Abstract
The heterotetrameric K+-channel KCNQ1/KCNE1 is expressed in heart, skeletal muscle, liver and several epithelia including the renal proximal tubule. In the heart, it contributes to the repolarization of cardiomyocytes. The repolarization is impaired in ischemia. Ischemia stimulates the AMP-activated protein kinase (AMPK), a serine/threonine kinase, sensing energy depletion and stimulating several cellular mechanisms to enhance energy production and to limit energy utilization. AMPK has previously been shown to downregulate the epithelial Na+ channel ENaC, an effect mediated by the ubiquitin ligase Nedd4-2. The present study explored whether AMPK regulates KCNQ1/KCNE1. To this end, cRNA encoding KCNQ1/KCNE1 was injected into Xenopus oocytes with and without additional injection of wild type AMPK (AMPKα1 + AMPKβ1 + AMPKγ1), of the constitutively active γR70QAMPK (α1β1γ1(R70Q)), of the kinase dead mutant αK45RAMPK (α1(K45R)β1γ1), or of the ubiquitin ligase Nedd4-2. KCNQ1/KCNE1 activity was determined in two electrode voltage clamp experiments. Moreover, KCNQ1 abundance in the cell membrane was determined by immunostaining and subsequent confocal imaging. As a result, wild type and constitutively active AMPK significantly reduced KCNQ1/KCNE1-mediated currents and reduced KCNQ1 abundance in the cell membrane. Similarly, Nedd4-2 decreased KCNQ1/KCNE1-mediated currents and KCNQ1 protein abundance in the cell membrane. Activation of AMPK in isolated perfused proximal renal tubules by AICAR (10 mM) was followed by significant depolarization. In conclusion, AMPK is a potent regulator of KCNQ1/KCNE1.
Keywords::
Introduction
The K+ channel KCNEx/KCNQ1 (KCNE1 was formerly called mink or IsK and KCNQ1 was also named KvLQT1 or Kv7.1) is expressed in a variety of tissues including the heart (Barhanin et al. Citation1996, Sanguinetti et al. Citation1996), skeletal muscle (Finsterer and Stollberger Citation2004) and several epithelia, such as the stria vascularis (Wangemann Citation2006), the renal proximal tubule (Vallon et al. Citation2001), the gastric parietal cells (Dedek and Waldegger Citation2001, Grahammer et al. Citation2001, Heitzmann et al. Citation2004), intestinal cells (Sugimoto et al. Citation1990, Schroeder et al. Citation2000, Dedek and Waldegger Citation2001, Nicolas et al. Citation2001, Vallon et al. Citation2001, Heitzmann et al. Citation2004) and hepatocytes (Demolombe et al. Citation2001, Lan et al. Citation2005, Lan et al. Citation2006).
Genetic defects of KCNE1 or KCNQ1 lead to Romano Ward syndrome, a disorder characterized by Long QT syndrome and cardiac arrhythmia predisposing to sudden cardiac death (Chiang and Roden Citation2000). Severe genetic loss-of-function defects in KCNQ1/KCNE1 lead to the Jervell and Lange-Nielson syndrome in humans comprising Long QT syndrome and deafness (Barhanin et al. Citation1996, Sanguinetti et al. Citation1996, Neyroud et al. Citation1997). KCNQ1 polymorphisms have further been associated with diabetes (Unoki et al. Citation2008, Yasuda et al. Citation2008).
KCNQ1 knockout mice are deaf and display a shaker/waltzer phenotype (Lee et al. Citation2000, Casimiro et al. Citation2001), defective gastric acid secretion (Scarff et al. Citation1999, Lee et al. Citation2000), vitamin B12 deficiency with anemia, blunted stimulation of intestinal Cl- secretion by cAMP, intestinal loss of Na+ and K+, as well as impaired renal and intestinal substrate transport (Vallon et al. Citation2005). Moreover, KCNQ1 participates in cell volume regulation (Grunnet et al. Citation2003, Lan et al. Citation2005, Citation2006, Bachmann et al. Citation2007, vanTol et al. Citation2007).
KCNQ1/KCNE1 activity is decreased and thus action potential duration enhanced by ischemia (Liu et al. Citation2007). Cellular mechanisms accounting for the downregulation of KCNQ1/KCNE1 activity during ischemia have remained elusive. Candidates include the AMP-activated protein kinase (AMPK), which is activated upon cellular energy depletion. The kinase senses the cytosolic AMP/ATP concentration ratio and thus the energy status of the cell (Towler and Hardie Citation2007, Winder and Thomson Citation2007). AMPK stimulates cellular glucose uptake, glycolysis, fatty acid oxidation and enzymes required for ATP production (Ojuka et al. Citation2000, Winder et al. Citation2000, Zheng et al. Citation2001, MacLean et al. Citation2002, Jessen et al. Citation2003, Li et al. Citation2004, Luiken et al. Citation2004, Lei et al. Citation2005, Walker et al. Citation2005, Carling Citation2007, Jensen et al. Citation2007, Natsuizaka et al. Citation2007, Winder and Thomson Citation2007, Guan et al. Citation2008, Horie et al. Citation2008, Park et al. Citation2009). AMPK thus enhances the cellular ATP generation (McGee and Hargreaves Citation2008). It further inhibits several energy-utilizing mechanisms, such as protein synthesis, gluconeogenesis and lipogenesis (Carling Citation2007, Winder and Thomson Citation2007, McGee and Hargreaves Citation2008). AMPK stimulates glucose uptake (Carling Citation2007, Winder and Thomson Citation2007), an effect largely due to activation of the facilitative glucose carriers GLUT1, GLUT2, GLUT3 and GLUT4 (Ojuka et al. Citation2000, Winder et al. Citation2000, Zheng et al. Citation2001, MacLean et al. Citation2002, Jessen et al. Citation2003, Li et al. Citation2004, Luiken et al. Citation2004, Lei et al. Citation2005, Walker et al. Citation2005, Natsuizaka et al. Citation2007, Guan et al. Citation2008, Park et al. Citation2009) and of the secondary active SGLT1 carrier (Sopjani et al. Citation2010). Accordingly, AMPK confers some protection against cell death during energy depletion (Hardie Citation2004, McGee and Hargreaves Citation2008, Foller et al. Citation2009).
AMPK has been shown to control the membrane abundance of the epithelial Na+ channel ENaC, an effect mediated by the ubiquitin ligase Nedd4-2 (Hallows et al. Citation2003a, Carattino et al. Citation2005, Bhalla et al. Citation2006, Almaca et al. Citation2009).
The present study explored whether AMPK regulates KCNQ1/KCNE1 channels. To this end, voltage-gated current was determined in Xenopus oocytes expressing KCNQ1/KCNE1 with or without wild type, constitutively active and inactive AMPK variants. Moreover, the KCNQ1 protein abundance at the cell membrane was determined by immunohistochemistry and confocal microscopy. Additional experiments explored whether the effect of AMPK is mimicked by coexpression of Nedd4-2. Finally, the potential difference across the basolateral membrane in the proximal renal tubule was studied without and upon activation of AMPK.
