
Abstract
Sulfoxaflor (CAS# 946578-00-3) is a novel active substance with insecticidal properties mediated via its agonism on the highly abundant insect nicotinic acetylcholine receptor (nAChR). In developmental and reproductive toxicity studies, gestational exposure caused fetal abnormalities (primarily limb contractures) and reduced neonatal survival in rats, but not rabbits, following high-dose dietary exposure. Sulfoxaflor induced these effects via a novel mode of action (MoA) mediated by the fetal-type muscle nAChR with the following key events: (1) binding to the receptor, (2) agonism on the receptor, causing (3) sustained muscle contracture in the near-term fetus and neonatal offspring. This sustained muscle contracture results in misshapen limbs, bent clavicles, and reduced diaphragm function, which compromises respiration in neonatal rats at birth, reducing their survival. This review evaluates the weight of evidence for this MoA based upon the Bradford Hill criteria, includes a cross-comparison of applied and internal doses in a variety of in vitro, ex vivo, and in vivo study designs, examines alternative MoAs, and applies a Human relevance framework (HRF) to ascertain human risk for this rat MoA. The review indicated, with a high level of confidence, that the sulfoxaflor-induced fetal abnormalities and neonatal death in rats occur via a single MoA comprising sustained activation of the rat fetal-type muscle nAChR resulting in a sustained muscle contracture. This MoA is considered not relevant to humans, given fundamental qualitative differences in sulfoxaflor agonism on the rat versus the human muscle nAChR. Specifically, sulfoxaflor does not cause agonism on either the human fetal- or adult-type muscle nAChR.
Introduction
Sulfoxaflor (XDE-208, X11422208, XR-208, [1-(6-Trifluormethylpyridin-3-yl)ethyl)](methyl)-oxido-l4-sulfanylidenecyanamide) is an active substance with insecticidal properties mediated via its agonism on the highly abundant insect nicotinic acetylcholine receptor (nAChR). During the regulatory guideline mammalian toxicology studies, developmental effects were observed in rats following dietary sulfoxaflor exposure, including offspring death and near-term fetal abnormalities (forelimb flexure, hindlimb rotation, and bent [misshapen] clavicle bones; ; CitationRasoulpour et al. 2012). More specifically, the rat limb effects were without changes in the associated skeletal bone structure while the offspring death occurred prior to postnatal day (PND) 4. Henceforth, these effects will together be classified as occurring in neonatal offspring. A subsequent cross-fostering study demonstrated that the neonatal offspring death was due to gestational, and not lactational, exposure. Additional non-guideline investigative toxicity studies demonstrated that the rat effects were inducible with one or two days of exposure before birth, with the limb effects being rapidly reversible upon withdrawal of sulfoxaflor exposure in surviving pups. Similar effects were not observed in a rabbit developmental toxicity or perinatal survival study. Similar maternal and fetal blood data showed that the interspecies difference between rats and rabbits was not due to toxicokinetics; therefore, the species difference was due to toxicodynamic factors.
Table 1. Summary of findings from developmental and reproductive toxicity studies on sulfoxalfor.
The mode of action (MoA) for several adverse developmental outcomes has been determined (CitationCorley et al. 2005, CitationFoster 2005, CitationHolson et al. 2005, CitationKavlock and Cummings 2005). The regulatory guideline and MoA toxicity studies with sulfoxaflor described above revealed findings for which an MoA had not previously been described (CitationRasoulpour et al. 2012). In the past decade, frameworks to evaluate the MoA of adverse toxicological effects have been developed through the International Programme on Chemical Safety (IPCS) (CitationBoobis et al. 2006) and by the International Life Sciences Institute Risk Science Institute (CitationMeek et al. 2003). These frameworks evaluate the weight of evidence for the MoA based upon the Bradford Hill criteria (CitationHill 1965) followed by subsequent application in a Human relevance framework (HRF). Specifically as it relates to active substances to be contained within plant protection products (PPPs), it is becoming more common for testing to be conducted above the “standard” regulatory toxicity requirements and incorporate higher tier investigative studies such as MoA work into MoA/HRF assessments to enable registration of the new PPPs. This increase in higher tier and customized study testing is driven by a general trend to obtain mechanistic understanding of the underlying hazard characteristics of molecules to inform risk management decisions. As the MoA/HRF approach is becoming an accepted component of PPP human health assessment, companion papers are included in this journal issue to more broadly discuss the application of the approach to sulfoxaflor, one such active substance (CitationTerry et al. 2014, CitationLeBaron et al. 2014, CitationRasoulpour et al. 2014). An interesting aspect to these case studies is that in some cases the experimental work was conducted prospectively, that is, before the definitive studies had identified the particular hazard, as an approach which minimised the delay to develop the active substance and regulatory submissions. The advantages and disadvantages of this approach are discussed (CitationTerry et al. 2014).
The MoA/HRF assessment approach has been utilized in the analysis of a number of carcinogen MoAs and their human relevance, as specifically for sulfoxaflor-induced Leydig cell tumors in rats (CitationRasoulpour et al. 2014) and for sulfoxaflor- induced liver tumors in rodents (CitationLeBaron et al. 2014).
Recent efforts have also been directed toward extending the MoA approach to non-cancer MoA analyses (CitationCorley et al. 2005, CitationFoster 2005, CitationHolson et al. 2005, CitationKavlock and Cummings 2005), with several addressing human relevance. Standardization of the approach has also been recommended by the IPCS (CitationBoobis et al. 2008).
No preceeding MoA evaluations or HRF assessments are available for the fetal-type muscle contracture abnormalities and associated neonatal offspring death induced by sufoxaflor. Therefore, the remainder of this review evaluates the weight of evidence for this MoA based upon the Bradford Hill criteria and includes data from a variety of different study designs in different settings, including in vivo, ex vivo, and in vitro studies, thereby providing a robust demonstration of the consistency of MoA of sulfoxaflor in the rat. Ten independent studies are included, many including dose–response data allowing for strong evidence of causality through the demonstration of clear dose–response relationships with each of the key events. Toxicokinetic analysis was incorporated in the majority of the in vivo toxicology studies in order to provide a measure of internal dose, which allows direct comparison between the in vivo data to the generated in vitro and ex vivo MoA data. In order to evaluate specificity and strength of the association of the postulated MoA, reversibility tests and extensive evaluations of a structurally similar agent, which is inactive at the insect nAChR, have been included, respectively. This evaluation includes consideration of alternative MoAs which can also result in limb abnormalities and/or death in neonatal offspring and a HRF analysis that addresses the relevance of the rat developmental effects to humans.
MoA for induction of muscle contracture and death in neonatal offspring
As discussed above, dietary exposure to high sulfoxaflor doses (≥ 400 ppm) during gestation in rats results in treatment-related limb contracture and death in neonatal offspring. Given that the rat limb abnormalities appeared as part of a generalized muscle contracture, and the changes in limb and shoulder girdle skeletal bone structure were inconsistent (i.e., limb bones were normal but the clavicles were occasionally bent), it was considered likely that the observed effects resulted from the action of sulfoxaflor on the neonatal skeletal muscle. Contracture is defined as an abnormal contraction of muscle tissue rendering it highly resistant to passive stretching (CitationMiller-Keane and O’Toole 2005). In the case of sulfoxaflor, this abnormal contraction ultimately results in the limb contractures and diaphragm-related deaths observed in neonatal offspring. The observation that fetuses in the developmental toxicity study exhibited a hunched posture, consistent with generalized muscle contracture, supports this hypothesis and the fact is that the clavicle (collar) bone depends upon the normal shoulder muscle function for proper development during the late fetal period (CitationPai 1965, CitationTran and Hall 1989). Similar to those of the limb and the shoulder muscles, action of sulfoxaflor on the main muscle involved in breathing, the diaphragm (CitationVander et al. 2001), could result in abnormal neonatal respiration and mediate the neonatal deaths.
A single MoA is considered to be responsible for the neonatal offspring findings as the sulfoxaflor-induced limb contracture and clavicle abnormalities and the neonatal death occurred at the same dose (1000 ppm) with similar incidences. In order for a single MoA to be plausible, the findings should have a similar incidence across dose levels from all studies. The similarity in the R2 values between the non-linear regression analyses of the individual dose–response curves for death (R2 = 0.93) and limb abnormalities (R2 = 0.91), respectively, support a single MoA responsible for different developmental effects rather than separate MoAs for each of the two major effects in rats (CitationRasoulpour et al. 2012).
