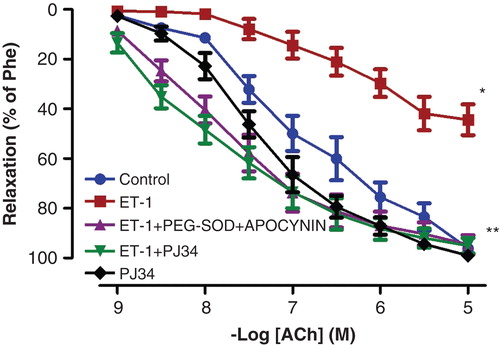
Figure 2. Effects of PEG-SOD plus apocynin, or PJ34 incubation on ET-1-induced vascular hyper-responsiveness to Phe in rat aortic rings. All values are expressed as mean ± standard error of the mean; *p < 0.05 as compared with controls; **p < 0.05 as compared with ET-1-incubated rings; n = 10 for all groups.
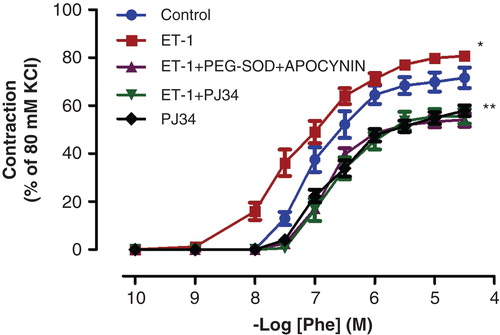
Figure 3. Endothelium-independent relaxation responses to SNP control, ET-1-incubated, ET-1 and PEG-SOD plus apocynin-incubated, ET-1 and PJ34-incubated, and PJ34-incubated thoracic aorta rings. All values are expressed as mean ± standard error of the mean; n = 10 for all groups.
Effect of PEG-SOD plus apocynin or PJ34 treatment on altered vascular reactivity induced by ET-1 incubation
The inhibitory effect of ET-1 on ACh-induced relaxation was reduced by the presence of PEG-SOD plus apocynin (; p < 0.05). Similarly, PARP-inhibitor treatment with PJ34 normalized the endothelial function in the ET-1-incubated thoracic aorta rings (; p < 0.05). However, incubation of aortic rings with PJ34 alone had no effect on ACh-induced relaxation (; p > 0.05). Otherwise, the potentiation of Phe-induced contractions observed in the presence of ET-1 was also inhibited by the presence of either PEG-SOD plus apocynin, or PJ34 (; p < 0.05).
Expressions of PAR and PARP-1 proteins in thoracic aorta
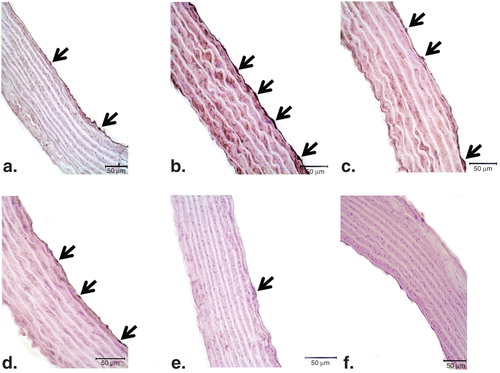
There was weak PARP-1 and PAR expression in endothelial cells of control vessels ( and , respectively). Conversely, in the endothelium of ET-1-incubated aorta rings, strong staining for both proteins was observed ( and ). In ET-1 and PEG-SOD plus apocynin-incubated thoracic aorta tissues, the expressions of PARP-1 and PAR were similar to that in controls ( and ). In accordance with these results, cross-sections of aortic rings co-incubated with ET-1 and PJ34 showed decreased PAR expression ( and ) when compared with vessels incubated with ET-1. PJ34 treatment alone did not cause a significant change in PARP-1 and PAR expression ( and ).
Figure 4. Representative photomicrographs of PARP-1 (a–f) immunohistochemistry in controls (a), ET-1-incubated (b), ET-1 and PEG-SOD plus apocynin-incubated (c), ET-1 and PJ34-incubated (d), and PJ34-incubated vessels (e). Strong immunostaining of PARP-1 was observed in ET-1-incubated endothelial cells (arrows). Expression of PARP-1 was minimal in both ET-1 and PEG-SOD plus apocynin-incubated, and ET-1 and PJ34-incubated vessel endothelium. Negative control sections with PARP-1 (f) primary antibodies. Note the absence of PARP-1 immunostainings. Original magnifications ×50.