Methods
Constructs
For generation of cRNA, constructs were used encoding wild type human KCNQ1/KCNE1 (Seebohm et al. Citation2008, Henrion et al. Citation2009), wild type AMPKα1-HA, AMPKβ1-Flag, AMPKγ1-HA (Fraser et al. Citation2007), constitutively active R70QAMPKγ1-HA (Hamilton et al. Citation2001), kinase dead mutant K45RAMPKα1-HA (Hallows et al. Citation2003a), wild type AMPK α2-HA (Steinberg and Kemp Citation2009), wild type Nedd4-2 (Boehmer et al. Citation2008a) and Nedd4-2S795A lacking an AMPK phosphorylation site [refer to “site directed mutagenesis”]. The AMPK inhibitor compound C (Calbiochem, Bad Soden, Germany) was used at a concentration of 10 μM, dibutyril-cAMP (Sigma, Schnelldorf, Germany) at a concentration of 1 mM.
Voltage clamp in Xenopus oocytes
Xenopus oocytes were prepared as previously described (Boehmer et al. Citation2008b, Laufer et al. Citation2009). cRNA encoding KCNQ1 (1.5 ng) and 1.5 ng cRNA encoding KCNE1 were injected with or without 4.6 ng of cRNA encoding either AMPKα1-HA + AMPKβ1-Flag + AMPKγ1-HA (WTAMPK), or AMPKα1-HA + AMPKβ1-Flag + R70QAMPKγ1-HA (γR70QAMPK) or K45RAMPKα1KD-HA + AMPKβ1-Flag + AMPKγ1-HA (αK45RAMPK) or AMPKα2-HA + AMPKβ1-Flag + R70QAMPKγ1-HA (γR70QAMPKα2) and with or without 5 ng cRNA encoding Nedd4-2 or Nedd4-2S795A on the day of preparation of the Xenopus oocytes. All experiments were performed at room temperature 3 or 4 days (Nedd4-2) after injection. In two-electrode voltage-clamp experiments KCNQ1/KCNE1 channel currents were elicited every 20 s with 5 s depolarizing pulses to +80 mV applied from a holding potential of −120 mV. Pulses were applied in 20 mV increments. The data were filtered at 1 kH and recorded with a Digidata 1322A A/D-D/A converter and Chart V.4.2 software for data acquisition and analysis (Axon Instruments) (Ureche et al. Citation2008). The analysis of the data was performed with Clampfit 8 (Axon Instruments) software. Activating current traces were fitted using the simplex algorithm to one exponential function: y = A0 + A1 * exp (−t/τ).
Immunohistochemistry
After 4% paraformaldehyde fixation for at least 12 h, oocytes were cryoprotected in 30% sucrose, frozen in mounting medium and placed on a cryostat (Gehring et al. Citation2009). Sections were collected at a thickness of 8 μm on coated slides and stored at −20°C. For immunostainings, sections were dehydrated at room temperature, fixated in acetone/methanol (1:1) for 15 min at room temperature, washed in PBS and pre-incubated for 1 h in 5% bovine serum albumin in PBS. The primary antibody used was rabbit anti-KCNQ1 antibody (diluted 1:500, Abcam, Cambridge, UK). Incubation was performed in a moist chamber overnight at 4°C. Binding of primary antibody was visualised with a goat anti-rabbit conjugated FITC antibody (diluted 1:500, Invitrogen, United States). Then, oocytes were analyzed by a fluorescence laser scanning microscope (LSM 510, Carl Zeiss MicroImaging GmbH, Germany) with A-Plan 40x/1.2W DICIII. Brightness and contrast settings were kept constant during imaging of all oocytes in each injection series.
Western blot
For western blotting, 20 intact healthy oocytes were homogenized with a pestle in 400 μl Buffer-H (100mM NaCl, 20mM Tris-HCl, pH 7.4, 1% Triton X-100, and Complete Protease Inhibitor [Roche Diagnostics GmbH, Mannheim, Germany]). The samples were kept at 4°C for 1 h on a rotator, then centrifuged for 2 min at 13,000 rpm. After measurement of the total protein concentration (Bradford assay), 50 μg of protein were solubilized in Roti-Load1 Buffer (Carl Roth GmbH, Karlsruhe, Germany) at 95°C for 10 min and resolved by 10% SDS-PAGE. For immunoblotting proteins were electro-transferred onto a nitrocellulose membrane and blocked with 5% non-fat milk in TBS-0.10% Tween 20 at room temperature for 1 h. The membrane was then incubated with rabbit anti-KCNQ1 antibody (diluted 1:500, Abcam, Cambridge, UK) at 4°C overnight. After washing (TBST), the blot was incubated with secondary anti-rabbit HRP antibody (diluted 1:1000, Cell Signaling Technology, Danvers, MA, USA) for 1 h at room temperature. For loading control the blot was stripped in stripping buffer (Carl Roth GmbH, Karlsruhe, Germany) at 56°C for 30 min. After washing with TBST the blot was blocked with 5% non-fat milk in TBST for 1 h at room temperature. The blot was then incubated with a rabbit anti-GAPDH antibody (diluted 1:1000, Cell Signaling Technology, Danvers, MA, USA) at 4°C overnight. After washing with TBST, the blot was incubated with anti-rabbit HRP antibody (diluted 1:1000, Cell Signaling Technology, Danvers, MA, USA) for 1 h at room temperature. Antibody-binding was detected with the ECL detection reagent (Amersham, Freiburg, Germany). Bands were quantified with Quantity One Software (Biorad, München, Germany).
Site-directed mutagenesis
The mutated human Nedd4-2S795A was generated by site-directed mutagenesis (QuikChange II Site-Directed Mutagenesis Kit; Stratagene, Heidelberg, Germany) according to the manufacturer's instructions. The following primers were used: Nedd4-2S795A s: 5′ GGATTTGAAGCCCAATGGGGCAGAAATAATGGTCACAAA 3′ and Nedd4-2S795A as: 5′ TTTGTGACCATTATTTCTGCCCCATTGGGCTTCAAATCC 3′. The mutant was sequenced to verify the presence of the desired mutation.
where: Φ is Hydrophobic residue; B is Basic residue; and ST is Phosphorylated serine residue.
Potential difference across the basolateral cell membrane of isolated perfused proximal straight tubules
The potential difference across the basolateral cell membrane (PDbl) was determined following incubation of isolated renal tubules from C57 BL/6 mice for 1 h at 22°C in the absence or presence of AMPK stimulator AICAR (10 mM). The bath and luminal perfusates were composed of (all numbers mmol/l): 120 NaCl, 5 KCl, 20 NaHCO3, 1.3 CaCl2, 1 MgCl2, 2 Na2HPO4. PDbl was measured by a high impedance electrometer (FD223, WPI, Science Trading, Frankfurt, Germany) connected with the electrode via an Ag/AgCl half cell. An Ag/AgCl reference electrode was connected to the bath. Entry of positive charge by electrogenic transport is expected to depolarize the basolateral cell membrane. The magnitude of the depolarization depends on the magnitude of the induced current on the one hand and on the resistances of cell membranes and shunt on the other.
Statistical analysis
Data are provided as means ± SEM, n represents the number of experiments. All oocyte experiments were repeated with at least two batches of oocytes; in all repetitions qualitatively similar data were obtained. Data were tested for significance using ANOVA or t-test, as appropriate, and results with p < 0.05 were considered statistically significant.