Muscle nAChR is hypothesized to mediate developmental effects of sulfoxaflor
As the insecticidal MoA of sulfoxaflor is agonism on the insect nAChR, it was logical to investigate the mammalian muscle nAChR as a biologically plausible target responsible for the effects seen in rat. While metabolism of sulfoxaflor is negligible in all mammalian species tested, it can be metabolized in the soil. Interestingly, primary soil metabolite (X11719474, N-(Methyloxido(1-(6-(trifluoromethyl)-3-pyridinyl)ethyl)sulfanylidene) urea) of sulfoxaflor, despite being only one functional group different from the parent molecule (replacement of the cyanate with an amide), does not bind to the insect nAChR and, when tested in developmental or reproductive toxicity studies, did not produce limb contracture abnormalities or neonatal offspring death at dose levels 5–10 times higher than the sulfoxaflor effect levels (data not shown). In both invertebrate and vertebrate species, nAChRs are important neurotransmitter receptors (CitationSattelle 1980, CitationMillar and Gotti 2009) and comprise a diverse family of oligomeric cell-surface receptors assembled from five (of many) subunits that co- assemble in a doughnut-shaped arrangement (CitationMillar and Denholm 2007, CitationMillar and Gotti 2009). In the center of the pentameric arrangement of subunits is a cation-selective ion channel, whereby binding of the endogenous neurotransmitter, acetylcholine (Ach), or other agonists stabilizes the open conformation allowing the influx of cations into the cell (agonism). In mammalian muscle cells, nAChRs are expressed at the neuromuscular junction (NMJ) in skeletal muscles and are composed of five nAChR subunits (α1, β1, γ, δ, and ϵ). Transcription of the γ and ϵ subunits is differentially regulated during development, with the γ subunit expressed in “fetal” muscle and the ϵ subunit expressed in “adult” muscle (CitationGu and Hall 1988). Muscle nAChRs contain two agonist binding sites, one at the interface of the α1 and δ subunits and another at the interface of the α1 and γ (or ϵ) subunits (CitationArias 2000).
NMJ development requires signaling from the motor neuron and the muscle cells. During the development, AChRs are expressed in the central region of muscle cell through a prepatterning process, then undergo a AChR clustering and stabilization reinforced by celluar signaling and postsynaptic potentials induced by ACh-release (CitationWitzemann 2006). In rats, the muscle nAChR develops functional subunit expression at the NMJ between GD 15 and 17 (CitationKues et al. 1995) resulting in synchronized fetal limb movements (CitationRobinson and Smotherman 1988) and diaphragmatic responsiveness between GD 16 and 17 (CitationBennett and Pettigrew 1974), the latter being critical for the transition to extrauterine respiration. Replacement of the γ subunit by the ϵ subunit initiates late during the first postnatal week in rodents and is largely complete by the end of the second postnatal week in limb and respiratory muscles (CitationMissias et al. 1996, CitationGu and Hall 1988).
In contrast to skeletal muscles, smooth muscle does not contain nAChRs but rather ACh signaling occurs through muscarinic AChRs of five genomic subtypes (M1–M5) (CitationHoffman and Taylor 2001). Although smooth muscle is not believed to be involved in developmental toxicity MoA of sulfoxaflor, it is briefly described here because involvement of the muscarinic AChR as an alternative MoA is examined within this paper. Smooth muscle is found in various locations throughout the body where autonomic effector cells are present, including within the walls of blood and lymphatic vessels, the urinary bladder, and the uterus. Muscarinic AChRs in smooth muscles form G protein-coupled receptor complexes in neuronal cell membranes. Following ACh (or other ligand) binding, excitation–contraction coupling occurs through G protein-induced changes in the functions of distinct membrane-bound effector molecules that modulate Ca+2 release. Besides its structure, function, and location, smooth muscle also differs from skeletal muscle in that regulation of contractions is involuntary. The ontogeny of smooth muscle development and innervations is generally less well known compared to that of skeletal muscle.
Key events in experimental animals
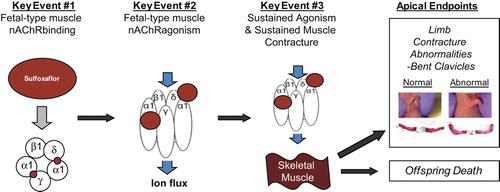
Given that sulfoxaflor targets the insect nAChR, and that functional expression of the fetal-type NMJ nAChR occurs in late gestation and is involved with limb muscle function, it was hypothesized that the rat neonatal offspring limb/clavicle abnormalities and death occur via a single MoA: sustained agonism of sulfoxaflor on the rat fetal-type muscle nAChR (the muscle nAChR subtype present at birth in rats) and subsequent sustained muscle contracture of the limb, shoulder girdle, and diaphragm, respectively. Based upon this hypothesis, a series of studies were designed and conducted (CitationRasoulpour et al. 2012). This MoA is proposed to progress through the following key events: (1) sulfoxaflor binding and (2) agonism on the rat fetal-type muscle nAChR, resulting in (3) sustained agonism/sustained muscle contracture in rat neonatal offspring (fetus and pup). This sustained muscle contracture results in limb contracture, bent clavicles, and abnormal neonatal respiration after birth resulting in neonatal offspring death ().
Figure 1. Key events for the postulated MoA of sulfoxaflor-induced muscle contracture abnormalities and associated death in neonatal rat offspring.
Concordance of dose–response relationship
Key Event #1: Specific binding to the rat fetal-type muscle nAChR
In mammals, nAChRs in muscle tissue are expressed postsynaptically, where they are responsible for contraction of muscle in response to the release of ACh from the presynaptic nerve terminal. In order to assess ability of sulfoxaflor to bind to the rat fetal-type muscle nAChR, competitive radioligand binding experiments were conducted using GD 21 forelimb muscle cell membrane preparations (containing fetal-type muscle NMJ nAChRs). Concentrations of sulfoxaflor causing half-maximal displacement (IC50 concentrations) of 30 nM [3H]-epibatidine were determined (). By fitting the data to a single binding site model (i.e., assuming the ligand would have similar affinity for both nAChR agonist binding sites; one being located at the α–γ interface and the other at the α–δ subunit interface) (solid lines in ), the IC50 estimate for sulfoxaflor is 2.3 mM. The fetal rat muscle binding data were not well fitted using this single binding-site model; therefore the data were fitted with a two-site model (dotted line in ) that revealed different affinities for the two nAChR binding sites (0.01 mM and 8.9 mM). The better fit of the rat fetal muscle nAChR experimental data to a two-site model would suggest that sulfoxaflor displaces [3H]-epibatidine from the two nAChR agonist-binding sites (located at the α–γ and α–δ subunit interfaces) with different affinities. The affinity of the individual binding sites for sulfoxaflor has not been further characterized.
Figure 2. Dose–response for MoA Key Event #1: Competition radioligand binding with sulfoxaflor. The ability of sulfoxaflor to displace binding of [3H]-epibatidine was examined in rat fetal muscle tissue. Samples were incubated with [3H]-epibatidine (30 nM) in the presence of a range of concentrations of sulfoxaflor. Data are means + SEM of 3–4 independent experiments, each performed with triplicate samples. Levels of radioligand binding are normalized to the level of specific binding observed in the absence of sulfoxaflor. Concentrations are plotted as log molar concentrations.
![Figure 2. Dose–response for MoA Key Event #1: Competition radioligand binding with sulfoxaflor. The ability of sulfoxaflor to displace binding of [3H]-epibatidine was examined in rat fetal muscle tissue. Samples were incubated with [3H]-epibatidine (30 nM) in the presence of a range of concentrations of sulfoxaflor. Data are means + SEM of 3–4 independent experiments, each performed with triplicate samples. Levels of radioligand binding are normalized to the level of specific binding observed in the absence of sulfoxaflor. Concentrations are plotted as log molar concentrations.](/cms/asset/b737d1f1-ced7-4e89-aa27-d832b2455b29/itxc_a_910752_f0002_b.gif)
The data obtained in this study provided clear and direct evidence that sulfoxaflor binds to fetal-type muscle nAChRs. Thresholded, dose-dependent binding was demonstrated with the lowest tested concentration having no apparent binding while incubation of the receptor pools with higher concentrations of sulfoxaflor completely displaced the high-affinity fetal-type muscle nAChR binding of [3H]-epibatidine, thereby showing specificity of sulfoxaflor to nAChR binding site(s). Indirect evidence for sulfoxaflor binding to the rat fetal-type muscle nAChR includes demonstrations of (1) functional agonism in Xenopus oocytes expressing recombinant rat fetal-type muscle nAChRs in vitro and (2) ex vivo muscle contracture in experiments using CD rat PND 0 phrenic nerve-hemidiaphragm muscle preparations. These studies will be discussed in more detail in relation to Key Events #2 (agonism on the rat fetal-type muscle nAChR) and #3 (sustained agonism on the rat fetal-type muscle nAChR and sustained muscle contracture), respectively, but these experiments showed that sulfoxaflor caused concentration-dependent agonism on the rat fetal-type muscle nAChR and contracture of the newborn rat diaphragm, responses which could only have occurred via binding and/or subsequent sustained agonism of sulfoxaflor at this receptor. When the direct and indirect evidence of sulfoxaflor binding to the fetal nAChR are taken together, there is support for nAChR binding being a key event operant in this MoA.
Key Event #2: Agonism on the rat fetal-type muscle nAChR
The radioligand-binding experiments described above demonstrate specific binding of sulfoxaflor to rat fetal-type muscle nAChRs, but do not indicate whether the binding of sulfoxaflor results in the functional activation of the receptor (i.e., it does not indicate whether sulfoxaflor acts as a functionalagonist on mammalian muscle nAChRs). In order to examine this question, rat fetal and adult muscle nAChRs were expressed as recombinant receptors through microinjection of cRNA in Xenopus oocytes. Recombinant rat nAChR subunits (five subunits total: fetal subtype α(2), β, δ, γ or adult subtype α(2), β, δ, ϵ) were expressed in Xenopus oocytes to determine the functional nAChR activity (two-electrode voltage-clamp recording of membrane currents as described in CitationRasoulpour et al. 2012). The nAChRs expressed in Xenopus oocytes form fully functional pentameric nAChRs expressed on the cell surface. Electrophysiological data obtained from such receptors have been demonstrated to be functionally similar to data obtained from native nAChRs expressed in muscle tissue (CitationMishina et al. 1986). Due to its correlation with in vivo functionality the method has been used in hundreds of research publications to examine the functional properties of ion channels such as the nAChR (CitationDascal 1987). It has also been previously demonstrated to confirm agonism of nAChR ligands (CitationCooper et al. 1996), some of which have been demonstrated to cause limb contracture abnormalities (CitationForsyth et al. 1996). In the current studies, functional responses (membrane currents) were confirmed via application of the endogenous agonist ACh. Clear dose-dependent agonist-evoked responses were observed with sulfoxaflor at the rat fetal-type nAChR; the lowest tested concentration having no agonism while incubation of the oocytes with higher concentrations of sulfoxaflor showed increasing agonism () (CitationRasoulpour et al. 2012). Data from the agonism experiments showed high concordance with the binding experiments thereby providing a demonstration of consistency across experimental designs at the same test concentrations.
