Abstract
In the last three decades the incidence of metabolic syndrome (MetS) has been growing worldwide along with an increase of obesity, type 2 diabetes mellitus, and non-alcoholic fatty liver disease (NAFLD). In children and adolescents such epidemics are particularly worrisome, since the metabolic consequences in adulthood will significantly burden the health care system.
Although the definition of MetS in childhood is still controversial, there is agreement with respect to NAFLD being the hepatic manifestation of MetS. However, the molecular pathogenesis of MetS and its contribution to NAFLD is complex and closely related to the pre- and postnatal environment as well as to genetic predisposing factors. The analysis of the possible relationships between NAFLD and MetS is particularly interesting, not only from an epidemiological point of view, but also to better understand the genetic and environmental factors contributing to the development of both diseases.
We here summarize the most recent epidemiological data on the incidence of both diseases in adolescents, and several aspects linking MetS with NAFLD, discussing the possible role played by genetics and intrauterine environment.
| Abbreviations | ||
| ATP III | = | Adult Treatment Panel III |
| BMI | = | body mass index |
| CRP | = | C-reactive protein |
| FFAs | = | free fatty acids |
| HDL | = | high-density lipoprotein |
| IDF | = | International Diabetes Federation |
| IL | = | interleukin |
| MetS | = | metabolic syndrome |
| MRI | = | magnetic resonance imaging |
| MRS | = | magnetic resonance spectroscopy |
| NAFLD | = | non-alcoholic fatty liver disease |
| NASH | = | non-alcoholic steatohepatitis |
| NCEP | = | National Cholesterol Education Program |
| PNPLA3 | = | patatin-like phospholipase domain-containing-3 |
| ROS | = | reactive oxygen species |
| TGF | = | transforming growth factor |
| TNF | = | tumour necrosis factor |
| VLDL | = | very-low-density lipoprotein |
Key messages
Metabolic syndrome and non-alcoholic fatty liver disease (NAFLD) in children and adolescents continue to grow in parallel with the outbreak of obesity.
Metabolic syndrome and NAFLD present common pathogenetic origins, and their understanding may facilitate the development of long-term, successful preventive strategies and treatment regimens.
Early identification of children and adolescents at risk (with genetic and/or intrauterine environment predisposition) of metabolic syndrome and NAFLD is required to prevent severe complications in adulthood.
Introduction
The metabolic syndrome (MetS) is a cluster of metabolic and cardiovascular risk factors including insulin resistance and diabetes, central obesity, elevated cholesterol levels, and high blood pressure (Citation1,Citation2). In particular, the latest worldwide definition of the MetS by the International Diabetes Federation (IDF) includes central obesity defined by increased waist circumference (with ethnicity and sex-specific values) and two or more of the following features: raised triglyceride concentrations (≥150 mg/dL); reduced high-density lipoprotein (HDL) cholesterol (<40 mg/dL in males, <50 mg/dL in females); elevated blood pressure (systolic blood pressure ≥130 and/or diastolic blood pressure ≥85 mmHg); and raised fasting glucose (≥100 mg/dL) (Citation3). Although this IDF definition was initially used also for children and adolescents, there is now a new definition of MetS that is more easily applicable in clinical practice. Waist circumference, considered as percentiles rather than absolute values, represents the main component of MetS in children and adolescents (Citation4). As discussed below, MetS in children is defined differently with respect to three age-groups: 6–10 years, 10–16 years, and ≥16 years.
In the last 20 years, incidence of MetS has increased dramatically in the adult population, and evidence is emerging of wide-spread MetS in children and adolescents (Citation6,Citation7). This escalation is partly due to genetic predisposition and intrauterine events, and partly to over-nutrition and sedentary life-style that characterize the adolescence of urban children (Citation8,Citation9). Unhealthy life-style and consequent obesity during childhood (2–11 years) strongly contribute to making adolescents (12–18 years) prone to develop MetS and related diseases during the teenage period (Citation10).
In addition to the classical components of the MetS, there is increasing alarm regarding the emergence of non-alcoholic fatty liver disease (NAFLD) (Citation11,Citation12), which comprises variable degrees of simple steatosis, non-alcoholic steatohepatitis (NASH), and fibrosis in obese children and adolescents (Citation13). Epidemiological studies support a close association between MetS and NAFLD both in adults and children (Citation14,Citation15). Furthermore, NAFLD, due to its frequent association with obesity, insulin resistance, and alterations of glucose and lipid metabolism, is considered the hepatic ‘component’ of MetS. Nevertheless, the aetiology of MetS and its contribution to NAFLD is complex and closely related both to genetic predisposing factors and life-style (Citation16–20).
We here review the most recent epidemiological data on the incidence of both diseases in adolescents, and, furthermore, we provide a general idea of several aspects of the link between MetS and NAFLD, discussing the pathogenetic role of genetics and intrauterine environment.
Definition and epidemiology
MetS
In recent years several studies have been performed to achieve a proper definition of MetS in children and adolescents, mainly based on the criteria approved for adults, adjusted for gender and age (Citation18–20). However, some obstacles have been found in applying, to paediatric subjects, the parameters used in adults. For example, the lack of a paediatric central obesity score linked to MetS, the paucity of imbalanced metabolic indicators, the lack of a normal range for insulin levels during childhood, the physiological reduction of insulin sensitivity during puberty, and the ethnic differences in lipid profiles, body composition, and other metabolic features—all limit the application of adult criteria to childhood and adolescence. In a recent systematic review, Ford and Li found 40 different definitions of paediatric MetS in 27 publications (Citation21). These definitions were mainly children-adapted variations of National Cholesterol Education Program (NCEP)/Adult Treatment Panel III (ATP III) criteria (Citation22). A recent IDF consensus has achieved an agreement on the definition of MetS in children older than 6 years and adolescents (Citation5). Two of the NCEP/ATP III-based descriptions of paediatric MetS criteria have been used in large paediatric population studies but, as shown in , different cut-off values for some components of the MetS have been proposed (Citation4,Citation23,Citation24). The use of different definitions to estimate MetS in children and adolescents has provided broadly variable data on the incidence of the disease. Cook et al. (Citation23) in the Third National Health and Nutrition Examination Survey (NHANES III), conducted from 1988 to 1994, estimated that 1 million 12–19-year-old adolescents in the United States had the MetS. In particular, this study reported that the incidence of MetS was 6.8% among overweight adolescents and 28.7% among obese adolescents. However, these results differ from those by de Ferranti et al. (Citation24), in which the incidence among adolescents was 31.2%. In the last 5 years, the general incidence of 4.2% reported by NHANES III has risen to 6.4% in NHANES 1999–2000, and up to 8.6% in the most recent survey (Citation25,Citation26).
Table I. Diagnostic criteria for MetS in children and adolescents.