Results
AMPK inhibited voltage-gated outward currents in KCNQ1/KCNE1-expressing Xenopus oocytes
In KCNQ1/KCNE1-expressing, but not in water-injected Xenopus oocytes, depolarization triggered a slowly activating current (IKs) with strong outward rectification (). Coexpression of the AMP-activated protein kinase (AMPKα1 + AMPKβ1 + AMPKγ1) was followed by a significant decrease of IKs by 41 ± 7% (n = 4 batches of 25–36 oocytes) at +80 mV. Furthermore, coexpression of the constitutively active R70QAMPK (AMPKα1 + AMPKβ1 + R70QAMPKγ1) similarly decreased the slowly activating outward current of KCNQ1/KCNE1. In contrast, coexpression of the inactive K45RAMPK mutant [K45RAMPKα1 + AMPKβ1 + AMPKγ1] did not significantly modify the slowly activating outward current of KCNQ1/KCNE1-expressing Xenopus oocytes. In addition, pharmacological inhibition of AMPK by compound C (10 μM) significantly blocked the AMPK effect on KCNQ1/E1-mediated currents (). Thus, kinase activity is required for the effect of AMPK on KCNQ1/KCNE1 activity.
Figure 1. Co-expression of AMPK decreased voltage-gated outward current in KCNQ1/KCNE1 expressing Xenopus oocytes. (A) Original tracings of the current induced by depolarization from −60 mV to −40, −20, 0, 20, 40, 60, and 80 mV in Xenopus oocytes injected with water (a), expressing KCNQ1/KCNE1 without (b) or with (c) additional co-expression of wild type AMPK, of kinase dead mutant αK45RAMPK (d) or of constitutively active γR70QAMPK (e). (B) Arithmetic means ± SEM (n = 22–36) of depolarization-induced K+ current at +80 mV in Xenopus oocytes injected with water (1st bar), expressing KCNQ1/KCNE1 without (2nd bar) or with additional coexpression of wild type AMPK (3rd bar), of kinase dead mutant K45RAMPK (4th bar) or of constitutively active R70QAMPK (5th bar). ***(p < 0.001) indicates statistically significant difference from the values obtained in oocytes expressing KCNQ1/KCNE1 alone. (C) Arithmetic means ± SEM of depolarization-induced current (Ig) as a function of the potential in Xenopus oocytes injected as in B. (D) Arithmetic means ± SEM (n = 12–24) of depolarization-induced K+ current at +80 mV in Xenopus oocytes injected with water (1st bar), expressing KCNQ1/KCNE1 without (2nd bar) or with additional co-expression of constitutively active R70QAMPK (3rd bar). The oocytes were incubated in the absence (white bars) or presence of 10 μM AMPK inhibitor compound C (black bars) for the indicated number of hours prior to the experiment. ***(p < 0.001) indicates statistically significant difference from the values obtained in oocytes expressing KCNQ1/KCNE1 alone. #, ### (p < 0.05, p < 0.001) indicate significant difference from the absence of compound C. (E) Arithmetic means ± SEM (n = 8–17) of depolarization-induced K+ current at +80 mV in Xenopus oocytes injected with water (1st bar), expressing KCNQ1/KCNE1 without (2nd bar) or with additional coexpression of constitutively active R70QAMPKα2 (3rd bar). ### (p < 0.001) indicates significant difference from the absence of R70QAMPKα2.
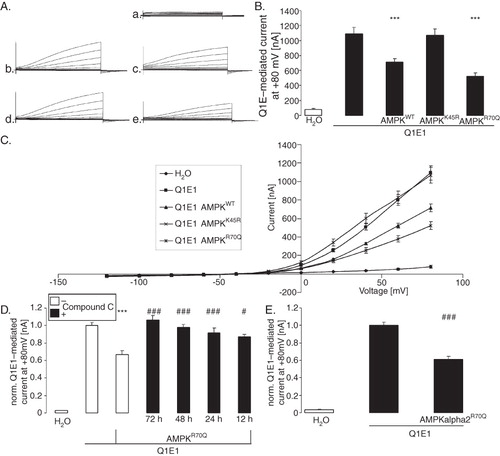
Another series of experiments tested whether AMPKα2 similarly decreases KCNQ1/KCNE1 activity. As shown in , the constitutively active R70QAMPKα2 indeed also decreases KCNQ1/KCNE1-mediated currents.
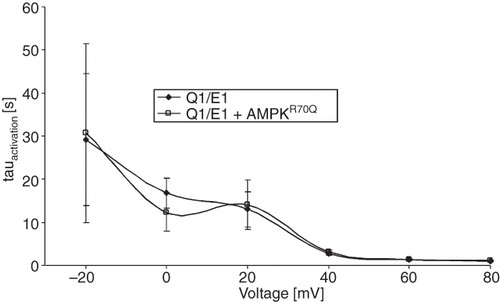
To test, whether AMPK changes the activation kinetics of the channel the activation constant τ was determined. As shown in , coexpression of constitutively active R70QAMPK did not significantly modify τ. Further experiments aimed to investigate whether PKA-dependent stimulation of KCNQ1/KCNE1 was modified by AMPK. To this end, KCNQ1/KCNE1-dependent currents were measured in the presence or absence of dibutyril-cAMP (1 mM) for three days. As a result, exposure to dibutyril-cAMP significantly increased the normalized current to 1.29 ± 0.06 rel. units (n = 25 oocytes) whereas coexpression of constitutively active R70QAMPK significantly reduced the normalized current to 0.72 ± 0.03 rel. units (n = 23 oocytes). Most importantly, dibutyril-cAMP failed to significantly modify the normalized KCNQ1/KCNE1-mediated current in oocytes co-expressing constitutively active R70QAMPK (0.73 ± 0.04, n = 22 oocytes). Thus, PKA fails to stimulate KCNQ1/KCNE1-dependent currents in oocytes expressing constitutively active R70QAMPK.
Figure 2. Co-expression of AMPK did not significantly modify activation kinetics of voltage-gated outward currents in KCNQ1/KCNE1-expressing Xenopus oocytes. Arithmetic means ± SEM (n = 31–36) of the time constant (τ) plotted vs. the depolarizing potential in Xenopus oocytes expressing KCNQ1/KCNE1 alone (closed symbols) or KCNQ1/KCNE1 together with constitutively active R70QAMPK (open symbols).
AMPK decreased the KCNQ1 protein abundance in the cell membrane
A decrease of the slowly activating outward current could have resulted from a decrease of KCNQ1/KCNE1 protein abundance in the cell membrane. To test this possibility, the KCNQ1 protein abundance was determined by confocal microscopy in Xenopus oocytes injected with water and in oocytes expressing KCNQ1/KCNE1 alone or together with AMPK. As shown in , the KCNQ1 cell surface expression of the channel protein in Xenopus oocytes injected with cRNA encoding KCNQ1/KCNE1 was indeed decreased by the co-expression of wild-type or constitutively active AMPK. The total protein abundance was not affected by AMPK ().