Table 2. Dose–response for MoA Key Event #2: Fetal-type muscle nAChR agonism.
Key Event #3: Sustained agonism on the fetal-type muscle nAChR and sustained muscle contracture
For the proposed MoA to be plausible, the agonism caused by sulfoxaflor would have to be sustained, as opposed to the brief agonism caused by the endogenous nAChR agonist, ACh, for example. In support of this, toxicokinetic blood analyses across in vivo study types strongly suggest that blood concentrations of sulfoxaflor are maintained at the steady-state levels due to continuous exposure via the diet and the previously mentioned negligible mammalian metabolism. As sulfoxaflor readily perfuses into muscle from the blood (data not shown), it is predicted that muscle sulfoxaflor concentrations would also be maintained at a steady-state as long as dietary treatment continued. At the location of the NMJ nAChR, sulfoxaflor would remain at the nAChR synaptic cleft due to its lack of hydrolysis by acetylcholinesterase (AChE). This differs from ACh which undergoes tightly regulated synaptic vesicle release followed by rapid hydrolysis by AChE. Thus receptor occupancy of sulfoxaflor would only be limited by association/dissociation kinetics of the molecule and not by removal from the receptor endplate region (as with ACh). Thus, upon fetal-type muscle nAChR activation, a sulfoxaflor- induced muscle contracture would be sustained for as long as sufficient sulfoxaflor molecules remain available for receptor binding, which is consistent with the observed experimental evidence.
Neonatal diaphragm
In order to directly assess muscle contracture at the neonatal diaphragm, sulfoxaflor was tested for agonist action on the isolated phrenic nerve-hemidiaphragm preparation (CitationBülbring 1946) from newborn rats. Since its introduction, the isolated phrenic nerve-hemidiaphragm preparation has become established as the standard nerve–muscle preparation for mechanistic investigations of drug action at the mammalian NMJ (e.g., CitationLiley and North 1953, CitationHubbard and Wilson 1973, CitationGibb and Marshall 1984, Citation1986, Citation1987, CitationWareham et al. 1994, CitationFortier et al. 2001). The value of the preparation rests with the fact that it is amenable to both muscle tension and electrophysiological measurements and, as the main muscle involved in breathing (CitationVander et al. 2001), the preparation is routinely used to investigate responses of respiratory muscle (including impairment) to pharmacological test materials (e.g. muscle relaxant drugs used in surgery; CitationGibb and Marshall 1986, Citation1987, CitationBowman 1990, CitationFortier et al. 2001). The phrenic nerve hemi-diaphragm experiments conducted with sulfoxaflor demonstrated a consistent, concentration-dependent contracture of the fetal-type diaphragm muscle () and prolonged (7 min) application of sulfoxaflor caused a sustained muscle contracture (CitationRasoulpour et al. 2012).
Table 3. Dose–response and reversibility for MOA Key Event #3: Sustained agonism and sustained muscle contracture and associated death in neonatal offspring.
Sustained agonism via the muscle nAChR could potentially lead to contracture and dependent on dose, reduced responsiveness, or rigidity (CitationMurray et al. 2009). A simplified version of the possible underlying chain of events following sustained activation of the muscle nAChR by sulfoxaflor can be summarized in the following steps: (1) depolarisation of muscle, (2) release of Ca++ from sarcoplasmic reticulum to muscle filaments, (3) elevated Ca++ levels in muscle filaments promote contraction, (4) muscle Ca++/ATPase membrane pump mediates muscle relaxation by reducing Ca++ levels in sarcoplasm, (5) sustained contraction reduces available ATP, (6) reduced ATP compromises active pump and Ca++ levels rise, (7) normal relaxation of the muscle cannot occur, and (8) contracture may occur.
Neonatal offspring limb contracture
The end result of this chain of events of muscle contracture is consistent with the apical endpoints observed in rat fetuses at GD 21 C-section and in neonatal offspring shortly after birth (). The limb contracture abnormalities of forelimb flexure and hindlimb rotation observed in the guideline rat developmental toxicity and critical window MoA studies (), and the neonatal offspring death observed in the critical window MoA and reproduction studies (), directly demonstrate a dose-dependent increase in sustained muscle contracture with increasing internal dose. In the assessment table for this key event () and the summary table (), blood sulfoxaflor concentrations from the in vivo studies have been converted from microgram per gram plasma to micromolar, to bridge in vitro, ex vivo, and in vivo data. While quantitative responses across these study types are not equivalent, this conversion facilitates qualitative comparisons of these responses for biological dose–response concordance. Using this approach, ex vivo diaphragm muscle contracture is observed to occur at exposures which result in demonstrated in vivo developmental effects. Indirect evidence of the sustained agonism/sustained muscle contracture of the shoulder girdle was also evident with the skeletal finding of bent clavicle, a finding earlier discussed as dependent upon increased muscle contraction ().
Table 4. Neonatal offspring limb and clavicle abnormalities resulting from sulfoxaflor-induced sustained muscle contracture.
Table 5. Neonatal offspring death resulting from sulfoxaflor-induced sustained muscle contracture.
Table 6. Sulfoxaflor: Summary of dose–response and reversibility for MOA Key events related to rat muscle contracture and associated death in neonatal offspring.
Neonatal offspring death
The neonatal offspring death observed in the critical window, reproduction, and developmental neurotoxicity studies directly demonstrated a dose-dependent increase with increasing internal and/or applied dose (). The incidence of this apical end point largely paralleled that of the limb and clavicle findings supporting a single MoA for these effects. Treatment-related pup deaths occurred at high sulfoxaflor doses (400, 500, or 1000 ppm) in all six studies where this endpoint was evaluated. Three of these were regulatory guideline studies (OECD 421, OECD 416, and OECD 426) and three were MoA studies (Critical Window Studies 1 and 2, and Cross-foster Study). In these studies, all treatment-related deaths occurred by PND 4 with the majority occurring within the first 48 hours of life (87% by PND 2; ).
Table 7. Daily observations of pup death between PNDs 1 and 4. Data are expressed as a cumulative percent per study.
Biological plausibility, temporal association, and coherence of developmental response with key events
Biological plausibility and temporal association
The observed sulfoxaflor-induced developmental effects are entirely biologically consistent with the functional ontogeny of the fetal-type muscle nAChR in the rat. In rats, the muscle nAChR develops functional subunit expression at the NMJ between GD 15 and 17 (CitationKues et al. 1995) resulting in synchronized fetal limb movements (CitationRobinson and Smotherman 1988) and diaphragmatic responsiveness between GD 16 and 17 (CitationBennett and Pettigrew 1974), the latter being critical for the transition to extrauterine respiration. Functional expression of the γ (fetal-type) subunit continues through the first postnatal week and is largely complete by the end of the second postnatal week in limb and respiratory muscles (CitationMissias et al. 1996). Importantly, this muscle receptor subtype is highly expressed during late gestation in the distal limbs muscles and diaphragm, with impairment of diaphragmatic maintenance of respiration at birth implicated in neonatal death from sulfoxaflor exposure.
Early critical window studies that were conducted in rats demonstrated that the critical period of developmental susceptibility to sulfoxaflor-induced offspring limb-contracture abnormalities and reduced survival was between GD 16 and 21 with follow-up studies further narrowing this window to shortly before birth (GD 20–21; CitationRasoulpour et al. 2012). Furthermore, the cross-fostering study demonstrated that the neonatal offspring death requires prenatal exposure, but not postnatal sulfoxaflor exposure, indirectly providing support for the critical nature of proper fetal-type nAChR diaphragm function prior to birth (data not shown). Also supporting the biological plausibility of the hypothesized MoA is the observation that fetuses in the developmental toxicity study exhibited a hunched posture, consistent with generalized muscle contracture and had bent clavicles at the skeletal exam (CitationRasoulpour et al. 2012), a finding consistent with the fact that the clavicle (collar) bone is highly dependent upon regulated muscle function for proper growth during the late fetal period, and alterations in muscle function having been demonstrated to affect clavicle development (CitationPai 1965, CitationTran and Hall 1989).
The temporality of the neonatal offspring death was further assessed through a more precise examination of the timing of the deaths. In the reproduction screening (OECD 421) and two generation reproductive toxicity (OECD 416) regulatory guideline studies, live pups are counted within 24 hours of parturition (PND 1) and on Day 4 post-partum (PND 4). Litters with missing pups or pups found dead in the cage on the interim days were noted in the study file but are not routinely reported. In the OECD 426 and Critical Window MoA studies, live pups were counted daily. This more detailed evaluation shows that the majority (87%) of all sulfoxaflor-induced offspring deaths occurred within the first 48 hours of life (50 and 37% on PND 0–1 and 1–2, respectively; ). The high incidence of pup death in such a short period following birth (72–100% by PND 2 for dams exposed to 1000 ppm sulfoxaflor during the majority of gestation), along with the demonstration in the cross-foster study that pup death was solely due to gestational exposure and not to exposure to sulfoxaflor during lactation, provide substantial support for a specific MoA for this effect.