These findings highlight that the cluster of metabolic disorders characterizing MetS in children and adolescents is still under debate, but there is wide consensus on the hypothesis that various components of disease may predispose for an early onset of the cardiovascular diseases in adulthood. In this context, a definition of MetS in the paediatric population may be a useful tool to establish the incidence of disease and also to estimate its potential evolution in adulthood (Citation6). Adolescents, due to their profound changes in body composition, are individuals particularly at risk of developing MetS, with many adverse consequences for health, including greater rates of mortality as young adults. At the same time, these changes may partly explain the difficulty in identifying earlier characterizations of the metabolic syndrome, being still so unstable during and beyond the teenage years.
The incidence of MetS in adolescents increases in parallel to body mass index (BMI), and it appears to be greater in boys than in girls (Citation15,Citation26). Moreover, the ethnic differences in adolescents reflected those observed in adults, with the greatest incidence in Hispanic-Americans, followed by non-Hispanic whites and then Afro-Americans (Citation27).
In the absence of a consensus on the definition of MetS in children and adolescents, the IDF definitions, within their limitations, operatively represent the best diagnostic tool for identifying the condition in young people. The IDF criteria are applicable to three age-groups: 6–9 years, 10–15 years, and 16 years and older (). In all three age-groups central obesity is an essential condition for the diagnosis. In adolescents aged 16 years or more the IDF criteria take also into account the different ethnicities.
Table II. IDF definition for paediatric MetS.
NAFLD
The term NAFLD refers to a spectrum of liver diseases that are characterized by the accumulation of excess fat in the liver (>5%–10% by weight) in the absence of significant alcohol consumption or other specific cause of liver disease. The most common form of NAFLD is the simple fatty liver, which is characterized by fat accumulation without liver damage, while the most advanced form of NAFLD is NASH, in which fat accumulation is associated with liver cell inflammation and different degrees of scarring. Simple fatty liver remains a benign process in most affected children, whereas the presence of necro-inflammation, typical of NASH, may be the driving force for the development of severe fibrosis and cirrhosis. Currently, only the histology discriminates among the different features of NAFLD, hence it appears to be critical for the diagnosis and management of paediatric NAFLD. The histological hall-mark in children with NAFLD is steatosis, but ballooning, inflammation, and fibrosis may be present (Citation28,Citation29). Therefore, liver biopsy is considered the ‘gold standard’ in establishing the diagnosis of NASH as well as in assessing disease severity (i.e. fibrosis). However, liver biopsy has several limitations particularly in children (i.e. invasiveness), and even though it represents the best methodology for estimating the real incidence of NAFLD in the paediatric population, it is not feasible in population-based studies (Citation30). To solve this problem and avoid liver biopsy, numerous non-invasive methods have been proposed as alternative screening/diagnostic tests (Citation31).
Most studies use surrogate markers for NAFLD, such as BMI and serum aminotransferase levels. Alternatively, ultrasound scan can be used, although the diagnostic accuracy of this approach is limited (i.e. the sensitivity drops sharply when the degree of steatosis decreases below 30%, and, in addition, this approach cannot rule out fibrosis), and it is not able to distinguish between simple steatosis and NASH.
In contrast to ultrasound, magnetic resonance spectroscopy (MRS) and magnetic resonance imaging (MRI) are able to accurately quantify intrahepatic lipid content, demonstrating a large potential, especially in longitudinal and cross-sectional studies (Citation32).
Even though MRS and MRI today represent the most viable alternative to liver biopsy for evaluating the presence of steatosis, they fail to detect fibrotic tissue. Recent reports indicate the ability of transient elastography to assess liver fibrosis in a large Italian paediatric series with NAFLD (Citation33). Several on-going studies aim to combine different non-invasive tests (i.e. serum markers and imaging) in order to achieve a diagnostic power very close to that obtained with histology.
Nowadays, NAFLD is recognized as the main cause of liver disease worldwide both in adults and children. Greatly variable incidence rates of paediatric NAFLD (from 2% to 10% in all individuals, up to 80% in obese subjects) have been reported in North and South America, Europe, Australia, and Asia (Citation34). This wide variability depends on the type of diagnostic tools but is also influenced by the age, sex, and race of the study population, as well as by the differences in ethnic composition and metabolic risk factors. In particular, hypertriglyceridaemia and/or hypercholesterolaemia, which are frequently associated with both obesity and type 2 diabetes, have been reported in 20%–80% of children with NAFLD. The prevalence of NAFLD rises in hyperglycaemic patients, and insulin resistance is more severe in individuals with NASH than in those with steatosis, thus explaining the significant association of hyperglycaemia with NASH (Citation34,Citation35). The pathogenetic role of these metabolic factors has been demonstrated by several observations, suggesting that NAFLD might be considered the hepatic manifestation of the MetS (Citation36–38). However, recent studies suggest that insulin resistance and MetS might represent a consequence rather than the cause of NAFLD (Citation39).
Risk factors and pathogenetic mechanisms
MetS
Despite a strong correlation between obesity, insulin resistance, and development of MetS, this relationship is nevertheless complex and only partially understood (Citation40). Moreover, besides obesity and insulin resistance, other factors are actively involved in the pathogenesis, including genetic predisposition, adipocytokines, inflammatory molecules, oxidative stress, life-style, and intrauterine events (Citation6,Citation41–44).
Obesity represents the major risk factor for MetS in both children and adolescents. Between 1999 and 2003, the percentage of obese American adolescents increased from 14.8% to 17.4%, implying a higher risk for MetS in young adulthood (Citation6,Citation19,Citation41). Several studies support the importance of insulin resistance in the link between obesity and MetS (Citation45). The pivotal role of insulin resistance in the metabolic syndrome was originally recognized in 1988 by Reaven, with subsequent studies further strengthening this concept (Citation46,Citation47). The relation between insulin resistance, obesity, and MetS is complex, but the presence of visceral obesity and reduced insulin sensitivity seem to be the main mechanisms implicated in the development of the syndrome both in adults and children. A relationship between obesity and insulin resistance, and the prevalence of the MetS has been reported in the paediatric population (Citation6,Citation47,Citation48). The close association of MetS with insulin resistance led investigators to consider these conditions as components of the same syndrome, with a common pathogenetic origin. Insulin resistance is caused by a complex interplay between excess of nutrients, systemic fatty acid, inflammation, hypoadiponectinaemia, and oxidative and endoplasmic reticulum (ER) stress.
The fact that the increasing incidence of MetS results from excess of nutrients secondary to increased food consumption and/or sedentary life-style is widely accepted and demonstrated by diet-induced MetS in several animal models (Citation49). Moreover, Westernized diets and high intake of carbohydrate are associated with the increased risk of the MetS in children and adolescents (Citation50–52).
Nevertheless, MetS has a multifactorial aetiology involving genetic background and hormonal balance.
Several lines of evidence have shown that common variants at candidate genes for glucose homeostasis, lipid metabolism, inflammation, and obesity are associated with altered plasma levels of MetS biomarkers (Citation53). Children with at least one parent with MetS are at higher risk of becoming overweight/obese and developing insulin resistance (Citation54). Polymorphisms in the insulin promoter gene have been shown to correlate with the risk of MetS in Italian obese adolescents (Citation55).