Figure 3. Co-expression of AMPK decreased the KCNQ1 protein abundance within the plasma membrane of oocytes. (A) Confocal images of KCNQ1 protein abundance in the plasma membrane of Xenopus oocytes injected with water (2nd upper panel), expressing KCNQ1/KCNE1 without (3rd upper panel) or with additional co-expression of wild type AMPK (1st lower panel), of kinase dead mutant αK45RAMPK (2nd lower panel) or of constitutively-active γR70QAMPK (3rd lower panel). The cells were subjected to immunofluorescent staining using FITC-conjugated antibody (grey/green). The 1st upper panel serves as control (absence of primary antibody). (B) Original Western Blots of total KCNQ1 (upper panel) and GAPDH (lower panel) in Xenopus oocytes injected with water (1st lane), expressing KCNQ1/KCNE1 without (2nd lane) or with additional co-expression of wild-type AMPK (3rd lane), of kinase dead mutant αK45RAMPK (4th lane) or of constitutively-active γR70QAMPK (5th lane). The lower bar diagram displays the densitometric analysis of the Western blots (arithmetic means ± SEM [n = 3]).
![Figure 3. Co-expression of AMPK decreased the KCNQ1 protein abundance within the plasma membrane of oocytes. (A) Confocal images of KCNQ1 protein abundance in the plasma membrane of Xenopus oocytes injected with water (2nd upper panel), expressing KCNQ1/KCNE1 without (3rd upper panel) or with additional co-expression of wild type AMPK (1st lower panel), of kinase dead mutant αK45RAMPK (2nd lower panel) or of constitutively-active γR70QAMPK (3rd lower panel). The cells were subjected to immunofluorescent staining using FITC-conjugated antibody (grey/green). The 1st upper panel serves as control (absence of primary antibody). (B) Original Western Blots of total KCNQ1 (upper panel) and GAPDH (lower panel) in Xenopus oocytes injected with water (1st lane), expressing KCNQ1/KCNE1 without (2nd lane) or with additional co-expression of wild-type AMPK (3rd lane), of kinase dead mutant αK45RAMPK (4th lane) or of constitutively-active γR70QAMPK (5th lane). The lower bar diagram displays the densitometric analysis of the Western blots (arithmetic means ± SEM [n = 3]).](/cms/asset/a8c6fe6f-e214-43c9-b721-166ffd71f0c3/imbc_a_520037_f0003_b.jpg)
KCNQ1 protein abundance is decreased by the ubiquitin ligase Nedd4-2
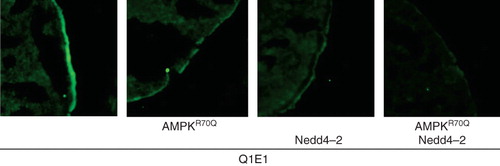
To test whether KCNQ1/KCNE1 is regulated by the AMPK-sensitive ubiquitin ligase Nedd4-2, KCNQ1/KCNE1 was expressed together with or without Nedd4-2, with or without constitutively active R70QAMPK. As shown in and 4B, Nedd4-2 indeed decreased KCNQ1/KCNE1-dependent currents. In another series of experiment, the effect of Nedd4-2 on KCNQ1/KCNE1-dependent currents was significantly reduced by co-expression of constitutively active R70QAMPK (). Nedd4-2S795A which lacks an AMPK phosphorylation site similarly decreased KCNQ1/KCNE1-dependent currents (). Constitutively active R70QAMPK, however, failed to significantly modify the Nedd4-2S795A action on KCNQ1/KCNE1-dependent currents (). Additional experiments were performed to determine whether Nedd4-2 is effective through altering KCNQ1 protein abundance in the cell membrane. The KCNQ1 protein abundance in the cell membrane was determined in oocytes expressing KCNQ1/KCNE1 together with or without Nedd4-2, with or without constitutively R70QAMPK. As shown in , the AMPK effect on KCNQ1 protein cell membrane abundance was indeed mimicked by co-expression of Nedd4-2.
Figure 4. Similar to γR70QAMPK the ubiquitin ligase Nedd4-2 downregulated KCNQ1/KCNE1. (A) Original tracings of the current induced by depolarization from −60 mV to −40, −20, 0, 20, 40, 60, and 80 mV in Xenopus oocytes injected with water (a), expressing KCNQ1/KCNE1 without (b,d) or with (c,e) additional coexpression of constitutively active γR70QAMPK in the absence (b,c) or presence (d,e) of Nedd4-2. (B) Arithmetic means ± SEM (n = 10–19) of depolarization-induced current at +80 mV in Xenopus oocytes injected with water (H2O), expressing KCNQ1/KCNE1 (Q1E1) without or with additional co-expression of constitutively active R70QAMPK (AMPKR70Q) in the absence (open bars) or presence (closed bars) of Nedd4-2. *** (p < 0.001) indicates statistically significant difference from the values obtained in oocytes expressing KCNQ1/KCNE1 alone. (C) Arithmetic means ± SEM (n = 11–14) of depolarization-induced current at +80 mV in Xenopus oocytes injected with KCNQ1/KCNE1 without (white bars) or with additional coexpression of constitutively active R70QAMPK (black bars) in the absence (2 left bars) or presence of Nedd4-2 (2 middle bars) or of Nedd4-2S795A (2 right bars). *** (p < 0.001) indicates statistically significant difference from the values obtained in oocytes expressing KCNQ1/KCNE1 alone. # (p < 0.05) indicates statistically significant difference from the absence of R70QAMPK.
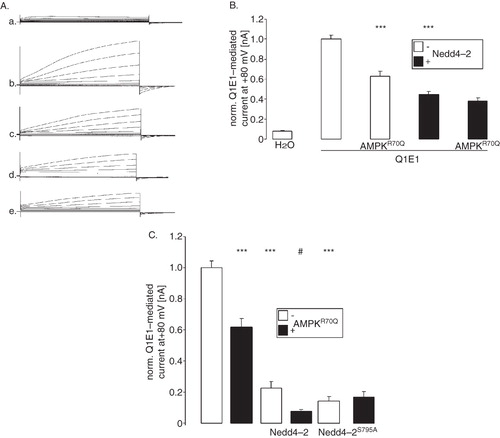
Figure 5. Similar to γR70QAMPK the ubiquitin ligase Nedd4-2 decreased the KCNQ1 protein abundance in the cell membrane. Confocal images of KCNQ1 protein abundance in the plasma membrane of Xenopus oocytes expressing KCNQ1/KCNE1 without (1st panel) or with additional coexpression of constitutively active γR70QAMPK (2nd panel), of the ubiquitin ligase Nedd4-2 (3rd panel) or of both, γR70QAMPK and Nedd4-2 (4th panel). The cells were subjected to immunofluorescent staining using FITC-conjugated antibody (grey/green).