The biological plausibility for the timing of the neonatal death is supported through an understanding of extrauterine respiration in neonatal offspring, where the nature of pup death is consistent with neonatal breathing difficulties associated with diaphragm muscle contracture. Extrauterine respiration is particularly dependent upon respiratory muscle responsiveness, characterized by rhythmic coordinated contractions of the muscles (primarily the diaphragm). Sustained disruption of diaphragm muscle responsiveness, as demonstrated with sulfoxaflor in isolated phrenic nerve-hemidiaphragm preparations, would be expected to directly impair extrauterine respiration and lead to breathing difficulties and death.
As noted above, 50% of the observed pup deaths occur between PND 0 and 1, with 87 and 99% taking place by PND 2 and 3, respectively (). The observation that some pups survive past PND 1 is not surprising given lack of full agonist activity of sulfoxaflor at the fetal-type muscle nAChR (CitationRasoulpour et al. 2012) and the significant but incomplete reduction in muscle twitch tension following sulfoxaflor- induced diaphragm contracture. The pups that survive beyond the first day of life, but succumb between PNDs 1 and 4, could have less severe breathing difficulties and are lost due to the consequences of persistent breathing difficulties and lack of oxygen, which could trigger respiratory and cardiac failure. Given that maturation of neuromuscular transmission is ongoing in the newborn rat, it is plausible that respiratory function in pups is initially less affected but may deteriorate over time and they too succumb. It has been shown, for example, that respiratory insufficiency until PND 3 in the neonatal rat results in less force being generated in the diaphragm when stimulated by the phrenic nerve (CitationKass and Bazzy 2001). In addition to pups succumbing from respiratory insufficiency, their compromised status may result in their inability to obtain sufficient maternal care (e.g., suckling of milk) resulting in death from inanition. Evidence of their comprised status was indicated using clinical observations in affected litters of pups blue in coloration and having decreased activity. Pups were also observed as being cold to touch or without milk in their stomach. The timing of these observations was coincident with the timing of the early pup death (data not shown). Thus, it is biologically plausible that the affected pups die due to sulfoxaflor-induced reductions in diaphragm responsiveness either during the first day of life from direct effects of breathing difficulties or by PND 4 due to a combination of reduced respiratory function and inanition.
In summary, the relationship between the pup limb abnormalities and the early postnatal deaths has been clearly demonstrated. A high correlation exists between the incidence of neonatal deaths and the incidence of limb abnormalities (CitationRasoulpour et al. 2012), and together with the evidence of the same critical window for induction of these two effects and the same short period in which they are manifested postnatally, the evidence points to the biological plausibility of a specific MoA of sulfoxaflor in causing both neonatal death and limb abnormalities. The MoA operates prenatally, resulting in effects that are manifested postnatally within a day or so of the critical exposure period for their induction. Thus the occurrence data are compatible with a single MoA for both effects.
Coherence
Coherence refers to the relationship of the postulated MOA with substance-specific findings in a broader sense. This may take into consideration information on structural analogs, species differences in sensitivity and their relationship to the key events, and whether or not data on the substance is internally consistent in supporting the proposed MoA. The results from the studies with sulfoxaflor demonstrate clear coherence of the biological effects to the postulated MoA (). The finding that the primary effects of the reproduction and developmental studies can all be aligned with agonist effects on muscle is important. While neonatal offspring death can result from a variety of complicating factors, the demonstration of diaphragm contracture ex vivo in the same strain, species, and at exposure concentrations similar to those achieved in vivo, supports the coherence of this MoA. The coherence of negative effects across end points and studies is also important to examine. As discussed earlier, exposure to a biologically inactive metabolite of sulfoxaflor, X11719474, resulted in no agonism on the fetal-type muscle nAChR and no developmental effects at 5–10 times the effect concentration of sulfoxaflor (). Developmental toxicity and perinatal mortality studies have been conducted with sulfoxaflor in the New Zealand white rabbit and with both studies being negative for limb abnormalities or neonatal offspring death. In summary, these in vivo studies provide critical linkages of biological plausibility, temporality of association and, to the extent possible, coherence for this MoA.
Strength, consistency, and specificity of association of developmental response with key events
A dose-dependent response with a clear threshold was demonstrated within each key event across both applied and internal dose and this response was consistent across the key events, with no MoA-related effects at or below an applied dose of 100 ppm (three in vivo studies), thereby providing strong evidence of causality. With nine studies providing pertinent data, one of which being an in vitro and one being an ex vivo study, the association of effect was very robust (the seven in vivo studies had an R2 value of 0.91–0.93 across the dose-ranges tested; CitationRasoulpour et al. 2012). Specificity was demonstrated through ability of sulfoxaflor to completely, and dose-dependently, displace a high-affinity nAChR ligand from nAChR binding sites (). Also, our experiments confirm that compounds known to be negative for these developmental effects have little to no agonism on the fetal-type muscle nAChR. For example, a structurally related sulfoxaflor soil metabolite (X11719474) was previously demonstrated to produce no neonatal pup loss or developmental effects at dose levels 5–10 times higher than the sulfoxaflor effect levels. Imidacloprid, a neonicotinoid insecticide which has not been demonstrated to cause contracture-related limb abnormalities, also induced very little agonism on the fetal-type muscle nAChR (data not shown). Specific to sulfoxaflor, no agonism could be demonstrated at the adult-type muscle nAChR (even at the limit of solubility; ), consistent with a lack of contracture-related effects in adult rats in vivo at similar systemic sulfoxaflor levels. Specificity at the fetal-type muscle nAChR was further demonstrated by showing that sulfoxaflor-induced diaphragm contracture could be blocked by co-exposure with a nAChR-specific antagonist, tubocurarine, thereby ruling out sulfoxaflor action via a postreceptor mechanism (CitationRasoulpour et al. 2012).
While agonism and muscle contracture was sustained in the presence of sulfoxaflor, its receptor-mediated pharmacologic agonist action in live offspring was rapidly reversible following removal of the sulfoxaflor. This was demonstrated both in the ex vivo diaphragm contracture experiments and more importantly in vivo in live offspring with muscle contracture-related limb abnormalities ( and ; CitationRasoulpour et al. 2012). Lastly, the incidence of the apical end point of neonatal death largely paralleled that of the limb and clavicle findings regarding its dose–response relationship, consistency, and specificity across studies thereby supporting a single muscle contracture-mediated MoA for these effects.
Consideration of alternative modes of action
In the process of evaluating and conducting experiments aimed at elucidating and testing the proposed MoA for sulfoxaflor, a number of alternative MoAs were ruled out. These included consideration of sulfoxaflor-induced limb abnormalities and neonatal offspring deaths due to: agonism on other AChR types (adult-type muscle nAChR, neuronal nAChR, and muscarinic AChR), action downstream of the fetal-type muscle nAChR, antagonism on the fetal-type muscle nAChR, maternally mediated fetal immobilization, maternal neglect, congenital diaphragmatic hernia, inhibition of AChE, inhibition of angiotensin-converting enzyme (ACE), and deficiency or disruption of pulmonary surfactant ().
Table 8. Summary evaluation for other possible MoAs in the rat.
Agonism on other ACh receptor types (adult-type muscle nAChR, neuronal nAChRs, muscarinic AChR)
Adult-type muscle nAChRs
The plausibility of agonism of sulfoxaflor on other ACh receptor subtypes was addressed through a combination of examination of the published literature and laboratory experiments. This alternative MoA was directly addressed by examining the ability of sulfoxaflor to act as an agonist recombinant adult-type muscle nAChRs (α1(2), β1, δ, and ϵ) expressed in the previously described Xenopus oocyte system where up to limit of solubility of sulfoxaflor, no agonism was demonstrated. Additionally, in the in vivo studies that demonstrated offspring effects, no clinical signs were observed which would be consistent with an effect on the adult muscle nAChR, such as muscle fasciculations or tonic or clonic limb contractions. The lack of agonism and agonist-associated clinical observations in these studies provides solid evidence to rule out agonism on the adult-type muscle nAChR as a MoA for the effects of sulfoxaflor on the fetal limb contracture abnormalities and neonatal death.
Neuronal nAChRs
An alternative AChR to assess for plausibility as a MoA is the potential action of sulfoxaflor at neuronal nAChRs. Direct evaluations of sulfoxaflor agonism on individual neuronal nAChR subtypes have not been conducted because in the studies demonstrating developmental effects during sulfoxaflor exposure there have been no indications of neuronal nAChR-mediated clinical signs in the adults or offspring. A notable hallmark which follows neuronal nAChR activation is desensitization of the receptors resulting in observations of muscle weakness or flaccid paralysis (CitationGermiller et al. 1998). Of important distinction is that the sulfoxaflor-induced limb contractures consistently resulted in rigid, contracted limbs, an observation consistent with the sustained diaphragm contracture seen with sulfoxaflor in the isolated phrenic nerve-hemidiaphragm preparations without any evidence of desensitization. These findings further suggest that neuronal nAChRs were not involved in effects of sulfoxaflor in rats.