In the pathogenesis of MetS, adipose tissue plays a crucial role, particularly determining the excessive release of free fatty acids (FFAs), and pro-inflammatory cytokines that contribute to insulin resistance in muscle and liver (Citation56). Hepatic insulin resistance, in turn, favours the increase of glucose production, very-low-density lipoprotein (VLDL) secretion, and production of pro-inflammatory factors such as C-reactive protein (CRP), and increased production of thrombotic factors such as fibrinogen. Tumour necrosis factor (TNF)-alpha is a well known factor linking obesity, diabetes, and chronic inflammation; however, several other inflammatory mediators and cytokines are over-expressed and involved in the pathogenesis of MetS in children and adolescents (Citation56,Citation57–59).
In the last years there is some experimental and clinical evidence for a causal link between pathogenesis of MetS and oxidative and ER stress and molecules and pathways that regulates these processes (Citation49,Citation60–62). Experimental models have demonstrated that increased oxidative stress is associated with the metabolic pattern of MetS, such as hypertriglyceridaemia, hyperglycaemia, hyperinsulinaemia, and hypertension (Citation60,Citation61). The presence of MetS components in overweight children associates with increased plasma levels of 8-isoprostane, a marker of systemic oxidative stress (Citation62).
Urbanization, unhealthy diet, and sedentary life-style increase the risk of MetS for the coming generations. Sedentary behaviour, such as television watching coupled with scarce physical activity, and increased hypercaloric diet regimens (high-fat and/or high-carbohydrate diets) represent today the major risk factors for obesity, MetS, and its co-morbidities in adolescents (Citation6). Thus, an adequate nutritional programme and exercise represent not only the most effective preventive strategy for MetS but also the first-line treatment in obese subjects with metabolic disorders (Citation63).
NAFLD
The pathogenesis of NAFLD is still unclear. In 1998, Day and James proposed a ‘two hit hypothesis’, subsequently replaced by the ‘multiple hits’ hypothesis (Citation64). Accordingly, liver fat accumulation and insulin resistance represent the ‘first hits’ and lead to fatty liver that is vulnerable to ‘second hits’ which trigger the progression to NASH (see ). ‘Second hits’ include oxidative stress, mitochondrial dysfunction, and imbalance of production/release of hormones derived from adipose tissue (adipocytokines) (Citation65). More recently, the gut/liver hypothesis has been proposed. This suggests that gut bacterial endotoxins activate molecules of innate immune response, acting as possible triggers of the primary fat accumulation in the liver and necro-inflammatory lesions in the progression of steatosis to NASH and severe fibrosis (see ) (Citation65,Citation66).
Figure 1. Schematic representation of pathogenetic mechanisms leading to NAFLD. Genetic predisposition and environmental factors (i.e. diet) are determinant in the early events leading to fatty liver, including insulin resistance and fatty liver accumulation. Gut-derived endotoxaemia contributes to the early onset of steatosis as well as to the progression to NASH concomitantly to the oxidative stress and adipocytokines. Cell and tissue-specific alterations that characterize NASH are determined by all these events during NAFLD pathogenesis.
Figure supplied by A. Alisi.
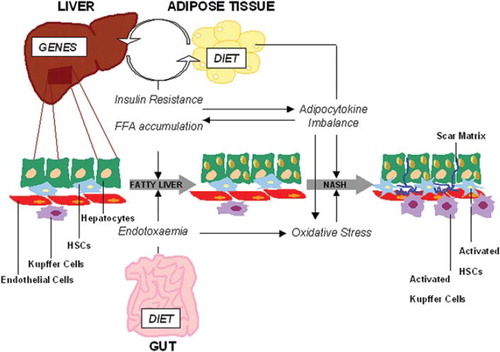
The development of fatty liver, occurring when the accumulation of lipids exceeds 5% of the liver tissue, is directly associated to disorders of lipid metabolism that may depend on several factors: excessive consumption of foods enriched in fat and fructose, increased release of free fatty acids (FFAs) from adipocytes (lipolysis), increased endogenous synthesis of lipids (de novo lipogenesis) or reduction of FFAs oxidation (mainly due to insulin resistance) (Citation67–69). FFAs introduced by the diet, or released by fat cells, are stored in the liver and for the most part are rapidly incorporated into complex lipids (e.g. triglycerides, phospholipids, glycolipids) and partially oxidized and converted into energy. When the FFAs intake exceeds the storage and oxidative capacity of peripheral tissues, FFAs are diverted to metabolic pathways that cause intracellular accumulation of toxic lipid-derived metabolites, which in turn might induce insulin resistance and activate oxidative stress signalling and inflammatory pathways (Citation69). On the other hand, hepatic insulin resistance, due to systemic factors interfering with insulin signalling, might induce the de novo lipogenesis and/or increase the triglyceride export via very-low-density lipoprotein, leading to fatty liver (Citation69–71). Insulin resistance is thought to be a critical factor in the pathogenesis of fatty liver and NASH in children and adolescents (Citation72). Hyperinsulinaemia/insulin resistance associates significantly with elevated levels of ALT and biopsy-proven paediatric NAFLD (Citation73).
Several experimental and clinical studies have highlighted the role played by oxidative stress in the development of NASH from fatty liver (Citation74,Citation75). Saturation of the oxidative processes, due to intrahepatic fat accumulation, induces the release of H2O2 and other reactive oxygen species (ROS). When ROS exceed the defensive capacity of intracellular antioxidants, they may induce NASH and fibrosis through lipid peroxidation and induction of adipocytokines (Citation74,Citation76). Lipid peroxidation leads to hepatocyte apoptosis/necrosis and proliferation and activation of hepatic stellate cells triggering intracellular signalling cascade and consequent gene expression of molecules involved in fibrogenesis. Oxidative stress influences the production and release of cytokines and adipokines, such as TNF-alpha, transforming growth factor (TGF)-beta, interleukin (IL)-6, leptin, adiponectin, and resistin, from hepatocytes, Kupffer cells, and adipocytes (Citation77,Citation78). Adipocytokines, that are preferentially secreted by adipose tissue, contribute to the inflammation, apoptosis/necrosis of hepatocytes, and onset of fibrosis, but some of them (TNF-alpha and adiponectin) also play important roles in the associations between obesity, insulin resistance, and liver fat accumulation in the preliminary phases of fatty liver development (Citation69,Citation76).
Increased serum levels of leptin and decreased adiponectin, strongly associated with insulin resistance regulation, have been found in children and adolescents with NAFLD (Citation79,Citation80).
A role of the gut/liver axis has recently been proposed as a critical factor in the pathogenesis of NAFLD. In particular, gut-derived endotoxins may increase in the blood of patients with NAFLD and activate lipopolysaccharide (LPS)-related sensors, such as Toll-like receptor 4, thus contributing to necro-inflammation and oxidative stress (Citation65,Citation66). A key role of systemic endotoxaemia in the pathogenesis of NAFLD has been suggested, and recently an association between endotoxin plasma levels and severity of disease has been demonstrated in paediatric NAFLD (Citation81).