Stimulation of AMPK depolarizes proximal renal tubule cells
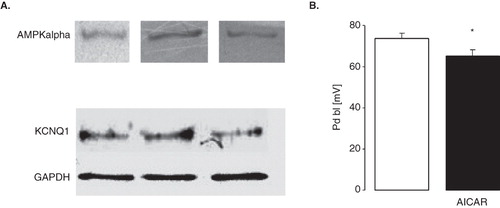
Isolated proximal renal tubules from C57 BL/6 mice express both, AMPK and KCNQ1 (). To test whether AMPK activity depolarizes proximal tubular cells as suggested by AMPK-dependent inhibition of KCNQ1/KCNE1, the potential difference across the basolateral cell membrane (PD bl) of isolated perfused proximal tubular cells was determined following incubation in the presence and absence of the AMPK stimulator AICAR (10 mM) for 1 h. As shown in , AICAR indeed depolarized proximal tubular cells.
Figure 6. Depolarization of proximal renal tubule cells by stimulation of AMPK (A) Original Western Blots demonstrating expression of AMPKα (upper panel) and KCNQ1 (lower panel; GAPDH was used as loading control) in isolated proximal tubules of C57 BL/6 mice. (B) Arithmetic means ± SEM (n = 7–8) of the potential difference across the basolateral membrane (PD) of isolated perfused proximal renal tubules from C57 BL/6 mice incubated for 1 h at 22°C in the absence (left bar) or presence (right bar) of AMPK stimulator AICAR (10 mM). * (p < 0.05) indicates statistically significant difference from the values obtained in absence of AICAR.
Discussion
The present study reveals a novel regulator of the slowly activating outward current generated by the heterotetrameric K+ channel KCNQ1/KCNE1. The AMP-activated protein kinase AMPK downregulates the channel and thus decreases K+ conductance and repolarization.
The AMPK-dependent downregulation of KCNQ1 is at least partially due to stimulation of the ubiquitin ligase Nedd4-2. AMPK has previously been shown to phosphorylate Nedd4-2 (Bhalla et al. Citation2006) thus influencing the interaction of the ubiquitin ligase with the epithelial Na+ channel ENaC (Carattino et al. Citation2005, Bhalla et al. Citation2006, Almaca et al. Citation2009).
According to the present observations, AMPK further disrupts the well known (Boucherot et al. Citation2001, Marx et al. Citation2002, Dilly et al. Citation2004, Nicolas et al. Citation2008, Dai et al. Citation2009) stimulation of KCNQ1 by cAMP. The AMPK-dependent regulation of the Cl- channel CFTR (Hallows et al. Citation2000, Citation2003a, Citation2003b, Citation2006, Citation2010, Crawford et al. Citation2006, Muimo et al. Citation2006, Mehta Citation2007) involves the phosphorylation of the R domain of the channel thus decreasing the activation of CFTR by protein kinase A (Walker et al. Citation2003, King et al. Citation2009, Kongsuphol et al. Citation2009a, Citation2009b).
In contrast to KCNQ1/KCNE1 activity, AMPK stimulates the activity of the facilitative glucose carriers GLUT1, GLUT2, GLUT3 and GLUT4 thus increasing cellular glucose uptake (Ojuka et al. Citation2000, Winder et al. Citation2000, Zheng et al. Citation2001, MacLean et al. Citation2002, Jessen et al. Citation2003, Li et al. Citation2004, Luiken et al. Citation2004, Lei et al. Citation2005, Walker et al. Citation2005, Natsuizaka et al. Citation2007, Guan et al. Citation2008, Park et al. Citation2009). The glucose uptake serves to provide the cell with fuel. Beyond that AMPK stimulates glycolysis, fatty acid oxidation and expression of enzymes required for ATP production (Carling Citation2007, Winder and Thomson Citation2007). All those functions counteract ATP depletion.
Inhibition of KCNQ1 by AMPK may be considered a double-edged sword. On the one hand, inhibition of K+ channels depolarizes the cell membrane, fostering Cl- entry and potentially deleterious cell swelling (Lang et al. Citation1986, Citation1998). In the heart, inhibition of KCNQ1 is expected to delay repolarization thus jeopardizing cardiac function (Peroz et al. Citation2008). On the other hand, inhibition of K+ channels could decrease energy expenditure. In the proximal renal tubule, for instance, inhibition of K+ channels decreases the driving force for Na+-coupled transport of glucose and other substrates across the apical membrane and at the same time decreases electrogenic HCO3 - exit across the basolateral cell membrane leading to cytosolic alkalinization and subsequent inhibition of the apical Na+/H+ exchanger (Lang and Rehwald Citation1992). Thus, depolarization curtails Na+ entry and thus decreases the requirement for energy-consuming Na+ extrusion by the Na+/K+ ATPase (Lang and Rehwald Citation1992). Inhibition of KCNQ1/KCNE1 may further limit the cellular K+ loss during impaired function of Na+/K+ ATPase in energy-depleted cells. Cellular K+ loss may foster suicidal cell death (Bortner and Cidlowski Citation2004, Foller et al. Citation2006, Shimizu et al. Citation2006, Becker et al. Citation2007, Schneider et al. Citation2007). By counteracting HCO3 - exit depolarization may prevent cytosolic acidification, which accelerates the death of apoptotic cells (Lupescu et al. Citation2009) and compromises glycolysis (Boiteux and Hess Citation1981).
The present observations may not only be relevant for ischemia and energy depletion. AMPK is further stimulated by an increase in the cytosolic Ca2+ activity (Towler and Hardie Citation2007), by a decrease of O2 levels (Evans et al. Citation2005) and by exposure to nitric oxide (Lira et al. Citation2007).
Moreover, the AMPK-dependent regulation of KCNQ1 is not only important for cardiac repolarization and maintenance of cell membrane potential in proximal renal tubules of the kidney. In addition to the heart (Barhanin et al. Citation1996, Sanguinetti et al. Citation1996, Neyroud et al. Citation1997) and kidney (Vallon et al. Citation2001) KCNQ1 is expressed in the liver (Demolombe et al. Citation2001, Lan et al. Citation2005, Citation2006), skeletal muscle (Finsterer and Stollberger Citation2004) and several epithelia (Sugimoto et al. Citation1990, Schroeder et al. Citation2000, Dedek and Waldegger Citation2001, Grahammer et al. Citation2001, Nicolas et al. Citation2001, Vallon et al. Citation2001, Citation2005, Heitzmann et al. Citation2004). In the liver, for instance, KCNQ1 governs cell volume and thus cell volume-sensitive functions including glucose uptake (Boini et al. Citation2009). Beyond that KCNQ1 is important for a variety of functions including hearing (Lee et al. Citation2000, Casimiro et al. Citation2001), gastric acid secretion (Scarff et al. Citation1999, Lee et al. Citation2000), as well as intestinal and renal transport (Vallon et al. Citation2005). AMPK-dependent regulation of KCNQ1 could thus participate in the pleotropic functional consequences of energy depletion in those tissues.
Conclusion
The present observations unravel a powerful inhibitory effect of the AMP-activated kinase AMPK on the slowly activating K+ channels KCNQ1/KCNE1. The effect is likely to profoundly affect cellular functions during energy depletion, hypoxia, excessive cytosolic Ca2+ activity, and exposure to nitric oxide.
Acknowledgements
The authors acknowledge the technical assistance of E. Faber. The manuscript was meticulously prepared by L. Subasic.
Declaration of interest: This study was supported by the Deutsche Forschungsgemeinschaft (GK 1302) and by a IZKF-Nachwuchsgruppe of the Medical Faculty of the University of Tübingen (No. 1889-0-0). The authors of this manuscript declare that they have neither financial nor any other conflicts of interests and they alone are responsible for the content and writing of the paper.