Fetal breathing movements are also known to be partially regulated by neuronal nAChRs in the brainstem. While respiratory rhythmogenesis is controlled primarily by neurons in the pre-Bötzinger complex through the neuromodulators (5-HT, substance P, catecholamines, high K+, and morphine), α4 and α7 containing neuronal nAChRs are present in the pre-Bötzinger complex and in motor nuclei innervating the respiratory muscles. The development of the respiratory rhythm generator must be well established and functionally robust by birth and fetal breathing movements are necessary for the proper maturation of the lungs with compounds which alter respiratory rhythm commonly resulting in lung hypoplasia (CitationDornan et al. 1984, CitationHarding 1995, CitationKobayashi et al. 2001). Notably, while prenatal nicotine exposure has been demonstrated to alter fetal lung development in rats (CitationMaritz and van Wyk 1997) and associated neuronal nAChR expression in primates (CitationSekhon et al. 1999), it paradoxically does not cause neonatal death during normoxic conditions in rats (CitationGeller 1959, CitationSobrian et al. 1995). In a histopathologic examination of GD 21 fetal rat lungs collected following sulfoxaflor maternal exposure no alterations were observed (data not shown). This suggests that neonatal offspring death of sulfoxaflor does not occur via a neuronal nAChR mechanism and is fundamentally different in its pharmacologic activity from nicotine, a molecule primarily active at neuronal nAChRs which causes neither neonatal death nor limb contracture abnormalities in rodents (CitationEugenín et al. 2008).
Muscarinic AChRs
Agonism on muscarinic AChRs (mAChRs) has also been explored as a possible alternative MoA. As opposed to nAChRs which are ion channel receptors and are found within striated muscle, muscarinic AChRs are found within smooth muscle and are G-protein coupled receptors of which there are five subtypes known with M1, M3, and M5 receptors having stimulatory and M2 and M4 receptors having inhibitory characteristics. The most plausible target by which a compound could cause neonatal offspring death would be directly via muscarinic activity at the mAChRs in the heart or lung. It has been previously proposed that fetal immobilization due to uterine constraint (CitationGordon 1998), which theoretically could result from contraction of maternal uterus, may indirectly result in limb contracture effects in fetuses and offspring. However, activity at these receptors would be accompanied by systemic clinical signs of mAChR activity and none of which have been observed in our studies. Also, gestational survival was unaffected (i.e., there were no pregnancy disruptions associated with uterine contraction), fetal hearts were grossly normal on GD 21 visceral examination (including internal structures), and as discussed previously, lungs were histologically normal.
Agonism downstream of the fetal-type muscle nAChR
It was considered that sulfoxaflor might exert its sustained muscle-contracture effects not directly at the nAChR, but rather downstream of the receptor. This MoA (in adults) has been demonstrated with the herbicide, cartap, which causes respiratory failure in adult rabbits (CitationLiao et al. 1998) and marked irreversible contracture of adult-type mouse phrenic-nerve diaphragm preparations (CitationLiao et al. 2000) in addition to its modulation the neuronal nAChR in rats (CitationNagata et al. 1997). Rather than acting at the muscle nAChR directly, the diaphragm contracture MoA of cartap has been demonstrated be via post-receptor induction of extracellular Caa2+ influx, release of internal Ca2+, and an inhibition of [3H]ryanodine binding to the Ca2+ release channel of sarcoplasmic reticulum (CitationLiao et al. 2000). In the phrenic-nerve hemidiaphragm experiments conducted with sulfoxaflor where sustained muscle contracture was demonstrated, a post-nAChR MoA would have been manifested by continued contraction in the presence of co-application with the nAChR antagonist tubocurarine. The sulfoxaflor-induced diaphragm contracture was completely eliminated with co-incubation with tubocurarine, thereby discounting a postreceptor MoA in the induction of the developmental effects of sulfoxaflor.
Fetal-type muscle nAChR inactivation
The most studied alternative MoA for decreased neonatal survival and neonatal limb abnormalities have come from studies in which the fetal-type muscle nAChR is either genetically absent or antagonized using various methods including: depolarizing and non-depolarizing blockade, autoimmune myasthenia gravis, or as previously discussed secondary desensitization following neuronal stimulation. Knockout of the gamma subunit from the muscle nAChR results in decreased prenatal and neonatal survival in gamma nAChR knockout mice (CitationTakahashi et al. 2002), the forelimbs are functional while the hindlimbs are not, and in humans an inactive gamma subunit results in severe limb contractures, pterygia, and increased intrauterine lethality (CitationMichalk et al. 2008).
While knockout of the gamma subunit is an extreme example of nAChR inactivity, nongenetic, pharmacologic antagonism of the fetal-type muscle nAChR also results in neonatal offspring death and limb abnormalities. A historical example of this includes the use of tubocurarine as a nondepolarizing neuromuscular blocking agent. Prenatal tubocurarine exposure during the last week of gestation in rats causes direct fetal paralysis capable of causing forelimb contracture, hindlimb rotation and death (CitationShoro 1977). While this may appear similar to the effects seen with sulfoxaflor there are several important distinctions. Firstly, known neuromuscular blocking agents do not readily cross the placenta (CitationEvans and Waud 1973). While this makes them particularly useful for pregnant women undergoing surgical operations, it is necessary to directly inject these agents into the fetus in order to exert these effects. Secondly, the limb contracture effects result in skeletal alterations in the limb cartilage, something that was not observed with sulfoxaflor in the prenatal developmental toxicity study. Finally, fetal paralysis additionally results in pulmonary hypoplasia, which has previously been mentioned to not occur following sulfoxaflor exposure.
Another historical example of agent-induced fetal paralysis resulting in neonatal death and limb contractures is plant alkaloid exposure in livestock. Examples include studies in cows, sheep, pigs, and goats following anabasine or conine exposure (CitationLee et al. 2006). Additionally, conine has been demonstrated to cause limb contractures in rabbits (CitationForsyth and Frank 1993). There are two important findings that distinguish these agents from that of sulfoxaflor. Ultrasound studies using a goat model demonstrate decreased or eliminated fetal activity when the mother goat is fed anabasine (CitationWeinzweig et al. 1999) that was associated with the observed muscle contractures, as well as cleft palate presumably resulting from inactive glossal muscle during palate closure. While the MoA of the plant alkaloids on the fetus is not known, it could be speculated that the previously noted sedation or biphasic stimulation-depression seen in adults may be operant in the fetus and responsible for the inhibition of fetal movement. There are currently no published data for this supposition. Sulfoxaflor exposure did not result in cleft palate but did result in decreased offspring activity, presumably an effect of respiratory difficulties associated with a sustained contracture of the diaphragm muscle and other skeletal muscles. The other distinguishing finding of the alkaloids is that a hallmark of plant alkaloid toxicity includes symptoms of acute cholinergic poisoning in the mother including fasiculations of muscle, clonic and tonic contractions of separate limbs, and convulsions followed by weakening and slowed heart rate, coma, and death (CitationBowman and Sanghvi 1963). As discussed earlier these symptoms are consistent with neuronal nAChR stimulation and subsequent depression, which are not observed in either adults or neonates in the developmental and reproductive toxicity studies with sulfoxaflor in which developmental effects were observed. The best-known plant alkaloid, nicotine, which causes increased prenatal death and no limb contractures in rodents, has a relatively low adult LD50 due to its central convulsant action (CitationSheveleva et al. 1983, Citation1984).
The last form of fetal-type muscle nAChR inactivation, which will be discussed is fetal paralysis, and limb abnormalities induced by maternally produced anti-AChR antibodies directed at functional fetal-type muscle AChRs. Blockade of ACh signaling at the fetal-type muscle nAChR causes multiple joint contractures that are associated with hypotonia, lung hypoplasia, and perinatal death (CitationJacobson et al. 1999, CitationPolizzi et al. 2000). As stated before, this MoA is not relevant to the findings in rats with sulfoxaflor as no hypotonia or lung hypoplasia was observed in the GD 21 fetal rat lung gross and histopathological analysis.
Maternal neglect
Maternal neglect is a possible MoA which could result in pup death, but not limb abnormalities, and could result from excessive maternal toxicity preventing the dam from caring for the pups. The loss of a large number of whole litters occurring within a short time period after birth, as seen at a dietary dose of 1000 ppm sulfoxaflor, is not a generalized feature of maternal toxicity, unless dams are very adversely affected by the toxicity and unable to care for their young. Clinical observations in affected sulfoxaflor litters across the six studies with pup deaths were examined to determine whether additional insights could be gained into the MoA. Although these findings are observational by nature, they can help in clarifying the nature of the physical condition of the dams proximate to the timing of the pup deaths. Clinical observations were noted in dams in only two of the six studies and in general these were minor findings (e.g., hairloss), of low incidence (pale skin color in 1 of 12 dams), and/or were without a dose-dependent response (data not shown). While some maternal toxicity at high sulfoxaflor doses was evident (i.e., liver toxicity), the clinical observations on the lactating dams clearly show that the dams were not severely affected in these studies. Maternal toxicity-associated neglect is therefore very unlikely to account for the pup deaths in the studies on sulfoxaflor.