In adolescents, NAFLD appears closely related to several risk factors of the MetS, especially obesity and insulin resistance (Citation12,Citation13). Mechanisms linking NAFLD to insulin resistance and MetS are still largely unknown; however, two main hypotheses have been proposed. Insulin resistance development is extremely complex because it involves both genetic polymorphisms, which influence the synthesis and action of insulin, and the action of environmental factors that promote obesity and NASH. On the other hand, in subjects with a genetic predisposition, environment and life-style interact with thrifty genes, favouring the development of insulin resistance and the inappropriate accumulation of fat in liver and muscle (Citation53,Citation69).
Genetic origins of NAFLD
Genetic predisposition to NAFLD is suggested by documented familial clustering of NAFLD and NASH and by the racial and ethnic differences in the prevalence of these disorders (Citation82). It has been reported that children from certain ethnicities, including Hispanics, Asians, and indigenous Americans, are more predisposed than others to develop NAFLD/NASH (Citation83). Ethnic/racial disparities might be a predictable effect of the heritability of NAFLD's risk factors (i.e. obesity and insulin resistance), but also a direct consequence of the susceptibility to NAFLD rather than to its risk factors. In this case, family members of children with NAFLD should be considered at high risk for the disease (Citation82).
The search for potential candidate genes associated with NAFLD has been based on data from animal models suggesting the involvement of specific genes, microRNAs, and proteins, and on the selection of de novo candidate genes found by genome-wide association (GWA), microarray, and proteomic studies in tissues from patients and animals (Citation82,Citation84,Citation85).
Interestingly, specific gene polymorphisms have been associated with NAFLD. Several polymorphisms have been described in genes that encode for products involved in molecular pathogenesis of NAFLD. In fact, susceptibility to NAFLD/NASH has been associated with genes influencing insulin sensitivity or regulating fatty acid metabolism (i.e. hepatic lipid synthesis, storage, and export), oxidative stress, immune regulation, and fibrosis development (see (Citation82,Citation84,Citation85) for references).
These candidate genes include: TNF-alpha, microsomal triglyceride transfer protein, methylenetetrahydrofolate reductase, adiponectin, peroxisome proliferator-activated receptor gamma coactivator 1alpha, peroxisome proliferator-activated receptor alpha, leptin receptor, and hepatic lipase. Several polymorphisms associated with NAFLD have been related also with MetS, indicating common genetic origins of these diseases () (Citation86–98).
Table III. Gene polymorphisms found in NAFLD and their correlation with MetS.
Romeo et al. have recently described the adiponutrin/patatin-like phospholipase domain-containing-3 (PNPLA3), which could be considered the first NAFLD gene. The variation in PNPLA3 contributes to racial differences in hepatic fat content and influences the susceptibility to NAFLD (Citation99). The hepatic protein expression of adiponutrin is increased by carbohydrate feeding and Western-type diet (Citation100). Moreover, it is may be involved in energy mobilization and storage of lipid droplets (Citation101). The rs738409 PNPLA3 SNP is strongly associated with severe steatosis, NASH, and the progression of liver fibrosis in a large series of Italian and UK patients with NAFLD (Citation102). More recently, the rs738409 PNPLA3 variant has been found to be associated with the severity of steatosis, hepatocellular ballooning, lobular inflammation, and perivenular fibrosis in paediatric NAFLD (Citation103).
Intrauterine environment, early nutrition, MetS, and NAFLD
Epidemiological studies in humans have shown that impaired intrauterine growth is associated with an increased incidence of insulin resistance, type 2 diabetes, and MetS in adulthood (Citation104–106). To explain this association the concept of programming was introduced. Foetal exposure to suboptimal intrauterine conditions, particularly malnutrition, during critical stages of development would lead to adaptive responses that the foetus makes to environmental cues, permanently programming tissue structure and functions (Citation107,Citation108). Once the organism is programmed in response to an adverse uterine environment, the exposure to different postnatal environmental conditions, such as over-nutrition, will determine a mismatch between intra- and extrauterine environments, eventually leading to the increased risk of metabolic disease (Citation109).
According to the ‘thrifty phenotype hypothesis’, when intrauterine foetal under-nutrition occurs during ‘critical periods’ of embryo–foetal development it permanently modifies the endocrine and metabolic pathways in an attempt to divert the limited nutrient supply to vital organs, such as the brain, at the expense of growth and the development of other organs such as the pancreas and liver. However, this adaptation to an adverse intrauterine environment, orchestrated to improve the chances of survival, may become detrimental if the organism is subsequently exposed to an extrauterine environment characterized by over-nutrition. This in utero programming may predispose to insulin resistance and metabolic syndrome, which, combined with the effects of obesity, aging, and physical inactivity, may result in cardiovascular and metabolic diseases (Citation110–112).
Another possible mechanism linking foetal environment with the risk of developing metabolic alterations in childhood and adulthood was proposed by Hattersley and Tooke (Citation113) who introduced the concept of the ‘foetal insulin hypothesis’, suggesting a strong contribution of genetic factors to the alterations of both insulin secretion and sensitivity. Polymorphisms or mutations in genes associated with insulin sensitivity result in impaired foetal growth, low birth-weight, and subsequent susceptibility to type 2 diabetes and cardiovascular disease in adult life. Mutations in the glucokinase gene determine beta-cell dysfunction, low birth-weight, and type 2 diabetes susceptibility (Citation114).
Furthermore, the structural and functional changes induced by programming may arise from epigenetic alterations of gene expression (Citation115). Animal models of intrauterine growth restriction have confirmed the role of epigenetic mechanisms in the development of long-term consequences for metabolism and blood pressure (Citation115,Citation116). In humans, periconceptional exposure to famine has been associated with persistent epigenetic changes of the insulin-like growth factor- 2 (IGF2) gene (Citation117).
A substantial body of evidence indicates that low birth-weight is associated with the components of MetS such as hyperinsulinaemia that may or may not be associated with impaired glucose tolerance and type 2 diabetes, dyslipidaemia, and hypertension (Citation118–123). These alterations may appear as early as in young adulthood, especially in subjects who experienced postnatal catch-up growth in weight (Citation124). Foetal exposure to the Dutch famine was associated with increased risk of obesity, glucose intolerance, and hypertension in adulthood (Citation125–128).