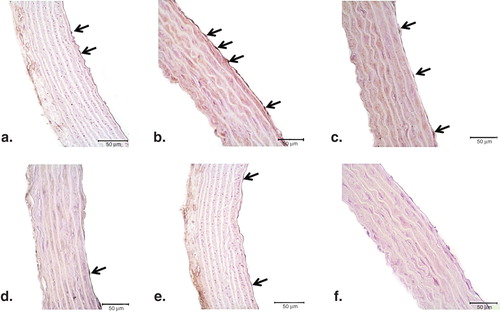
Figure 5. Representative photomicrographs of PAR (a–f) immunohistochemistry in controls (a), ET-1-incubated (b), ET-1 and PEG-SOD plus apocynin-incubated (c), ET-1 and PJ34-incubated (d), and PJ34-incubated vessels (e). Strong immunostaining of PAR was observed in ET-1-incubated endothelial cells (arrows). Expression of PAR was minimal in both ET-1 and PEG-SOD plus apocynin-incubated, and ET-1 and PJ34-incubated vessel endothelium. Negative control sections with PAR (f) primary antibodies. Note the absence of PAR immunostaining. Original magnifications ×50.
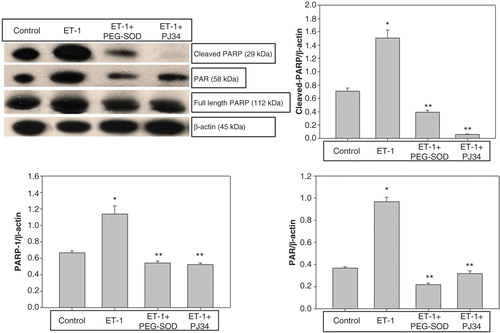
In order to elucidate the expression levels of PARP-1 and PAR, we performed Western blot analysis of these proteins extracted from thoracic aorta of control, ET-1-incubated, ET-1 and PEG-SOD plus apocynin-incubated, and ET-1 and PJ34-incubated aortic rings. In the control group, weak PARP-1, cleaved PARP-1, and PAR expressions were detected. However, the expressions of PARP-1, cleaved PARP-1, and PAR increased in ET-1-incubated thoracic aorta rings (; p < 0.05). The PEG-SOD plus apocynin incubation reduced the expression levels of PARP-1, cleaved PARP-1, and PAR in the thoracic aorta incubated with ET-1. Similarly, in ET-1 and PJ34-incubated aortic rings, the expressions of PARP-1, cleaved PARP-1, and PAR remained similar to that in controls.
Figure 6. Evaluation of PARP-1 and PAR expression in thoracic aorta rings by Western blot analysis in controls, ET-1-incubated, ET-1 and PEG-SOD plus apocynin-incubated, and ET-1 and PJ34-incubated specimens. *p < 0.05 as compared with controls; **p < 0.05 as compared with ET-1-incubated rings.
Discussion
This is the first study examining whether ET-1-induced ED is associated with changes in the activity of PARP-1. Thus, we clearly showed that endothelium-dependent relaxation was significantly inhibited by NADPH oxidase-mediated oxidative stress, and subsequent PARP-1 activation as a result of ET-1 incubation. Endothelium-independent relaxant response in thoracic arteries however, was not affected by the ET-1 administration. Hence, these results suggest that incubation of thoracic aorta rings with ET-1 leads to impairment mainly in the endothelium.
The mechanism(s) of ET-1-induced ED has not yet been fully established. There is evidence in the literature of the possible role of oxidative stress in the pathophysiology of ED caused by increased ET-1 levels (Citation7). NADPH oxidase is the most important source of ROS in the artery wall (Citation16). The upregulation of ROS, which are produced after activation of NADPH oxidases in response to cardiovascular risk factors, appears to be a key factor related to development of ED (Citation17). Accordingly, in our study, incubation of isolated thoracic aorta rings with PEG-SOD plus apocynin significantly improved ET-1- induced ED. Therefore, NADPH-oxidase-mediated ROS production seems to participate in the decreased endothelium-dependent relaxation.