Structural malformation induced pup death (e.g., Nitrofen)
The apical endpoint of neonatal pup death is a key feature of nitrofen exposure in rodents. Neonatal offspring from dams treated with the herbicide nitrofen exhibit symptoms of labored breathing, cyanosis, and litter survival can be reduced to zero within the first 48 h of life (CitationCostlow and Manson 1981). Nitrofen exposure does not result in any other external observations, particularly no limb abnormalities or hunched appearance as seen with sulfoxaflor. Probably the most distinctive difference between the nitrofen MoA and that of sulfoxaflor is the observation of structural malformations in nitrofen-exposed offspring, including cardiac malformations and congenital diaphragmatic hernia, both deemed incompatible with life (CitationCostlow and Manson 1981), neither of which occur with sulfoxaflor treatment. The peak period of susceptibility for nitrofen-induced pup deaths is GD 9–12 (CitationGreer 2013), a treatment period in which no deaths were observed with sulfoxaflor. The critical distinctions mentioned above between the nitrofen MoA and observations in sulfoxaflor-exposed pups make it clear that this MoA is not operant with sulfoxaflor.
AChE inhibition
Inhibition of AChE during pregnancy results in cholinergic signs of toxicity, as previously discussed, and ultimately results in maternal death (CitationFarag et al. 2006). At sublethal exposure concentrations in rats cholinergic signs of AChE toxicity are generally observed as clinical signs in the absence of external, visceral, or skeletal abnormalities in fetuses, and thus do not fit the profile observed following sulfoxaflor exposure.
ACE inhibition
ACE inhibition presents another MoA that has been demonstrated to result in fetal limb contractures when exposure occurs during fetal development in humans (CitationButtar 1997). The limb contractures which occur are secondary to renal failure associated oligohydramniosis. Other secondary fetal anomalies including: potentially fatal hypotension, anuria, craniofacial deformities, and hypoplastic lung development. In a few cases, postnatal persistence of a patent ductus arteriosus has also been linked to intrauterine exposure to ACE inhibitors (CitationBarr 1994). In contrast to humans, rodents are relatively resistant to the teratogenic effects of ACE inhibitors and high doses of ACE inhibitors typically result only in fetal growth retardation and occasionally increased pup death. Based upon the lack of similarity in neonatal abnormalities and the relative resistance of rodents to ACE inhibition-induced limb contractures this alternative MoA was considered not relevant to the findings with sulfoxaflor and was not considered further.
Pulmonary surfactant insufficiency
Pulmonary surfactants form stable films at the air–water interface of the lung and maintain the low pulmonary surface tension for long intervals. This keeps fluid out of the air space, maintains optimal lung compliance, and lowers the work required to breathe (CitationTaeusch et al. 2005). Insufficiency of pulmonary surfactant levels is a hallmark of acute respiratory distress syndrome (RDS) in infants and treatment with surfactant is an important treatment modality to prevent RDS. Synthesis of pulmonary surfactants is initiated within the first 48 hours of neonatal life in rodents and peaks by PND 5 (CitationGross and Narine 1989), timing corresponding to the observed neonatal offspring death with sulfoxaflor.
Compounds with surfactant physical–chemical properties, such as perfluronooctane sulfonate (PFOS), have been shown to produce a high incidence of neonatal offspring death (CitationLau et al. 2003), interact with pulmonary surfactant components (CitationXie et al. 2007), and partition disproportionately into the lung relative to the maternal blood (CitationBorg et al. 2010). Sulfoxaflor does not have physical–chemical properties similar to surfactants (http://www.chemspider.com/Chemical-Structure.17626728.html), does not partition into the lungs at high levels (data not shown), did not have any indications of producing pulmonary edema in GD 21 fetus examined histopathologically in the prenatal developmental toxicity study (data not shown), and thus does not fit a profile that would be consistent with a pulmonary surfactant insufficiency MoA.
Conclusion of consideration of alternative MoAs
Following consideration of the presented alternative MoAs it is concluded that there is sufficient evidence to exclude these as plausible alternative MoAs for the observed neonatal offspring limb abnormalities and death.
A summary evaluation for the considered alternative MoAs is presented in ().
Uncertainties, inconsistencies, and data gaps
Uncertainties
The limb and shoulder girdle contracture effects have been demonstrated to be reversible in live offspring. However, experimental amelioration or prevention of the effect has not been conducted to date.
Inconsistencies
In the two-generation reproductive toxicity study, neonatal offspring limb contractures were not observed at doses which caused decreased pups survival. In addition, early studies (one-generation reproductive toxicity, dietary reproductive toxicity cross-fostering) did not examine this parameter.
Data gaps
The limb and shoulder girdle contracture effects have been demonstrated to be reversible in live offspring. However, experimental amelioriation or prevention of the effect has not been conducted to date. Direct assessment of fetal-type nAChR inhibition or neuronal nAChR agonism by sulfoxaflor could be considered a data gap. Neither of these assessments were conducted as they are inconsistent with the repeatable and robust observations in neonatal offspring for developmental effects of sulfoxaflor.
Assessment of postulated MoA
The data for sulfoxaflor are judged with a high degree of confidence to adequately explain the induction of neonatal offspring limb contractures and death following sustained pharmacologic agonism on the fetal-type muscle nAChR by dietary sulfoxaflor exposure at the end of gestation in rats. Based on the MoA analysis utilizing the Bradford Hill criteria for causality, there is a high degree of confidence that the observed sulfoxaflor-induced muscle contracture and associated death in neonatal offspring in rats occur via a single MoA through the following key events: (1) binding and (2) agonism on the fetal-type muscle nAChR by sulfoxaflor, thereby resulting in (3) sustained agonism and sustained muscle contracture in the fetus and neonatal pup. This sustained muscle contracture results in limb contractures, bent clavicles, and abnormal neonatal respiration after birth resulting in reductions in neonatal survival.
This novel MoA analysis demonstrates that the proposed MoA is plausible and has strong consistency, dose–responsiveness, and specificity across study types and dose ranges. The critical period of induced effects of sulfoxaflor at the rat fetal-type muscle nAChR, and an absence of the effects in animals after PND 4 are consistent with this ontogeny/ maturational transitioning of nAChR and spatial expression pattern which correlate with the timing and location of limb movement onset and fetal respiratory practice.
Human applicability of the proposed MoA
Question 1. Is the weight of evidence sufficient to establish the MoA in animals?
Yes. Based on this MoA analysis utilizing the Bradford Hill criteria for causality, there is a high level of confidence that the observed sulfoxaflor-induced muscle contracture and associated death in neonatal offspring in rats occur via a singular MoA through the following key events: (1) binding (2) agonism on the fetal-type muscle nAChR by sulfoxaflor, thereby resulting in and (3) sustained agonism and sustained muscle contracture in the fetus and neonatal pup. This sustained muscle contracture results in limb contractures, bent clavicles, and abnormal neonatal respiration after birth resulting in reductions in neonatal survival.
This novel MoA analysis demonstrates that the described MoA is plausible and has strong consistency, dose– responsiveness, and specificity across study types and dose ranges. The critical period of induced effects of sulfoxaflor at the rat fetal-type muscle nAChR, and an absence of the effects in animals after PND 4, are consistent with this ontogeny/maturational transitioning of nAChR and spatial expression pattern which correlate with the timing and location of limb movement onset and fetal respiratory practice.
Question 2. Can human relevance of the MoA be reasonably excluded based on fundamental qualitative differences in key events between experimental animals and humans?
Yes. This MoA is not relevant to humans based upon data demonstrating fundamental qualitative differences in the agonism of sulfoxaflor at the rat or human fetal-type muscle nAChR. Specifically, binding, but no agonism, was evident with sulfoxaflor at the human fetal-type or human adult- type muscle nAChR (, CitationRasoulpour et al. 2012). Both muscle receptor types were examined as the transition from the fetal-type to adult-type human muscle nAChR occurs prenatally (CitationHesselmans et al. 1993). The species-specificity of the effects in the rat is further supported by the finding that although sulfoxaflor binds to the fetal rabbit muscle nAChR (data not shown) it does not induce any developmental effects in this species despite similar systemic exposure.
Table 9. Sulfoxaflor: Human dose–response data for MoA Key events one and two related to muscle contracture and associated death in neonatal offspring.
Several precedents exist for species-selective agonist activity of nAChR ligands. Indeed, a single amino acid difference in the α3 subunit of rat and chick can account for the selective agonist effect of nicotine on rat and chick α3β2 nAChRs (CitationHussy et al. 1994). Similarly TMAQ, a nicotinic agonist that is selective for neuronal nAChRs containing a β4 subunit (CitationYoung et al. 2007), binds to both human and rat α3β4 nAChRs but acts only as an agonist on human α3β4 nAChRs (CitationYoung et al. 2007). The selective agonist activity of TMAQ for human α3β4 nAChRs can be explained by two amino acids differences between the human and rat b4 subunit (CitationYoung et al. 2007). Given that one or two amino acid differences can confer species-selective agonist activity for nicotinic ligands, it seems entirely plausible that the differences in agonist activity of sulfoxaflor are explainable by differences in the amino acid sequence of the rat and human nAChR γ subunits. Comparison of the amino acid sequence of the rat and human γ subunit revealed that although the two subunits are similar (approximately 90% identical), they contain 53 amino acid differences (CitationRasoulpour et al. 2012). The γ and ϵ subunits show even greater sequence differences than the human and rat γ subunit (even from the same species), where these subunits share only about 50% identity in amino acid sequence in the rat (data not shown).