The effects of in utero conditions on adult health and disease also comprise the risk of developing postnatal obesity in foetuses exposed to hyper-nutrition. The excessive weight gain in pregnancy may predispose offspring to altered energy balance and increased adiposity in adulthood (Citation129). Studies in animal models have shown that the hypothalamic systems implicated in the control of appetite may be permanently affected by intrauterine exposure to maternal over-nutrition, eventually leading to a hyperphagic and obese phenotype in adult offspring (Citation130). The metabolic susceptibility to hyper-nutrition extends further into early extrauterine life as rapid weight gain during infancy, often induced by formula feeding, is associated with increased risk of obesity in adulthood (Citation131). Interestingly, it has recently been reported that intra-uterine growth restriction (IUGR) is an important risk factor for paediatric NAFLD, whereas breast-feeding is protective for development of NASH and its clinical expression in children (Citation132,Citation133). Within this context, the preventive role of human milk and breast-feeding along with the delayed introduction of solids in the first year of life in preventing childhood obesity (Citation134,Citation135) should always be stressed. Whether the possible causes are related to some environmental conditions or specific human milk biofactors and nutrients (such as protein and fatty acid composition) is still a matter of debate (Citation136). Accordingly, birth-weight (as the main clinical indicator of intrauterine conditions) together with the type of infant feeding should be included in clustering estimation of the metabolic risk in children (Citation137).
Concluding remarks and clinical implications
The definition of MetS in childhood is still controversial. This lack of a consensus on the diagnostic criteria inevitably affects the knowledge of the incidence and the identification of causative factors. In this review we have described the close association between MetS and NAFLD. Increasing evidence suggests that they represent the clinical expression of a common metabolic rearrangement rather than being causally related to each other. In severely obese children with features suggesting MetS, the concomitant presence of NAFLD should be suspected. Unfortunately, there is no reliable clinical, biochemical, or radiological tool to establish the diagnosis of NAFLD which is still based on histological hall-marks, thus limiting the feasibility of early diagnosis. Therefore, the search for non-invasive reliable tests for identifying NAFLD in childhood represents a priority in paediatric research.
In both MetS and NAFLD the interaction between environment and genetic predisposition plays a pivotal role from the early phases of embryo–foetal development. Intrauterine programming, followed by a mismatch between intra- and extrauterine environments, permanently affects tissue structure and function, eventually leading, in genetically predisposed subjects, to the metabolic alterations underlying both MetS and NAFLD. In this context, late interventions such as the promotion of breast-feeding in infancy and a healthy diet and life-style in adolescence and young adulthood may be ineffective in preventing or reversing the metabolic abnormalities. The early identification of ‘at-risk’ children may lead to nutritional and/or pharmacological interventions aimed at de-programming the organism, taking advantage of the biological plasticity which characterizes the early phases of extrauterine life. Finally, there is increasing evidence indicating that the pre- and periconceptional period represents another critical time window for the metabolic outcome of the offspring, thus suggesting that the optimization of maternal metabolism could represent another target for an effective preventive strategy.
Acknowledgements
The authors are indebted to Mr Ron Gerson for the careful revision of the English style.
Declaration of interest: The paper has been completely funded by Bambino Gesu Children's Hospital—IRCCS, Rome, Italy. The authors state no conflicts of interest.
References
- Alberti KG, Zimmet P, Shaw J; IDF Epidemiology Task Force Consensus Group. The metabolic syndrome—a new worldwide definition. Lancet. 2005;366:1059–62.
- Alberti KG, Zimmet P, Shaw J. Metabolic syndrome-a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med. 2006;23:469–80.
- International Diabetes Federation, 2010. Available at: http://www.idf.org/idf-worldwide-definition-metabolic-syndrome. Accessed date: 13 October 2010.
- Zimmet P, Alberti G, Kaufman F, Tajima N, Silink M, Arslanian S, . The metabolic syndrome in children and adolescents. Lancet. 2007;369:2059–61.
- International Diabetes Federation, 2010. Available at: http://www.idf.org/node/1405?unode=4A7F23CB-FA35-4471-BB06-0294AD33F2FC. Accessed date: 13 October 2010.
- Steinberger J, Daniels SR, Eckel RH, Hayman L, Lustig RH, McCrindle B, . Progress and challenges in metabolic syndrome in children and adolescents: a scientific statement from the American Heart Association Atherosclerosis, Hypertension, and Obesity in the Young Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular Nursing; and Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2009;119:628–47.
- Kiess W, Blüher S, Kapellen T, Körner A. Metabolic syndrome in children and adolescents: prevalence, public health issue, and time for initiative. J Pediatr Gastroenterol Nutr. 2009;49:268–71.
- Guerrero-Romero F, Aradillas-García C, Simental-Mendia LE, Monreal-Escalante E, de la Cruz Mendoza E, Rodríguez-Moran M. Birth weight, family history of diabetes, and metabolic syndrome in children and adolescents. J Pediatr. 2010;156:719–23, 723.e1.
- Biro FM, Wien M. Childhood obesity and adult morbidities. Am J Clin Nutr. 2010;91:1499S–1505S.
- Edmison JM, Kalhan SC, McCullough AJ. Obesity, hepatic metabolism and disease. Nestle Nutr Workshop Ser Pediatr Program. 2009;63:163–72; discussion 172–6, 259–68.
- Sundaram SS, Zeitler P, Nadeau K. The metabolic syndrome and nonalcoholic fatty liver disease in children. Curr Opin Pediatr. 2009;21:529–35.
- Alisi A, Manco M, Vania A, Nobili V. Pediatric nonalcoholic fatty liver disease in 2009. J Pediatr. 2009;155:469–74.
- Alisi A, Locatelli M, Nobili V. Nonalcoholic fatty liver disease in children. Curr Opin Clin Nutr Metab Care. 2010;13: 397–402.
- Lobstein T, Jackson-Leach R. Estimated burden of paediatric obesity and co-morbidities in Europe. Part 2. Numbers of children with indicators of obesity-related disease. Int J Pediatr Obes. 2006;1:33–41.
- Farrell GC. The liver and the waistline: Fifty years of growth. J Gastroenterol Hepatol. 2009;24 Suppl 3:S105–18.
- Nobili V, Cianfarani S, Agostoni C. Programming, metabolic syndrome, and NAFLD: the challenge of transforming a vicious cycle into a virtuous cycle. J Hepatol. 2010;52:788–90.
- Nobili V, Alisi A, Panera N, Agostoni C. Low birth weight and catch-up-growth associated with metabolic syndrome: a ten year systematic review. Pediatr Endocrinol Rev. 2008; 6:241–7.
- Lambert M, Paradis G, O'Loughlin J, Delvin EE, Hanley JA, Levy E. Insulin resistance syndrome in a representative sample of children and adolescents from Quebec, Canada. Int J Obes Relat Metab Disord. 2004;28:833–41.
- Weiss R, Dziura J, Burgert TS, Tamborlane WV, Taksali SE, Yeckel CW, . Obesity and the metabolic syndrome in children and adolescents. N Engl J Med. 2004;350:2362–74.
- Rodríguez-Morán M, Salazar-Vázquez B, Violante R, Guerrero-Romero F. Metabolic syndrome among children and adolescents aged 10–18 years. Diabetes Care. 2004;27: 2516–7.
- Ford ES, Li C. Defining the metabolic syndrome in children and adolescents: Will the real definition please stand up. J Pediatr. 2008;152:160–4.
- National Institutes of Health. The third report of the national cholesterol education program expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). Bethesda: National Institutes of Health; 2001.