References
- Almaca J, Kongsuphol P, Hieke B, Ousingsawat J, Viollet B, Schreiber R, Amaral MD, Kunzelmann K. 2009. AMPK controls epithelial Na(+) channels through Nedd4-2 and causes an epithelial phenotype when mutated. Pflugers Arch 458:713–721.
- Bachmann O, Heinzmann A, Mack A, Manns MP, Seidler U. 2007. Mechanisms of secretion-associated shrinkage and volume recovery in cultured rabbit parietal cells. Am J Physiol Gastrointest Liver Physiol 292:G711–717.
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. 1996. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384:78–80.
- Becker S, Reinehr R, Graf D, vom DS, Haussinger D. 2007. Hydrophobic bile salts induce hepatocyte shrinkage via NADPH oxidase activation. Cell Physiol Biochem 19:89–98.
- Bhalla V, Oyster NM, Fitch AC, Wijngaarden MA, Neumann D, Schlattner U, Pearce D, Hallows KR. 2006. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J Biol Chem 281:26159–26169.
- Boehmer C, Laufer J, Jeyaraj S, Klaus F, Lindner R, Lang F, Palmada M. 2008a. Modulation of the voltage-gated potassium channel Kv1.5 by the SGK1 protein kinase involves inhibition of channel ubiquitination. Cell Physiol Biochem 22:591–600.
- Boehmer C, Palmada M, Klaus F, Jeyaraj S, Lindner R, Laufer J, Daniel H, Lang F. 2008b. The peptide transporter PEPT2 is targeted by the protein kinase SGK1 and the scaffold protein NHERF2. Cell Physiol Biochem 22:705–714.
- Boini KM, Graf D, Hennige AM, Koka S, Kempe DS, Wang K, Ackermann TF, Foller M, Vallon V, Pfeifer K, Schleicher ED, Ullrich S, Haring HU, Haussinger D, Lang F. 2009. Enhanced insulin sensitivity of gene targeted mice lacking functional KCNQ1. Am J Physiol Regul Integr Comp Physiol 296:R1695–1701.
- Boiteux A, Hess B. 1981. Design of glycolysis. Philos Trans R Soc Lond B Biol Sci 293:5–22.
- Bortner CD, Cidlowski JA. 2004. The role of apoptotic volume decrease and ionic homeostasis in the activation and repression of apoptosis. Pflugers Arch 448:313–318.
- Boucherot A, Schreiber R, Kunzelmann K. 2001. Regulation and properties of KCNQ1 (K(V)LQT1) and impact of the cystic fibrosis transmembrane conductance regulator. J Membr Biol 182:39–47.
- Carattino MD, Edinger RS, Grieser HJ, Wise R, Neumann D, Schlattner U, Johnson JP, Kleyman TR, Hallows KR. 2005. Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J Biol Chem 280:17608–17616.
- Carling D. 2007. The role of the AMP-activated protein kinase in the regulation of energy homeostasis. Novartis Found Symp 286:72–81.
- Casimiro MC, Knollmann BC, Ebert SN, Vary JC Jr, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K. 2001. Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc Natl Acad Sci USA 98:2526–2531.
- Chiang CE, Roden DM. 2000. The long QT syndromes: Genetic basis and clinical implications. J Am Coll Cardiol 36:1–12.
- Crawford RM, Treharne KJ, Best OG, Riemen CE, Muimo R, Gruenert DC, Arnaud-Dabernat S, Daniel JY, Mehta A. 2006. NDPK-A (but not NDPK-B) and AMPK alpha1 (but not AMPK alpha2) bind the cystic fibrosis transmembrane conductance regulator in epithelial cell membranes. Cell Signal 18:1595–1603.
- Dai S, Hall DD, Hell JW. 2009. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev 89:411–452.
- Dedek K, Waldegger S. 2001. Colocalization of KCNQ1/KCNE channel subunits in the mouse gastrointestinal tract. Pflugers Arch 442:896–902.
- Demolombe S, Franco D, de Boer P, Kuperschmidt S, Roden D, Pereon Y, Jarry A, Moorman AF, Escande D. 2001. Differential expression of KvLQT1 and its regulator IsK in mouse epithelia. Am J Physiol Cell Physiol 280:C359–372.
- Dilly KW, Kurokawa J, Terrenoire C, Reiken S, Lederer WJ, Marks AR, Kass RS. 2004. Overexpression of beta2-adrenergic receptors cAMP-dependent protein kinase phosphorylates and modulates slow delayed rectifier potassium channels expressed in murine heart: Evidence for receptor/channel co-localization. J Biol Chem 279:40778–40787.
- Evans AM, Mustard KJ, Wyatt CN, Peers C, Dipp M, Kumar P, Kinnear NP, Hardie DG. 2005. Does AMP-activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to calcium signaling in O2-sensing cells? J Biol Chem 280:41504–41511.
- Finsterer J, Stollberger C. 2004. Skeletal muscle involvement in congenital long QT syndrome. Neurol Sci 25:238–240.
- Foller M, Kasinathan RS, Duranton C, Wieder T, Huber SM, Lang F. 2006. PGE2-induced apoptotic cell death in K562 human leukaemia cells. Cell Physiol Biochem 17:201–210.
- Foller M, Sopjani M, Koka S, Gu S, Mahmud H, Wang K, Floride E, Schleicher E, Schulz E, Munzel T, Lang F. 2009. Regulation of erythrocyte survival by AMP-activated protein kinase. FASEB J 23:1072–1080.
- Fraser SA, Gimenez I, Cook N, Jennings I, Katerelos M, Katsis F, Levidiotis V, Kemp BE, Power DA. 2007. Regulation of the renal-specific Na+-K+-2Cl- co-transporter NKCC2 by AMP-activated protein kinase (AMPK). Biochem J 405:85–93.
- Gehring EM, Zurn A, Klaus F, Laufer J, Sopjani M, Lindner R, Strutz-Seebohm N, Tavare JM, Boehmer C, Palmada M, Lang UE, Seebohm G, Lang F. 2009. Regulation of the glutamate transporter EAAT2 by PIKfyve. Cell Physiol Biochem 24:361–368.
- Grahammer F, Herling AW, Lang HJ, Schmitt-Graff A, Wittekindt OH, Nitschke R, Bleich M, Barhanin J, Warth R. 2001. The cardiac K+ channel KCNQ1 is essential for gastric acid secretion. Gastroenterology 120:1363–1371.
- Grunnet M, Jespersen T, MacAulay N, Jorgensen NK, Schmitt N, Pongs O, Olesen SP, Klaerke DA. 2003. KCNQ1 channels sense small changes in cell volume. J Physiol 549:419–427.
- Guan F, Yu B, Qi GX, Hu J, Zeng DY, Luo J. 2008. Chemical hypoxia-induced glucose transporter-4 translocation in neonatal rat cardiomyocytes. Arch Med Res 39:52–60.