In conclusion, while sulfoxaflor demonstrates both clear binding and agonism to the rat fetal-type muscle nAChR, sulfoxaflor binds to, but does not induce any agonism to, the human fetal- or adult-type muscle nAChR. Furthermore, these findings would be expected to represent the human population as there are no known polymorphisms in the subunits which compose human muscle nAChRs. Without agonism, there can be no sustained agonism or contracture in humans. As sustained agonism resulting in contracture is the MoA in rats, the MoA cannot occur in humans.
Question 3. Can human relevance of the MoA be reasonably excluded based on quantitative differences in either kinetic or dynamic factors between experimental animals and humans?
As the human relevance of the experimental animal MoA was reasonably excluded on the basis of qualitative differences in key events (Question 2), a quantitative assessment of kinetic or dynamic factors is not necessary.
Conclusion: statement of confidence, analysis, and implications
Statement of confidence in the evaluation
This HRF evaluation for sulfoxaflor-induced fetal-type muscle contracture abnormalities and associated neonatal offspring death in rats follows the guideline established for this process (CitationBoobis et al. 2006, Citation2008, CitationMeek et al. 2003, CitationSeed et al. 2005). The extensive toxicological database for sulfoxaflor, including several focused in vitro, ex vivo, and in vivo MoA studies in rats are high-quality studies that provide the necessary data to determine the fetal-type muscle nAChR-mediated MoA for sulfoxaflor-induced muscle contracture abnormalities and neonatal offspring death. The key events for sulfoxaflor show clear, thresholded, dose–responsive alterations and provide informative, temporal-specific characterization of sulfoxaflor-induced developmental effects. Other possible MoAs for the observed fetal abnormalities and neonatal offspring death in rats have been evaluated with respect to sulfoxaflor and have been dismissed because they lack plausibility and coherence.
Implications for risk assessment
The concordance analysis points out clear differences for a muscle nAChR-mediated contracture MoA in rodents as compared to humans (). There is convincing evidence that the MoA for sulfoxaflor-induced fetal-type muscle contracture abnormalities and associated neonatal offspring death in rats poses no risk to humans based upon fundamental qualitative differences in sulfoxaflor agonism on the rat versus the human muscle nAChR where no agonism occurs at the human fetal-type or adult-type, muscle nAChR.
Table 10. Concordance of Key events for induction of skeletal muscle contracture and associated death in neonatal offspring in rats, rabbits, and humans.
Acknowledgments
The authors thank the Developmental and Reproductive Toxicology (DART) study team for all of their hard work in generating these data. In particular, thanks to Carol Zablotny, Valerie Marshall, Amanda Andrus, and Keith Brooks for lending their expertise in conducting several DART studies, Dr Shakil Saghir for lending his toxicokinetic expertise, and Lisa McFadden for statistical assistance. The authors also thank Drs. Neil Millar and Alasdair Gibb at the University of College London for their diligent effort in generating data for this MoA program.
Declaration of interest
The authors are employed by The Dow Chemical Company, the developer and producer of sulfoxaflor. The authors have sole responsibility for the writing and content of the paper.
References
- Arias HR. (2000). Localization of agonist and competitive antagonist binding sites on nicotinic acetylcholine receptors. Neurochem Int, 36, 595–645.
- Barr M Jr. (1994). Teratogen update: angiotensin-converting enzyme inhibitors. Teratology, 50, 399–409.
- Bennett MR, Pettigrew AG. (1974). The formation of synapses in striated muscle during development. J Physiol, 241, 515–45.
- Boobis AR, Cohen SM, Dellarco V, McGregor D, Meek ME, Vickers C, et al. (2006). IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol, 36, 781–92.
- Boobis AR, Doe JE, Heinrich-Hirsch B, Meek ME, Munn S, Ruchirawat M, et al. (2008). IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit Rev Toxicol, 38, 87–96.
- Borg D, Bogdanska J, Sundström M, Nobel S, Håkansson H, Bergman Å, et al. (2010). Tissue distribution of (35)S-labelled perfluorooctane sulfonate (PFOS) in C57Bl/6 mice following late gestational exposure. Reprod Toxicol, 30, 558–65.
- Bowman W. (1990). Pharmacology of Neuromuscular Function. 2nd ed. London: Wright.
- Bowman WC, Sanghvi IS. (1963). Pharmacological actions of hemlock (Conium maculatum) alkaloids. J Pharm Pharmacol, 15, 1–25.
- Bülbring E. (1946). Observations on the isolated phrenic nerve diaphragm preparation of the rat. Br J Pharmacol Chemother, 1, 38–61.
- Buttar HS. (1997). An overview of the influence of ACE inhibitors on fetal-placental circulation and perinatal development. Mol Cell Biochem, 176, 61–71.
- Cooper JC, Gutbrod O, Witzemann V, Methfessel C. (1996). Pharmacology of the nicotinic acetylcholine receptor from fetal rat muscle expressed in Xenopus oocytes. Eur J Pharmacol, 309, 287–98.
- Corley RA, Meek ME, Carney EW. (2005). Mode of action: oxalate crystal-induced renal tubule degeneration and glycolic acid-induced dysmorphogenesis—renal and developmental effects of ethylene glycol. Crit Rev Toxicol, 35, 691–702.
- Costlow RD, Manson JM. (1981). The heart and diaphragm: target organs in the neonatal death induced by nitrofen (2,4-dichlorophenyl-p-nitrophenyl ether). Toxicology, 20, 209–27.
- Dascal N. (1987). The use of Xenopus oocytes for the study of ion channels. CRC Crit Rev Biochem, 22, 317–87.
- Dornan JC, Ritchie JW, Meban C. (1984). Fetal breathing movements and lung maturation in the congenitally abnormal human fetus. J Dev Physiol, 6, 367–75.
- Eugenín J, Otárola M, Bravo E, Coddou C, Cerpa V, Reyes-Parada M, et al. (2008). Prenatal to early postnatal nicotine exposure impairs central chemoreception and modifies breathing pattern in mouse neonates: a probable link to sudden infant death syndrome. J Neurosci, 28, 13907–17.
- Evans CA, Waud DR. (1973). Do maternally administered neuromuscular blocking agents interfere with fetal neuromuscular transmission?Anesth Analg, 52, 548–52.
- Farag AT, Karkour TA, El Okazy A. (2006). Developmental toxicity of orally administered technical dimethoate in rats. Birth Defects Res Part B Dev Reprod Toxicol, 77, 40–6.
- Forsyth CS, Frank AA. (1993). Evaluation of developmental toxicity of coniine to rats and rabbits. Teratology, 48, 59–64.
- Forsyth CS, Speth RC, Wecker L, Galey FD, Frank AA. (1996). Comparison of nicotinic receptor binding and biotransformation of coniine in the rat and chick. Toxicol Lett, 89, 175–83.
- Fortier LP, Robitaille R, Donati F. (2001). Increased sensitivity to depolarization and nondepolarizing neuromuscular blocking agents in young rat hemidiaphragms. Anesthesiology, 95, 478–84.
- Foster PM. (2005). Mode of action: impaired fetal leydig cell function—effects on male reproductive development produced by certain phthalate esters. Crit Rev Toxicol, 35, 713–9.
- Geller LM. (1959). Failure of nicotine to affect development of offspring when administered to pregnant rats. Science, 129, 212–4.
- Germiller JA, Lerner AL, Pacifico RJ, Loder RT, Hensinger RN. (1998). Muscle and tendon size relationships in a paralyzed chick embryo model of clubfoot. J Pediatr Orthop, 18, 314–8.
- Gibb AJ, Marshall IG. (1984). Pre-and post-junctional effects of tubocurarine and other nicotinic antagonists during repetitive stimulation in the rat. J Physiol, 351, 275–97.
- Gibb AJ, Marshall IG. (1986). Nicotinic antagonists produce differing amounts of tetanic fade in the isolated diaphragm of the rat. Br J Pharmacol, 89, 619–24.
- Gibb AJ, Marshall IG. (1987). Examination of the mechanisms involved in tetanic fade produced by vecuronium and related analogues in the rat diaphragm. Br J Pharmacol, 90, 511–21.
- Gordon N. (1998). Arthrogryposis multiplex congenita. Brain Dev, 20, 507–11.
- Greer JJ. (2013). Current concepts on the pathogenesis and etiology of congenital diaphragmatic hernia. Resp Physiol Neurobi, 189, 232–40.
- Gross NJ, Narine KR. (1989). Surfactant proteins a and d and pulmonary host defense. J Appl Physiol, 67, 414–21.
- Gu Y, Hall ZW. (1988). Immunological evidence for a change in subunits of the acetylcholine receptor in developing and denervated rat muscle. Neuron, 1, 117–25.
- Harding R. (1995). Sustained alterations in postnatal respiratory function following sub-optimal intrauterine conditions. Reprod Fertil Develop, 7, 431–41.
- Hesselmans LF, Jennekens FG, Van den Oord CJ, Veldman H, Vincent A. (1993). Development of innervation of skeletal muscle fibers in man: relation to acetylcholine receptors. Anat Rec, 236, 553–62.
- Hill AB. (1965). The Environment and disease: association or causation?Proc R Soc Med, 58, 295–300.
- Hoffman BB, Taylor P. (2001). Neurotransmission. The autonomic and somatic motor nervous systems. In: Hardmam JG, Limbird LE, Gilman AG, Eds. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 10th ed. New York: McGraw-Hill.