- Cook S, Weitzman M, Auinger P, Nguyen M, Dietz WH. Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988–1994. Arch Pediatr Adolesc Med. 2003;157:821–7.
- de Ferranti SD, Gauvreau K, Ludwig DS, Neufeld EJ, Newburger JW, Rifai N. Prevalence of the metabolic syndrome in American adolescents: findings from the Third National Health and Nutrition Examination Survey. Circulation. 2004;110:2494–7.
- Duncan GE, Li SM, Zhou XH. Prevalence and trends of a metabolic syndrome phenotype among US adolescents, 1999–2000. Diabetes Care. 2004;27:2438–43.
- Johnson WD, Kroon JJ, Greenway FL, Bouchard C, Ryan D, Katzmarzyk PT. Prevalence of risk factors for metabolic syndrome in adolescents: National Health and Nutrition Examination Survey (NHANES), 2001–2006. Arch Pediatr Adolesc Med. 2009;163:371–7.
- Hoffman RP. Metabolic syndrome racial differences in adolescents. Curr Diabetes Rev. 2009;5:259–65.
- Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7:195–203.
- Loomba R, Sirlin CB, Schwimmer JB, Lavine JE. Advances in pediatric nonalcoholic fatty liver disease. Hepatology. 2009;50:1282–93.
- Pietrobattista A, Fruwirth R, Natali G, Monti L, Devito R, Nobili V. Is juvenile liver biopsy unsafe? Putting an end to a common misapprehension. Pediatr Radiol. 2009;39:959–61.
- Vizzutti F, Arena U, Nobili V, Tarquini R, Trappoliere M, Laffi G, . Non-invasive assessment of fibrosis in non-alcoholic fatty liver disease. Ann Hepatol. 2009;8:89–94.
- Springer F, Machann J, Claussen CD, Schick F, Schwenzer NF. Liver fat content determined by magnetic resonance imaging and spectroscopy. World J Gastroenterol. 2010;16:1560–6.
- Nobili V, Vizzutti F, Arena U, Abraldes JG, Marra F, Pietrobattista A, . Accuracy and reproducibility of transient elastography for the diagnosis of fibrosis in pediatric nonalcoholic steatohepatitis. Hepatology. 2008;48:442–8.
- Barshop NJ, Francis CS, Schwimmer JB, Lavine JE. Nonalcoholic fatty liver disease as a comorbidity of childhood obesity. Ped Health. 2009;3:271–81.
- Alisi A, Manco M, Panera N, Nobili V. Association between type two diabetes and non-alcoholic fatty liver disease in youth. Ann Hepatol. 2009;8 Suppl 1:S44–50.
- Nobili V, Marcellini M, Devito R, Ciampalini P, Piemonte F, Comparcola D, . NAFLD in children: a prospective clinical-pathological study and effect of lifestyle advice. Hepatology. 2006;44:458–65.
- Patton HM, Yates K, Unalp-Arida A, Behling CA, Huang TT, Rosenthal P, . Association between metabolic syndrome and liver histology among children with nonalcoholic Fatty liver disease. Am J Gastroenterol. 2010;105:2093–102.
- Almeda-Valdés P, Cuevas-Ramos D, Aguilar-Salinas CA. Metabolic syndrome and non-alcoholic fatty liver disease. Ann Hepatol. 2009;8(Suppl 1):S18–24.
- Vanni E, Bugianesi E, Kotronen A, De Minicis S, Yki-Järvinen H, Svegliati-Baroni G. From the metabolic syndrome to NAFLD or vice versa? Dig Liver Dis. 2010; 42:320–30.
- Pladevall M, Singal B, Williams LK, Brotons C, Guyer H, Sadurni J, . A single factor underlies the metabolic syndrome: a confirmatory factor analysis. Diabetes Care. 2006;29:113–22.
- Jasik CB, Lustig RH. Adolescent obesity and puberty: the ‘perfect storm’. Ann N Y Acad Sci. 2008;1135:265–79.
- Joy T, Lahiry P, Pollex RL, Hegele RA. Genetics of metabolic syndrome. Curr Diab Rep. 2008;8:141–8.
- Valsamakis G, Kanaka-Gantenbein C, Malamitsi-Puchner A, Mastorakos G. Causes of intrauterine growth restriction and the postnatal development of the metabolic syndrome. Ann N Y Acad Sci. 2006;1092:138–47.
- Funahashi T, Matsuzawa Y. Metabolic syndrome: clinical concept and molecular basis. Ann Med. 2007;39:482–94.
- Viner RM, Segal TY, Lichtarowicz-Krynska E, Hindmarsh P. Prevalence of the insulin resistance syndrome in obesity. Arch Dis Child. 2005;90:10–14.
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–607.
- Chiarelli F, Marcovecchio ML. Insulin resistance and obesity in childhood. Eur J Endocrinol. 2008;159 Suppl 1: S67–74.
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–28.
- Kennedy AJ, Ellacott KL, King VL, Hasty AH. Mouse models of the metabolic syndrome. Dis Model Mech. 2010;3: 156–66.
- O'Sullivan TA, Lyons-Wall P, Bremner AP, Ambrosini GL, Huang RC, Beilin LJ, . Dietary glycaemic carbohydrate in relation to the metabolic syndrome in adolescents: comparison of different metabolic syndrome definitions. Diabet Med. 2010;27:770–8.
- Aeberli I, Spinas GA, Lehmann R, l'Allemand D, Molinari L, Zimmermann MB. Diet determines features of the metabolic syndrome in 6- to 14-year-old children. Int J Vitam Nutr Res. 2009;79:14–23.
- Pan Y, Pratt CA. Metabolic syndrome and its association with diet and physical activity in US adolescents. J Am Diet Assoc. 2008;108:276–86.
- Sookoian S, Pirola CJ. Metabolic syndrome: From the genetics to the pathophysiology. Curr Hypertens Rep. 2010 Oct 19 (Epub ahead of print).
- Pankow JS, Jacobs DR Jr, Steinberger J, Moran A, Sinaiko AR. Insulin resistance and cardiovascular disease risk factors in children of parents with the insulin resistance (metabolic) syndrome. Diabetes Care. 2004;27:775–80.
- Santoro N, Cirillo G, Amato A, Luongo C, Raimondo P, D'Aniello A, . Insulin gene variable number of tandem repeats (Ins Vntr) genotype and metabolic syndrome in childhood obesity. J Clin Endocrinol Metab. 2006;91: 4641–4.
- Chu NF, Chang JB, Shieh SM. Plasma leptin, fatty acids, and tumor necrosis factor-receptor and insulin resistance in children. Obes Res. 2003;11:532–40.
- Körner A, Kratzsch J, Gausche R, Schaab M, Erbs S, Kiess W. New predictors of the metabolic syndrome in children—role of adipocytokines. Pediatr Res. 2007;61:640–5.
- Calcaterra V, De Amici M, Klersy C, Torre C, Brizzi V, Scaglia F, . Adiponectin, IL-10 and metabolic syndrome in obese children and adolescents. Acta Biomed. 2009;80:117–23.
- Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314:1–16.
- Túri S, Friedman A, Bereczki C, Papp F, Kovàcs J, Karg E, . Oxidative stress in juvenile essential hypertension. J Hypertens. 2003;21:145–52.
- Sinaiko A, Steinberger J, Moran A, Prineas RJ, Vessby B, Basu S, . Relation of body mass index and insulin resistance to cardiovascular risk factors, inflammatory factors, and oxidative stress during adolescence. Circulation. 2005;111:1985–91.
- Kelly AS, Steinberger J, Kaiser DR, Olson TP, Bank AJ, Dengel DR. Oxidative stress and adverse adipokine profile characterize the metabolic syndrome in children. J Cardiometab Syndr. 2006;1:248–52.
- McCall A, Raj R. Exercise for prevention of obesity and diabetes in children and adolescents. Clin Sports Med. 2009;28:393–421.
- de Alwis NM, Day CP. Non-alcoholic fatty liver disease: the mist gradually clears. J Hepatol. 2008;48 Suppl 1: S104–12.
- Tilg H, Moschen AR. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol Metab. 2008;19:371–9.
- Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. J Hepatol. 2009;51:212–23.
- Cheung O, Sanyal AJ. Abnormalities of lipid metabolism in non-alcoholic fatty liver disease. Semin Liver Dis. 2008;28:351–9.
- Tessari P, Coracina A, Cosma A, Tiengo A. Hepatic lipid metabolism and non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2009;19:291–302.
- Cusi K. Role of insulin resistance and lipotoxicity in non-alcoholic steatohepatitis. Clin Liver Dis. 2009;13:545–63.
- Musso G, Gambino R, Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog Lipid Res. 2009;48:1–26.
- Tsochatzis EA, Manolakopoulos S, Papatheodoridis GV, Archimandritis AJ. Insulin resistance and metabolic syndrome in chronic liver diseases: old entities with new implications. Scand J Gastroenterol. 2009;44:6–14.
- Manco M, Marcellini M, Devito R, Comparcola D, Sartorelli MR, Nobili V. Metabolic syndrome and liver histology in paediatric non-alcoholic steatohepatitis. Int J Obes (Lond). 2008;32:381–7.
- Schwimmer JB, Deutsch R, Rauch JB, Behling C, Newbury R, Lavine JE. Obesity, insulin resistance, and other clinicopathological correlations of pediatric nonalcoholic fatty liver disease. J Pediatr. 2003;143:500–5.
- Malaguarnera M, Di Rosa M, Nicoletti F, Malaguarnera L. Molecular mechanisms involved in NAFLD progression. J Mol Med. 2009;87:679–95.
- Duvnjak M, Leroti I, Barsi N, Tomasi V, Virovi Juki L, Velagi V. Pathogenesis and management issues for non-alcoholic fatty liver disease. World J Gastroenterol. 2007; 13:4539–50.
- Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–88.
- Kamada Y, Takehara T, Hayashi N. Adipocytokines and liver disease. J Gastroenterol. 2008;43:811–22.
- Polyzos SA, Kountouras J, Zavos C. Nonalcoholic fatty liver disease: the pathogenetic roles of insulin resistance and adipocytokines. Curr Mol Med. 2009;9:299–314.
- Louthan MV, Barve S, McClain CJ, Joshi-Barve S. Decreased serum adiponectin: an early event in pediatric nonalcoholic fatty liver disease. J Pediatr. 2005;147:835–8.
- Nobili V, Manco M, Ciampalini P, Diciommo V, Devito R, Piemonte F, . Leptin, free leptin index, insulin resistance and liver fibrosis in children with non-alcoholic fatty liver disease. Eur J Endocrinol. 2006;155:735–43.
- Alisi A, Manco M, Devito R, Piemonte F, Nobili V. Endotoxin and plasminogen activator inhibitor-1 serum levels associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr. 2010;50:645–9.
- Wilfred de Alwis NM, Day CP. Genes and nonalcoholic fatty liver disease. Curr Diab Rep. 2008;8:156–63.
- Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–93.
- Day CP. Genetic and environmental susceptibility to non-alcoholic fatty liver disease. Dig Dis. 2010;28:255–60.
- Duvnjak M, Barsi N, Tomasi V, Leroti I. Genetic polymorphisms in non-alcoholic fatty liver disease: clues to pathogenesis and disease progression. World J Gastroenterol. 2009;15:6023–7.
- Sookoian S, García SI, Gianotti TF, Dieuzeide G, González CD, Pirola CJ. The G-308A promoter variant of the tumor necrosis factor-alpha gene is associated with hypertension in adolescents harboring the metabolic syndrome. Am J Hypertens. 2005;18:1271–5.
- Sookoian SC, González C, Pirola CJ. Meta-analysis on the G-308A tumor necrosis factor alpha gene variant and phenotypes associated with the metabolic syndrome. Obes Res. 2005;13:2122–31.
- Zák A, Jáchymová M, Tvrzická E, Vecka M, Duffková L, Zeman M, . The influence of polymorphism of -493G/T MTP gene promoter and metabolic syndrome on lipids, fatty acids and oxidative stress. J Nutr Biochem. 2008;19:634–41.
- Vasseur F, Meyre D, Froguel P. Adiponectin, type 2 diabetes and the metabolic syndrome: lessons from human genetic studies. Expert Rev Mol Med. 2006;8:1–12.
- Ferguson JF, Phillips CM, Tierney AC, Pérez-Martínez P, Defoort C, Helal O, . Gene-nutrient interactions in the metabolic syndrome: single nucleotide polymorphisms in ADIPOQ and ADIPOR1 interact with plasma saturated fatty acids to modulate insulin resistance. Am J Clin Nutr. 2010;91:794–801.
- Sookoian S, García SI, Porto PI, Dieuzeide G, González CD, Pirola CJ. Peroxisome proliferator-activated receptor gamma and its coactivator-1 alpha may be associated with features of the metabolic syndrome in adolescents. J Mol Endocrinol. 2005;35:373–80.
- Vimaleswaran KS, Radha V, Deepa R, Mohan V. Absence of association of metabolic syndrome with PPARGC1A, PPARG and UCP1 gene polymorphisms in Asian Indians. Metab Syndr Relat Disord. 2007;5:153–62.
- Robitaille J, Brouillette C, Houde A, Lemieux S, Pérusse L, Tchernof A, . Association between the PPARalpha-L162V polymorphism and components of the metabolic syndrome. J Hum Genet. 2004;49:482–9.
- Phillips CM, Goumidi L, Bertrais S, Field MR, Ordovas JM, Cupples LA, . Leptin receptor polymorphisms interact with polyunsaturated fatty acids to augment risk of insulin resistance and metabolic syndrome in adults. J Nutr. 2010;140:238–44.
- Stancáková A, Baldaufová L, Javorský M, Kozárová M, Salagovic J, Tkác I. Effect of gene polymorphisms on lipoprotein levels in patients with dyslipidemia of metabolic syndrome. Physiol Res. 2006;55:483–90.