The PARP pathway has been recently established as a major downstream intracellular pathway of oxidative stress (Citation10). Oxidant species are endogenous inducers of DNA strand breakage and are required for PARP activation. Once activated, the enzyme rapidly catalyses the transfer of adenosine diphosphate (ADP)-ribose moieties from NAD+ to the acceptor protein and to itself (Citation18). Thus, activation of PARP results in a rapid depletion of intracellular NAD+. In an attempt to restore the NAD+ pools, NAD+ is resynthesized with a consumption of two to four molecules of ATP per molecule of NAD+ (Citation19,20). This slows the rate of glycolysis and mitochondrial respiration, eventually leading to cellular dysfunction and death via apoptosis or necrosis. Our immunohistochemical studies showed increased expression of both PARP-1 and PAR in endothelial cells after ET-1 administration. The expression patterns of PARP-1 and PAR were similar to each other. The increased PARP-1 and PAR expression in thoracic aorta incubated with ET-1 was also confirmed by the results of our Western blot analysis. In accordance with these results, PJ34 treatment reduced PARP-1 and PAR expression to control levels. Inhibition of PARP-1 and PAR immunoreactivities also paralleled with the reduction in ED induced by ET-1. Indeed, results of the present study have shown that ET-1-induced vascular hyporeactivity to ACh was improved by PJ34 incubation for 18 hours. Hence, the potentiating effect of PJ34 on endothelium-dependent relaxation responses may be related with its protective effects on the endothelium. By this route, after the ET-1 incubation, PARP activation in endothelial cells causes functional impairment of endothelial function at the cellular level and reduced ability of endothelial cells to produce nitric oxide when stimulated by an endothelium-dependent relaxant agonist, such as ACh.
Incubation of rat aortic rings with ET-1 produces not only endothelial cell dysfunction. It may also cause apoptotic cell death in the endothelium. Results of our Western blot studies clearly indicated that cleaved PARP (an apoptotic form of PARP) expression increased in ET-1-incubated aortic rings, which indicates endothelial cell apoptosis. It is known that PARP-1 is cleaved in fragments of 85 and 29 kDa during apoptosis, which has become a useful hallmark of this type of cell death (Citation21). Apoptotic cell death allows cells with irreparable DNA damage to become eliminated in a safe way. By means of our results, it may be suggested that ET-1 triggers PARP activation, resulting in cellular energetic impairment in the endothelial cells, which subsequently induces endothelial cell apoptosis. Importantly, incubation of isolated thoracic aorta rings with PJ34 has been shown to cause a significant decrease in cleaved PARP-1 expression induced by ET-1. Taken together, these results indicate that incubation of thoracic aorta with ET-1 may lead to endothelial cell apoptosis and ED by overactivation of the PARP pathway. Hence, PARP inhibition with PJ34 may exert its beneficial effects through inhibition of overactivated PARP, thus improving the cellular energy status and reducing endothelial cell apoptosis. However, more extensive studies are required to explain the exact mechanisms that are responsible for this protective effect.
In conclusion, the current study is the first one to directly demonstrate that PARP is activated in the endothelium after ET-1 incubation and that the PARP pathway may be implicated in ET-1-induced ED. The present study also provides the first evidence that inhibition of PARP activity was able to prevent ED induced by ET-1. In the future, therefore, PARP inhibitors may be interesting agents for the treatment of ED, especially when related with increased ET-1 levels.
Acknowledgements
This study was supported by Akdeniz University Research Foundation (Grant No. 2010.04.0103.012).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Perticone F, Sciacqua A, Maio R, Perticone M, Maas R, Boger RH, et al. Asymmetric dimethylarginine, l-arginine, and endothelial dysfunction in essential hypertension. J Am Coll Cardiol. 2005;46:518–23.
- De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–74.
- Treasure CB, Vita JA, Cox DA, Fish RD, Gordon JB, Mudge GH, et al. Endothelial-dependent dilation of the coronary microvasculature is impaired in dilated cardiomyopathy. Circulation. 1990;91:772–9.