- Hallows KR, Fitch AC, Richardson CA, Reynolds PR, Clancy JP, Dagher PC, Witters LA, Kolls JK, Pilewski JM. 2006. Up-regulation of AMP-activated kinase by dysfunctional cystic fibrosis transmembrane conductance regulator in cystic fibrosis airway epithelial cells mitigates excessive inflammation. J Biol Chem 281:4231–4241.
- Hallows KR, Kobinger GP, Wilson JM, Witters LA, Foskett JK. 2003a. Physiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cells. Am J Physiol Cell Physiol 284:C1297–1308.
- Hallows KR, McCane JE, Kemp BE, Witters LA, Foskett JK. 2003b. Regulation of channel gating by AMP-activated protein kinase modulates cystic fibrosis transmembrane conductance regulator activity in lung submucosal cells. J Biol Chem 278:998–1004.
- Hallows KR, Mount PF, Pastor-Soler NM, Power DA. 2010. Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am J Physiol Renal Physiol, in press.
- Hallows KR, Raghuram V, Kemp BE, Witters LA, Foskett JK. 2000. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J Clin Invest 105:1711–1721.
- Hamilton SR, Yao SY, Ingram JC, Hadden DA, Ritzel MW, Gallagher MP, Henderson PJ, Cass CE, Young JD, Baldwin SA. 2001. Subcellular distribution and membrane topology of the mammalian concentrative Na+-nucleoside cotransporter rCNT1. J Biol Chem 276:27981–27988.
- Hardie DG. 2004. The AMP-activated protein kinase pathway – new players upstream and downstream. J Cell Sci 117:5479–5487.
- Heitzmann D, Grahammer F, von Hahn T, Schmitt-Graff A, Romeo E, Nitschke R, Gerlach U, Lang HJ, Verrey F, Barhanin J, Warth R. 2004. Heteromeric KCNE2/KCNQ1 potassium channels in the luminal membrane of gastric parietal cells. J Physiol 561:547–557.
- Henrion U, Strutz-Seebohm N, Duszenko M, Lang F, Seebohm G. 2009. Long QT syndrome-associated mutations in the voltage sensor of I(Ks) channels. Cell Physiol Biochem 24:11–16.
- Horie T, Ono K, Nagao K, Nishi H, Kinoshita M, Kawamura T, Wada H, Shimatsu A, Kita T, Hasegawa K. 2008. Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J Cell Physiol 215:733–742.
- Jensen TE, Rose AJ, Hellsten Y, Wojtaszewski JF, Richter EA. 2007. Caffeine-induced Ca(2+) release increases AMPK-dependent glucose uptake in rodent soleus muscle. Am J Physiol Endocrinol Metab 293:E286–292.
- Jessen N, Pold R, Buhl ES, Jensen LS, Schmitz O, Lund S. 2003. Effects of AICAR and exercise on insulin-stimulated glucose uptake, signaling, and GLUT-4 content in rat muscles. J Appl Physiol 94:1373–1379.
- King JD Jr, Fitch AC, Lee JK, McCane JE, Mak DO, Foskett JK, Hallows KR. 2009. AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am J Physiol Cell Physiol 297:C94–101.
- Kongsuphol P, Cassidy D, Hieke B, Treharne KJ, Schreiber R, Mehta A, Kunzelmann K. 2009a. Mechanistic insight into control of CFTR by AMPK. J Biol Chem 284:5645–5653.
- Kongsuphol P, Hieke B, Ousingsawat J, Almaca J, Viollet B, Schreiber R, Kunzelmann K. 2009b. Regulation of Cl(-) secretion by AMPK in vivo. Pflugers Arch 457:1071–1078.
- Lan WZ, Abbas H, Lemay AM, Briggs MM, Hill CE. 2005. Electrophysiological and molecular identification of hepatocellular volume-activated K+ channels. Biochim Biophys Acta 1668:223–233.
- Lan WZ, Wang PY, Hill CE. 2006. Modulation of hepatocellular swelling-activated K+ currents by phosphoinositide pathway-dependent protein kinase C. Am J Physiol Cell Physiol 291:C93–103.
- Lang F, Busch GL, Ritter M, Volkl H, Waldegger S, Gulbins E, Haussinger D. 1998. Functional significance of cell volume regulatory mechanisms. Physiol Rev 78:247–306.
- Lang F, Messner G, Rehwald W. 1986. Electrophysiology of sodium-coupled transport in proximal renal tubules. Am J Physiol 250:F953–962.
- Lang F, Rehwald W. 1992. Potassium channels in renal epithelial transport regulation. Physiol Rev 72:1–32.
- Laufer J, Boehmer C, Jeyaraj S, Knuwer M, Klaus F, Lindner R, Palmada M, Lang F. 2009. The C-terminal PDZ-binding motif in the Kv1.5 potassium channel governs its modulation by the Na+/H+ exchanger regulatory factor 2. Cell Physiol Biochem 23:25–36.
- Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, Francis H, Gorelikow M, Gu H, Washington K, Argani P, Goldenring JR, Coffey RJ, Feinberg AP. 2000. Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest 106:1447–1455.
- Lei B, Matsuo K, Labinskyy V, Sharma N, Chandler MP, Ahn A, Hintze TH, Stanley WC, Recchia FA. 2005. Exogenous nitric oxide reduces glucose transporters translocation and lactate production in ischemic myocardium in vivo. Proc Natl Acad Sci USA 102:6966–6971.
- Li J, Hu X, Selvakumar P, Russell RR, III, Cushman SW, Holman GD, Young LH. 2004. Role of the nitric oxide pathway in AMPK-mediated glucose uptake and GLUT4 translocation in heart muscle. Am J Physiol Endocrinol Metab 287:E834–841.
- Lira VA, Soltow QA, Long JH, Betters JL, Sellman JE, Criswell DS. 2007. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. Am J Physiol Endocrinol Metab 293:E1062–1068.
- Liu XS, Jiang M, Zhang M, Tang D, Clemo HF, Higgins RS, Tseng GN. 2007. Electrical remodeling in a canine model of ischemic cardiomyopathy. Am J Physiol Heart Circ Physiol 292:H560–571.
- Luiken JJ, Coort SL, Koonen DP, van der Horst DJ, Bonen A, Zorzano A, Glatz JF. 2004. Regulation of cardiac long-chain fatty acid and glucose uptake by translocation of substrate transporters. Pflugers Arch 448:1–15.
- Lupescu A, Geiger C, Zahir N, Aberle S, Lang PA, Kramer S, Wesselborg S, Kandolf R, Foller M, Lang F, Bock CT. 2009. Inhibition of Na+/H+ exchanger activity by parvovirus B19 protein NS1. Cell Physiol Biochem 23:211–220.
- MacLean PS, Zheng D, Jones JP, Olson AL, Dohm GL. 2002. Exercise-induced transcription of the muscle glucose transporter (GLUT 4) gene. Biochem Biophys Res Commun 292:409–414.
- Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. 2002. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295:496–499.
- McGee SL, Hargreaves M. 2008. AMPK and transcriptional regulation. Front Biosci 13:3022–3033.
- Mehta A. 2007. The cystic fibrosis transmembrane recruiter the alter ego of CFTR as a multi-kinase anchor. Pflugers Arch 455:215–221.