- Zhou YJ, Li YY, Nie YQ, Yang H, Zhan Q, Huang J, . Influence of polygenetic polymorphisms on the susceptibility to non-alcoholic fatty liver disease of Chinese people. J Gastroenterol Hepatol. 2010;25:772–7.
- Chien KL, Hsu HC, Chen YC, Su TC, Lee YT, Chen MF. Association between sequence variant of c.553 G > T in the apolipoprotein A5 gene and metabolic syndrome, insulin resistance, and carotid atherosclerosis. Transl Res. 2009;154:133–41.
- Johansson LE, Johansson LM, Danielsson P, Norgren S, Johansson S, Marcus C, . Genetic variance in the adiponutrin gene family and childhood obesity. PLoS One. 2009;4:e5327.
- Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, . Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–5.
- Hoekstra M, Li Z, Kruijt JK, Eck MV, Berkel TJ, Kuiper J. The expression level of non-alcoholic fatty liver disease-related gene PNPLA3 in hepatocytes is highly influenced by hepatic lipid status. J Hepatol. 2010;52:244–51.
- Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. Characterization of the human patatin-like phospholipase family. J Lipid Res. 2006;47:1940–9.
- Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, . Homozygosity for the PNPLA3 / adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209–17.
- Valenti L, Alisi A, Galmozzi E, Bartuli A, Del Menico B, Alterio A, . I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology. 2010;52:1274–80.
- Hales C, Barker D, Clark P, Cox L, Fall C, Osmond C, . Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303:1019–22.
- Geremia C, Cianfarani S. Insulin sensitivity in children born small for gestational age (SGA). Rev Diabet Stud. 2004;1: 58–65.
- Lévy-Marchal C, Czernichow P. Small for gestational age and the metabolic syndrome: which mechanism is suggested by epidemiological and clinical studies? Horm Res. 2006;65 Suppl 3:123–30.
- Fowden A, Giussani D, Forhead A. Intrauterine programming of physiological systems: causes and consequences. Physiology (Bethesda). 2006;21:29–37.
- Lucas A. Programming by early nutrition in man. Ciba Found Symp. 1991;156:38–50.
- Godfrey K, Lillycrop K, Burdge G, Gluckman P, Hanson M. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr Res. 2007;61:5R–10R.
- Hales CN, Barker DJ. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5–20.
- Nair L, Nair MK, Chacko DS. Markers of fetal onset adult diseases. Indian Pediatr. 2009;46:48–54.
- Ross MG, Beall MH. Adult sequelae of intrauterine growth restriction. Semin Perinatol. 2008;32:213–8.
- Hattersley AT, Tooke JE. The fetal insulin hypothesis: an alternative explanation of the association of low birth-weight with diabetes and vascular disease. Lancet. 1999; 353:1789–92.
- Hattersley AT, Beards F, Ballantyne E, Appleton M, Harvey R, Ellard S. Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet. 1998;19: 268–70.
- Gluckman P, Hanson M, Buklijas T, Low F, Beedle A. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol. 2009;5:401–8.
- Goyal R, Goyal D, Leitzke A, Gheorghe C, Longo L. Brain renin-angiotensin system: fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod Sci. 2010;17:227–38.
- Pinney S, Simmons R. Epigenetic mechanisms in the development of type 2 diabetes. Trends Endocrinol Metab. 2010;21:223–9.
- Heijmans B, Tobi E, Stein A, Putter H, Blauw G, Susser E, . Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–9.
- Eriksson J, Osmond C, Kajantie E, Forsén T, Barker D. Patterns of growth among children who later develop type 2 diabetes or its risk factors. Diabetologia. 2006;49:2853–8.
- Barker D, Hales C, Fall C, Osmond C, Phipps K, Clark P. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia. 1993;36:62–7.
- Eriksson J, Forsén T, Tuomilehto J, Jaddoe V, Osmond C, Barker D. Effects of size at birth and childhood growth on the insulin resistance syndrome in elderly individuals. Diabetologia. 2002;45:342–8.
- Claris O, Beltrand J, Levy-Marchal C. Consequences of intrauterine growth and early neonatal catch-up growth. Semin Perinatol. 2010;34:207–10.
- Fagerberg B, Bondjers L, Nilsson P. Low birth weight in combination with catch-up growth predicts the occurrence of the metabolic syndrome in men at late middle age: the Atherosclerosis and Insulin Resistance study. J Intern Med. 2004;256:254–9.
- Bhargava S, Sachdev H, Fall C, Osmond C, Lakshmy R, Barker D, . Relation of serial changes in childhood body-mass index to impaired glucose tolerance in young adulthood. N Engl J Med. 2004;350:865–75.
- Ravelli G, Stein Z, Susser M. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;295:349–53.
- Ravelli A, van der Meulen J, Michels R, Osmond C, Barker D, Hales C, . Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351:173–7.
- Ravelli A, van Der Meulen J, Osmond C, Barker D, Bleker O. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr. 1999;70:811–6.
- Roseboom T, van der Meulen J, Ravelli A, van Montfrans G, Osmond C, Barker D, . Blood pressure in adults after prenatal exposure to famine. J Hypertens. 1999;17:325–30.
- Gluckman P, Hanson M. Developmental and epigenetic pathways to obesity: an evolutionary-developmental perspective. Int J Obes (Lond). 2008;32 Suppl 7:S62–71.
- Kirk S, Samuelsson A, Argenton M, Dhonye H, Kalamatianos T, Poston L, . Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS One. 2009;4:e5870.
- Baird J, Fisher D, Lucas P, Kleijnen J, Roberts H, Law C. Being big or growing fast: systematic review of size and growth in infancy and later obesity. BMJ. 2005;331:929.
- Nobili V, Marcellini M, Marchesini G, Vanni E, Manco M, Villani A, . Intrauterine growth retardation, insulin resistance, and nonalcoholic fatty liver disease in children. Diabetes Care. 2007;30:2638–40.
- Nobili V, Bedogni G, Alisi A, Pietrobattista A, Alterio A, Tiribelli C, . A protective effect of breastfeeding on the progression of non-alcoholic fatty liver disease. Arch Dis Child. 2009;94:801–5.
- Koletzko B, von Kries R, Closa R, Monasterolo RC, Escribano J, Subias JE, . Can infant feeding choices modulate later obesity risk? Am J Clin Nutr. 2009;89:1502S–8S.
- Schack-Nielsen L, Sorensen TI, Mortensen EL, Michaelsen KF. Late introduction of complementary feeding, rather than duration of breastfeeding, may protect against adult overweight. Am J Clin Nutr. 2010;91:619–27.
- Bartok CJ, Ventura AK. Mechanisms underlying the association between breastfeeding and obesity. Int J Pediatr Obes. 2009;4:196–204.
- Brambilla P, Lissau I, Flodmark CE, Moreno LA, Widhalm K, Wabitsch M, . Metabolic risk-factor clustering estimation in children: to draw a line across pediatric metabolic syndrome. Int J Obes. 2007;31:591–600.