- Poredos P. Endothelial dysfunction and cardiovascular disease. Pathophysiol Haemost Thromb. 2002;32:274–7.
- Böhm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res. 2007;76:8–18.
- Münzel T, Just H, Harrison DG. The physiology and pathophysiology of the nitric oxide/superoxide system. In Born GVR, Schwartz CJ, editors. Vascular endothelium: physiology, pathology and therapeutic options. Stuttgart: Schattauer; 1997. p 205–20.
- Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ, et al. Endothelin-1 increases vascular superoxide via endothelin(A)-NADPH oxidase pathway in low-renin hypertension. Circulation. 2003;107:1053–8.
- Gómez-Guzmán M, Jiménez R, Sánchez M, Zarzuelo MJ, Galindo P, Quintela AM, et al. Epicatechin lowers blood pressure, restores endothelial function, and decreases oxidative stress and endothelin-1 and NADPH oxidase activity in DOCA-salt hypertension. Free Radic Biol Med. 2012;52:70–9.
- Jiménez R, López-Sepúlveda R, Kadmiri M, Romero M, Vera R, Sánchez M, et al. Polyphenols restore endothelial function in DOCA-salt hypertension: role of endothelin-1 and NADPH oxidase. Free Radic Biol Med. 2007;43:462–73.
- Radovits T, Lin LN, Zotkina J, Gero D, Szabó C, Karck M, et al. Poly(ADP-ribose) polymerase inhibition improves endothelial dysfunction induced by reactive oxidant hydrogen peroxide in vitro. Eur J Pharmacol. 2007;564:158–66.
- Celik-Ozenci C, Tasatargil A, Tekcan M, Sati L, Gungor E, Isbir M, et al. Effects of abamectin exposure on male fertility in rats: potential role of oxidative stress-mediated poly(ADP-ribose) polymerase (PARP) activation. Regul Toxicol Pharmacol. 2011;61:310–17.
- Tekcan M, Koksal IT, Tasatargil A, Kutlu O, Gungor E, Celik-Ozenci C. Potential role of poly(ADP-ribose) polymerase activation in the pathogenesis of experimental left varicocele. J Androl. 2012;33:122–32.
- Tasatargil A, Dalaklioglu S, Sadan G. Poly(ADP-ribose) polymerase inhibition prevents homocysteine-induced endothelial dysfunction in the isolated rat aorta. Pharmacology. 2004;72:99–105.
- McDonald MC, Mota-Filipe H, Wright JA, Abdelrahman M, Threadgill MD, Thompson AS, et al. Effects of 5-aminoisoquinolinone, a water-soluble, potent inhibitor of the activity of poly(ADP-ribose) polymerase on the organ injury and dysfunction caused by haemorrhagic shock. Br J Pharmacol. 2000;130:843–50.
- Spina-Purrello V, Patti D, Giuffrida-Stella AM, Nicoletti VG. Parp and cell death or protection in rat primary astroglial cell cultures under LPS/IFNgamma induced proinflammatory conditions. Neurochem Res. 2008;33:2583–92.
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–71.
- Takac I, Schröder K, Brandes RP. The Nox family of NADPH oxidases: friend or foe of the vascular system? Curr Hypertens Rep. 2012;14:70–8.
- Homburg S, Visochek L, Moran N, Dantzer F, Priel E, Asculai E, et al. A fast signal-induced activation of poly(ADP-ribose) polymerase: a novel downstream target of phospholipase c. J Cell Biol. 2000;150:293–307.
- Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15.
- Schraufstatter IU, Hinshaw DB, Hyslop PA, Spragg RG, Cochrane CG. Oxidant injury of cells. DNA strand-breaks activate polyadenosine diphosphate-ribose polymerase and lead to depletion of nicotinamide adenine dinucleotide. J Clin Invest. 1986;77:1312–20.
- Sallmann FR, Bourassa S, Saint-Cyr J, Poirier GG. Characterization of antibodies specific for the caspase cleavage site on poly(ADP-ribose) polymerase: specific detection of apoptotic fragments and mapping of the necrotic fragments of poly(ADP-ribose) polymerase. Biochem Cell Biol. 1997;75:451–6.