- Holson JF, Stump DG, Pearce LB, Watson RE, DeSesso JM. (2005). Mode of action: yolk sac poisoning and impeded histiotrophic nutrition–HBOC-related congenital malformations. Crit Rev Toxicol, 35, 739–45.
- Hubbard JI, Wilson DF. (1973). Neuromuscular transmission in a mammalian preparation in the absence of blocking drugs and the effect of D-tubocurarine. J Physiol, 228, 307–25.
- Hussy N, Ballivet M, Bertrand D. (1994). Agonist and antagonist effects of nicotine on chick neuronal nicotinic receptors are defined by α and β subunits. J Neurophysiol, 72, 1317–26.
- Jacobson L, Polizzi A, Morriss-Kay G, Vincent A. (1999). Plasma from human mothers of fetuses with severe arthrogryposis multiplex congenita causes deformities in mice. J Clin Invest.103, 1031–8.
- Kass LJ, Bazzy AR. (2001). Chronic hypoxia modulates diaphragm function in the developing rat. J Appl Physiol, 90, 2325–9.
- Kavlock R, Cummings A. (2005). Mode of action: reduction of testosterone availability—molinate-induced inhibition of spermatogenesis. Crit Rev Toxicol, 35, 685–90.
- Kobayashi K, Lemke RP, Greer JJ. (2001). Ultrasound measurements of fetal breathing movements in the rat. J Appl Physiol, 91, 316–20.
- Kues WA, Sakmann B, Witzemann V. (1995). Differential expression patterns of five acetylcholine receptor subunit genes in rat muscle during development. Eur J Neurosci, 7, 1376–85.
- Lau C, Thibodeaux JR, Hanson RG, Rogers JM, Grey BE, Stanton ME, et al. (2003). Exposure to perfluorooctane sulfonate during pregnancy in rat and mouse. II: postnatal evaluation. Toxicol Sci.74, 382–392.
- LeBaron MJ, Gollapudi BB, Terry C, Billington R, Rasoulpour RJ. (2014). Human relevance framework for rodent liver tumors induced by the insecticide sulfoxaflor. Crit Rev Toxicol, 44, 15–24.
- Lee ST, Wildeboer K, Panter KE, Kem, WR, Gardner DR, Molyneux RJ, et al. (2006). Relative toxicities and neuromuscular nicotinic receptor agonistic potencies of anabasine enantiomers and anabaseine. Neurotoxicol Teratol, 28, 220–8.
- Liao JW, Kang JJ, Liu SH, Jeng CR, Cheng YW, Hu CM, et al. (2000). Effects of cartap on isolated mouse phrenic nerve diaphragm and its related mechanism. Toxicol Sci, 55, 453–9.
- Liao JW, Tsai SF, Lu SY, Liu SH, Kang JJ, Cheng YW, et al. (1998). The lethal effect of cartap via eye toxicity study in rabbits. J Soc Toxicol, 23, 398.
- Liley AW, North KA. (1953). An electrical investigation of effects of repetitive stimulation on mammalian neuromuscular junction. J Neurophysiol, 16, 509–27.
- Maritz GS, van Wyk G. (1997). Influence of maternal nicotine exposure on neonatal rat lung structure: protective effect of ascorbic acid. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol, 117, 159–65.
- Meek ME, Bucher JR, Cohen SM, DeMarco V, Hill RN, Lehman-McKeeman LD, et al. (2003). A framework for human relevance analysis of information on carcinogenic modes of action. Crit Rev Toxicol, 33, 591–654.
- Michalk A, Stricker S, Becker J, Rupps R, Pantzar T, Miertus J, et al. (2008). Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am J Hum Genet, 82, 464–76.
- Millar NS, Denholm I. (2007). Nicotinic acetylcholine receptors: targets for commercially important insecticides. Invert Neurosci, 7, 53–66.
- Millar NS, Gotti C. (2009). Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology, 56, 237–246.
- Miller-Keane O’Toole, MT. (2005). Miller-Keane Encyclopedia and Dictionary of Medicine, Nursing, and Allied Health. 7th ed. Philadelphia: W.B. Saunders.
- Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, et al. (1986). Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nature, 313, 364–9.
- Missias AC, Chu GC, Klocke BJ, Sanes JR, Merlie JP. (1996). Maturation of the acetylcholine receptor in skeletal muscle: regulation of the AChR γ-to-ϵ switch. Dev Biol, 179, 223–238.
- Murray RK, Bender DA, Botham KM, Kennelly PJ, Rodwell VW, Weil PA. (2009). Harper's Illustrated Biochemistry. Lange Medical Books, New York: McGraw-Hill.
- Nagata K, Iwanaga Y, Shono T, Narahash T. (1997). Modulation of the neuronal nicotinic acetylcholine receptor channel by imidacloprid and cartap. Pest Biochem Physiol, 59, 119–28.
- Pai AC. (1965). Developmental genetics of a lethal mutation, muscular dysgenesis (Mdg), in the mouse. I. genetic analysis and gross morphology. Dev Biol, 11, 82–92.
- Polizzi A, Huson SM, Vincent A. (2000). Teratogen update: maternal myasthenia gravis as a cause of congenital arthrogryposis. Teratology, 62, 332–41.
- Rasoulpour RJ, Ellis-Hutchings RG, Terry C, Millar NS, Zablotny CL, Gibb A, et al. (2012). A novel mode-of-action mediated by the fetal muscle nicotinic acetylcholine receptor resulting in developmental toxicity in rats. Toxicol Sci, 127, 522–534.
- Rasoulpour RJ, Terry C, LeBaron MJ, Stebbins K, Ellis-Hutchings RG, Billington R. (2014). Mode-of-action and human relevance framework analysis for rat Leydig cell tumors associated with Sulfoxaflor. Crit Rev Toxicol, 44, 25–44.
- Robinson SR, Smotherman WP. (1988). Behavior of the Fetus. The Telford Press: Caldwell, New Jersey.
- Sattelle DB. (1980). Acetylcholine receptors of insects. Adv Insect Physiol, 15, 215–315.
- Seed J, Carney EW, Corley RA, Crofton KM, DeSesso JM, Foster PM, et al. (2005). Overview: using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit Rev Toxicol, 35, 664–72.
- Sekhon HS, Jia Y, Raab R, Kuryatov A, Pankow JF, Whitsett JA, et al. (1999). Prenatal nicotine increases pulmonary alpha7 nicotinic receptor expression and alters fetal lung development in monkeys. J Clin Invest, 103, 637–47.
- Sheveleva GA, Sheina NI, Silant'eva IV. (1984). Postnatal development of rat progeny after antenatal nicotine exposure. Farmakol Toksikol, 47, 85–9.
- Sheveleva GA, Silant'ev IV, Sheina NI. (1983). [Effect of nicotine on embryogenesis and fetal development]. Akush Ginekol (Mosk), 10, 56–7.
- Shoro AA. (1977). Intra-uterine growth retardation and limb deformities produced by neuromuscular blocking agents in the rat fetus. J Anat, 123, 341–50.
- Sobrian SK, Ali SF, Slikker W Jr, Holson RR. (1995). Interactive effects of prenatal cocaine and nicotine exposure on maternal toxicity, postnatal development and behavior in the rat. Mol Neurobiol, 11, 121–43.
- Taeusch HW, Ballard RA, Gleason CA, Avery ME. (2005). Avery's Diseases of the Newborn. Philadelphia: Elsevier Health Sciences
- Takahashi M, Kubo T, Mizoguchi A, Carlson CG, Endo K, Ohnishi K. (2002). Spontaneous muscle action potentials fail to develop without fetal-type acetylcholine receptors. EMBO Rep, 3, 674–81.
- Terry C, Rasoulpour RJ, Saghir S, Marty S, Gollapudi BB, Billington R. (2014). Application of a Novel Integrated Toxicity Testing Strategy Incorporating “3R” Principles of Animal Research to Evaluate the Safety of a New Agrochemical, Sulfoxaflor. Crit Rev Tox 44, 1–14.
- Tran S, Hall BK. (1989). Growth of the clavicle and development of clavicular secondary cartilage in the embryonic mouse. Acta Anat (Basel), 135, 200–7.
- Vander A, Sherman J, Luciano D. (2001). Human Physiology: The Mechanisms of Body Function. London: McGraw-Hill.
- Wareham AC, Morton RH, Meakin GH. (1994). Low quantal content of the endplate potential reduces safety factor for neuromuscular transmission in the diaphragm of the newborn rat. Br J Anaesth, 72, 205–9.
- Weinzweig J, Panter KE, Pantaloni M, Spangenberger A, Harper JS, Lui F, et al. (1999). The fetal cleft palate: I. Characterization of a congenital model. Plast Reconstr Surg, 103, 419–28.
- Witzemann V. (2006). Development of the neuromuscular junction. Cell Tissue Res, 326, 263–71.
- Xie W, Kania-Korwel I, Bummer PM, Lehmler HJ. (2007). Effects of potassium perfluorooctanesulfonate, perfluorooctanoate, and octanesulfonate on the phase transition of dipalmitoylphostatidylcholine (DPPC) bilayers. Biochim Biophys Acta.1768, 1299–308.
- Young GT, Broad LM, Zwart R, Astles PC, Bodkin M, Sher E, Millar NS. (2007). Species selectivity of a nicotinic acetylcholine receptor agonist is conferred by two adjacent extracellular β4 amino acids that are implicated in the coupling of binding to channel gating. Mol Pharmacol, 71, 389–397.