- Muimo R, Crawford RM, Mehta A. 2006. Nucleoside diphosphate kinase A as a controller of AMP-kinase in airway epithelia. J Bioenerg Biomembr 38:181–187.
- Natsuizaka M, Ozasa M, Darmanin S, Miyamoto M, Kondo S, Kamada S, Shindoh M, Higashino F, Suhara W, Koide H, Aita K, Nakagawa K, Kondo T, Asaka M, Okada F, Kobayashi M. 2007. Synergistic up-regulation of Hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. Exp Cell Res 313:3337–3348.
- Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P. 1997. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 15:186–189.
- Nicolas CS, Park KH, El Harchi A, Camonis J, Kass RS, Escande D, Merot J, Loussouarn G, Le Bouffant F, Baro I. 2008. IKs response to protein kinase A-dependent KCNQ1 phosphorylation requires direct interaction with microtubules. Cardiovasc Res 79:427–435.
- Nicolas M, Dememes D, Martin A, Kupershmidt S, Barhanin J. 2001. KCNQ1/KCNE1 potassium channels in mammalian vestibular dark cells. Hear Res 153:132–1345.
- Ojuka EO, Nolte LA, Holloszy JO. 2000. Increased expression of GLUT-4 and hexokinase in rat epitrochlearis muscles exposed to AICAR in vitro. J Appl Physiol 88:1072–1075.
- Park S, Scheffler TL, Gunawan AM, Shi H, Zeng C, Hannon KM, Grant AL, Gerrard DE. 2009. Chronic elevated calcium blocks AMPK-induced GLUT-4 expression in skeletal muscle. Am J Physiol Cell Physiol 296:C106–115.
- Peroz D, Rodriguez N, Choveau F, Baro I, Merot J, Loussouarn G. 2008. Kv7.1 (KCNQ1) properties and channelopathies. J Physiol 586:1785–1789.
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. 1996. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384:80–83.
- Scarff KL, Judd LM, Toh BH, Gleeson PA, Van Driel IR. 1999. Gastric H(+),K(+)-adenosine triphosphatase beta subunit is required for normal function, development, and membrane structure of mouse parietal cells. Gastroenterology 117:605–618.
- Schneider J, Nicolay JP, Foller M, Wieder T, Lang F. 2007. Suicidal erythrocyte death following cellular K+ loss. Cell Physiol Biochem 20:35–44.
- Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R, Jentsch TJ. 2000. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature 403:196–199.
- Seebohm G, Strutz-Seebohm N, Ureche ON, Henrion U, Baltaev R, Mack AF, Korniychuk G, Steinke K, Tapken D, Pfeufer A, Kaab S, Bucci C, Attali B, Merot J, Tavare JM, Hoppe UC, Sanguinetti MC, Lang F. 2008. Long QT syndrome-associated mutations in KCNQ1 and KCNE1 subunits disrupt normal endosomal recycling of IKs channels. Circ Res 103:1451–1457.
- Shimizu T, Wehner F, Okada Y. 2006. Inhibition of hypertonicity-induced cation channels sensitizes HeLa cells to shrinkage-induced apoptosis. Cell Physiol Biochem 18:295–302.
- Sopjani M, Bhavsar SK, Fraser S, Kemp BE, Föller M, Lang F. 2010. Regulation of Na+ -coupled glucose carrier SGLT1 by AMP-activated protein kinase. Mol Membr Biol 27:137–144.
- Steinberg GR, Kemp BE. 2009. AMPK in health and disease. Physiol Rev 89:1025–1078.
- Sugimoto T, Tanabe Y, Shigemoto R, Iwai M, Takumi T, Ohkubo H, Nakanishi S. 1990. Immunohistochemical study of a rat membrane protein which induces a selective potassium permeation: Its localization in the apical membrane portion of epithelial cells. J Membr Biol 113:39–47.
- Towler MC, Hardie DG. 2007. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 100:328–341.
- Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, Ng DP, Holmkvist J, Borch-Johnsen K, Jorgensen T, Sandbaek A, Lauritzen T, Hansen T, Nurbaya S, Tsunoda T, Kubo M, Babazono T, Hirose H, Hayashi M, Iwamoto Y, Kashiwagi A, Kaku K, Kawamori R, Tai ES, Pedersen O, Kamatani N, Kadowaki T, Kikkawa R, Nakamura Y, Maeda S. 2008. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 40:1098–1102.
- Ureche ON, Baltaev R, Ureche L, Strutz-Seebohm N, Lang F, Seebohm G. 2008. Novel insights into the structural basis of pH-sensitivity in inward rectifier K+ channels Kir2.3. Cell Physiol Biochem 21:347–356.
- Vallon V, Grahammer F, Richter K, Bleich M, Lang F, Barhanin J, Volkl H, Warth R. 2001. Role of KCNE1-dependent K+ fluxes in mouse proximal tubule. J Am Soc Nephrol 12:2003–2011.
- Vallon V, Grahammer F, Volkl H, Sandu CD, Richter K, Rexhepaj R, Gerlach U, Rong Q, Pfeifer K, Lang F. 2005. KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc Natl Acad Sci USA 102:17864–17869.
- vanTol BL, Missan S, Crack J, Moser S, Baldridge WH, Linsdell P, Cowley EA. 2007. Contribution of KCNQ1 to the regulatory volume decrease in the human mammary epithelial cell line MCF-7. Am J Physiol Cell Physiol 293:C1010–1019.
- Walker J, Jijon HB, Churchill T, Kulka M, Madsen KL. 2003. Activation of AMP-activated protein kinase reduces cAMP-mediated epithelial chloride secretion. Am J Physiol Gastrointest Liver Physiol 285:G850–860.
- Walker J, Jijon HB, Diaz H, Salehi P, Churchill T, Madsen KL. 2005. 5-aminoimidazole-4-carboxamide riboside (AICAR) enhances GLUT2-dependent jejunal glucose transport: A possible role for AMPK. Biochem J 385:485–491.
- Wangemann P. 2006. Supporting sensory transduction: Cochlear fluid homeostasis and the endocochlear potential. J Physiol 576:11–21.
- Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. 2000. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol 88:2219–2226.
- Winder WW, Thomson DM. 2007. Cellular energy sensing and signaling by AMP-activated protein kinase. Cell Biochem Biophys 47:332–347.
- Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, Hirota Y, Mori H, Jonsson A, Sato Y, Yamagata K, Hinokio Y, Wang HY, Tanahashi T, Nakamura N, Oka Y, Iwasaki N, Iwamoto Y, Yamada Y, Seino Y, Maegawa H, Kashiwagi A, Takeda J, Maeda E, Shin HD, Cho YM, Park KS, Lee HK, Ng MC, Ma RC, So WY, Chan JC, Lyssenko V, Tuomi T, Nilsson P, Groop L, Kamatani N, Sekine A, Nakamura Y, Yamamoto K, Yoshida T, Tokunaga K, Itakura M, Makino H, Nanjo K, Kadowaki T, Kasuga M. 2008. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 40:1092–1097.
- Zheng D, MacLean PS, Pohnert SC, Knight JB, Olson AL, Winder WW, Dohm GL. 2001. Regulation of muscle GLUT-4 transcription by AMP-activated protein kinase. J Appl Physiol 91:1073–1083.